
EMT in Breast Carcinoma—A Review


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Impact of the EMT on Breast Cancer
EMT in the Progression to Metastatic Disease
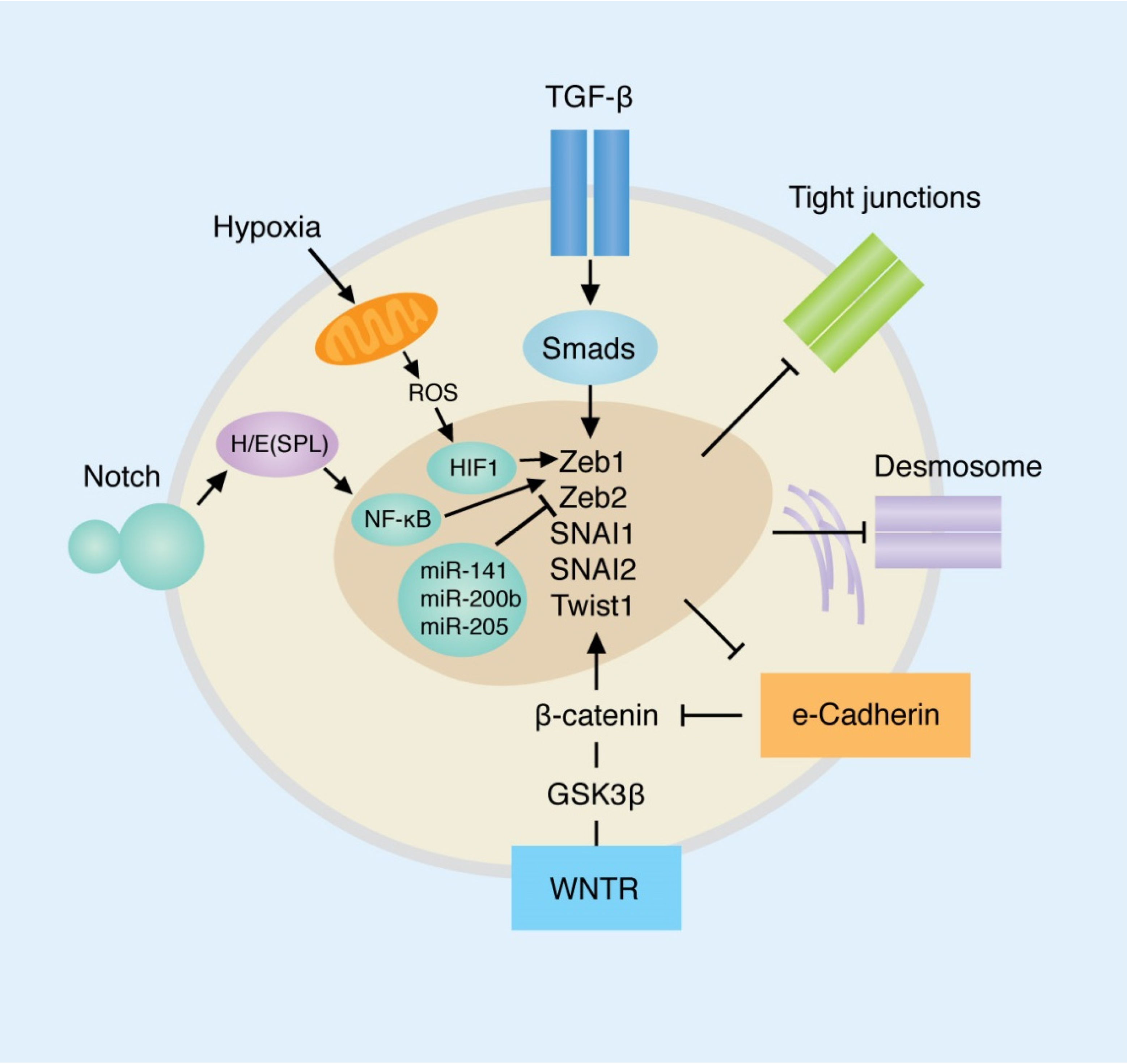
3. Primary Pathways Involved in EMT and Their Roles in Breast Cancer
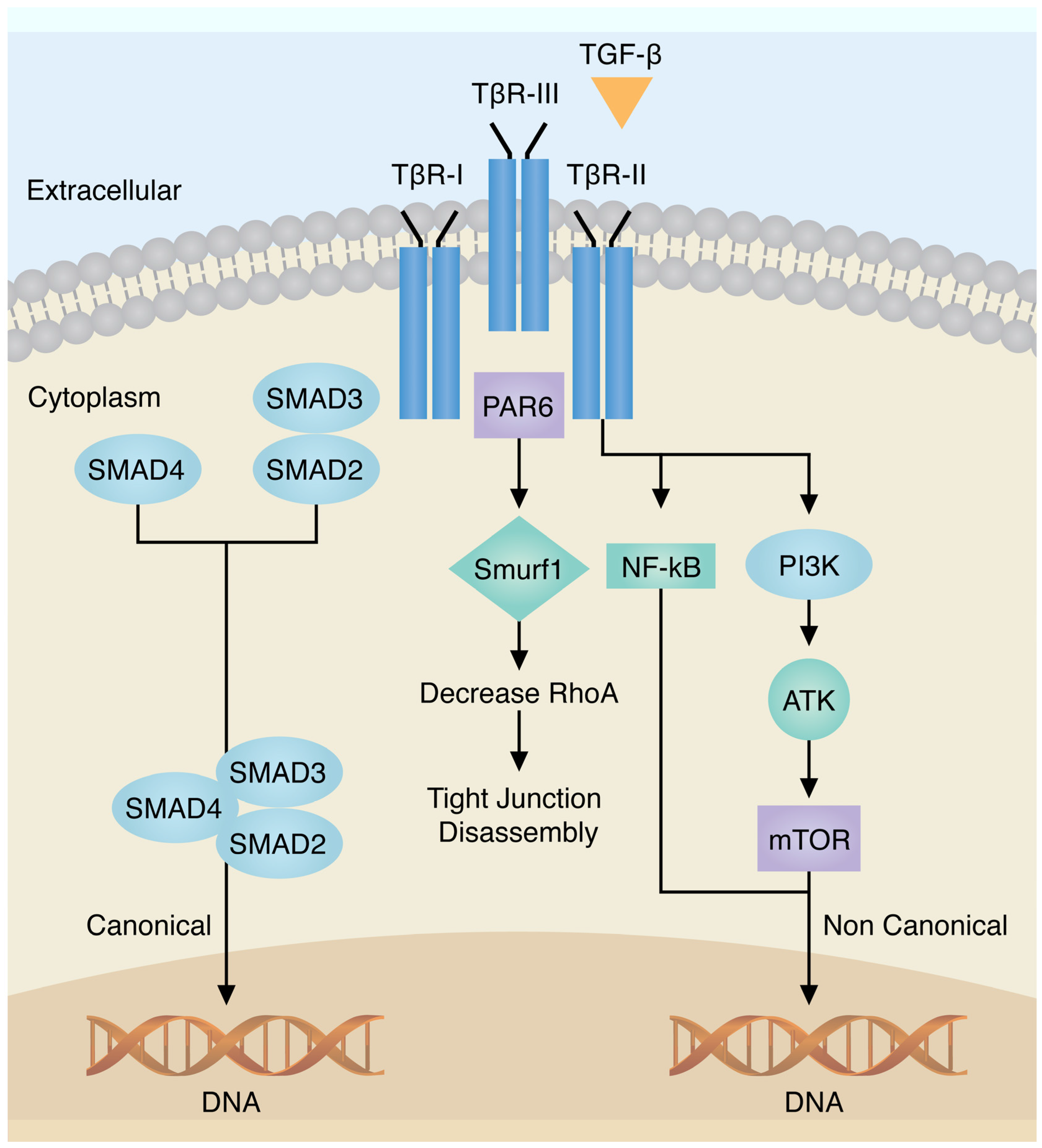
3.1. Transforming Growth Factor-β
3.2. E-Cadherin
3.2.1. E-Cadherin Loss in EMT Leads to Loss of Cell-Cell Adhesion
3.2.2. Migration and Invasion related to E-cadherin loss and changes in Metalloproteinases
3.3. WNT/β-Catenin Pathway
3.4. Notch
3.5. Hypoxia
3.6. Tumor Necrosis Factor-Alpha (TNF-α)
3.7. MicroRNAs
3.8. Cancer Stem Cell and EMT in Breast Cancer
4. EMT Clinical Behavior, Tumor Invasion and Metastasis
5. Potential Prognostic and Predictive Value of EMT in Breast Cancer
6. EMT Targeted Therapy
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Prat, A.; Perou, C.M. Desconstructing the molecular portraits of breast cancer. Mol. Oncol. 2011, 5, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.H.; Herschkowitz, J.I.; Komurov, K.; Zhou, A.Y.; Gupta, S.; Yang, J.; Hartwell, K.; Onder, T.T.; Gupta, P.B.; Evans, K.W.; et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc. Natl. Acad. Sci. USA 2010, 107, 15449–15454. [Google Scholar] [CrossRef] [PubMed]
- Micalizzi, D.; Ford, H. Epithelial to mesenchymal transition in development of cancer. Future Oncol. 2009, 8, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Scimeca, M.; Antonacci, C.; Colombo, D.; Bonfiglio, R.; Buonomo, O.C.; Bonanno, E. Emerging prognostic markers related to mesenchymal characteristics of poorly differentiated breast cancers. Tumor Biol. 2016, 37, 5427–5435. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. 2009, 9, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.A.; Parvani, J.G.; Schiemann, W.P. The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-β in normal and malignant mammary epithelial cells. J. Mammary Gland Biol. Neoplasia 2010, 15, 169–190. [Google Scholar] [CrossRef] [PubMed]
- Sarrió, D.; Rodriguez-Pinilla, S.M.; Hardisson, D.; Cano, A.; Moreno-Bueno, G.; Palacios, J. Epithelial-Mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008, 68, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Lien, H.C.; Hsiao, Y.H.; Lin, Y.S.; Yao, Y.T.; Juan, H.F.; Kuo, W.H.; Hung, M.C.; Chang, K.J.; Hsieh, F.J. Molecular signatures of metaplastic carcinoma of the breast by large-scale transcriptional profiling: Identification of genes potentially related to epithelial-mesenchymal transition. Oncogene 2007, 26, 7859–7871. [Google Scholar] [CrossRef] [PubMed]
- Al Saleh, S.; Al Mulla, F.; Luqmani, Y.A. Estrogen receptor silencing induces epithelial to mesenchymal transition in human breast cancer cells. PLoS ONE 2011, 6, e20610. [Google Scholar] [CrossRef] [PubMed]
- Pomp, V.; Leo, C.; Mauracher, A.; Korol, D.; Guo, W.; Varga, Z. Differential expression of epithelial-mesenchymal transition and stem cell markers in intrinsic subtypes of breast cancer. Breast Cancer Res. Treat. 2015, 154, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Czerwinska, P.; Kaminska, B. Regulation of breast cancer stem cell features. Contemp. Oncol. 2015, 19, A7–A15. [Google Scholar] [CrossRef] [PubMed]
- Coradini, D.; Boracchi, P.; Ambrogi, F.; Biganzoli, E.; Oriana, S. Cell polarity. Epithelial-mesenchymal transition and cell-fate decision gene expression in ductal carcinoma in situ. Int. J. Surg. Oncol. 2012, 2012, 984346. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, B.P. Epithelial-mesenchymal transition—A hallmark of breast cancer metastasis. Cancer Hallm. 2013, 1, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, E.S.; Ertel, A.; Davicioni, E.; Kline, J.; Schwartz, G.F.; Witkiewicz, A.K. Progression of ductal carcinoma in situ to invasive breast cancer is associated with gene expression programs of EMT and myoepithelial. Breast Cancer Res. Treat. 2012, 133, 1009–1024. [Google Scholar] [CrossRef] [PubMed]
- Bouris, P.; Skandalis, S.S.; Piperigkou, Z.; Afratis, N.; Karamanou, K.; Aletras, A.J.; Moustakas, A.; Theocharis, A.D.; Karamanos, N.K. Estrogen receptor alpha mediates epithelial to mesenchymal transition, expression of specific matrix effectors and functional properties of breast cancer cells. Matrix Biol. 2015, 43, 42–60. [Google Scholar] [CrossRef] [PubMed]
- Radisky, E.; Radisky, D. Matrix metalloproteinase-induced epithelial-mesenchymal transition in breast cancer. J. Mammary Gland Biol. Neoplasia 2012, 15, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Cichon, M.A.; Nelson, C.M.; Radisky, D.C. Regulation of epithelial-mesenchymal transition in breast cancer cells by cell contact and adhesion. Cancer Inform. 2015, 14 (Suppl. 3), 1–13. [Google Scholar] [PubMed]
- Faronato, M.; Nguyen, V.T.; Patten, D.K.; Lombardo, Y.; Steel, J.H.; Patel, N.; Woodley, L.; Shousha, S.; Pruneri, G.; Coombes, R.C.; et al. DMXL2 drives epithelial to mesenchymal transition in hormonal therapy resistant breast cancer through notch hyper-activation. Oncotarget 2015, 6, 22467–22479. [Google Scholar]
- Fischer, K.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Lombaerts, M.; van Wezel, T.; Philippo, K.; Dierssen, J.W.; Zimmerman, R.M.; Oosting, J.; van Eijk, R.; Eilers, P.H.; van de Water, B.; Cornelisse, C.J.; et al. E-cadherin transcriptional downregulation by promoter methylation but not mutation is related to epithelial-to-mesenchymal transition in breast cancer cell lines. Br. J. Cancer 2006, 94, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Onder, T.T.; Gupta, P.B.; Mani, S.A.; Yang, J.; Lander, E.S.; Weinberg, R.A. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008, 68, 3645–3654. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.; Petti, F.; Sujka-Kwok, I.; Mercado, P.; Bean, J.; Monaghan, M.; Seymour, S.L.; Argast, G.M.; Epstein, D.M.; Haley, J.D. A system view of epithelial-mesenchymal transition signaling states. Clin. Exp. Metastasis 2011, 28, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Abdulla, T.; Luna-Zurita, L.; de la Pompa, J.L.; Schleich, J.M.; Summers, R. Epithelial to mesenchymal transition—The roles of cell morphology, labile adhesion and junctional coupling. Comput Methods Progr. Biomed. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, E.; Dubois-Marshall, S.; Sims, A.H.; Gautier, P.; Caldwell, H.; Meehan, R.R.; Harrison, D.J. An in vitro model that recapitulates the epithelial to mesenchymal transition (EMT) in human breast cancer. PLoS ONE 2001, 6, e17083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, W.J.; Nusse, R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science 2004, 303, 1483–1487. [Google Scholar] [CrossRef] [PubMed]
- Geyer, F.C.; Lacroix-Triki, M.; Savage, K.; Arnedos, M.; Lambros, M.B.; MacKay, A.; Natrajan, R.; Reis-Filho, J.S. β-catenin pathway activation in breast cancer is associated with triple-negative phenotype but not with CTNNB1 mutation. Mod. Pathol. 2011, 24, 209–231. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, N.; Panda, C. Subtype-specific alterations of the Wnt signaling pathway in breast cancer. Clin. Progn. Signif. Cancer Sci. 2012, 103, 210–220. [Google Scholar]
- Li, Y.; Wang, Z. Regulation of EMT by Notch signaling pathway in tumor progression. Curr. Cancer Drug Targets 2013, 13, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Farnie, G.; Clarke, R. Mammary stem cells and breast cancer—Role of Notch signaling. Stem Cell Rev. 2007, 3, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Zhang, M.; Wu, H.; Xu, H.; Han, N.; Chu, Q.; Yu, S.; Chen, Y.; Wu, K. Expression of Notch 1 correlates with breast cancer progression and prognosis. PLoS ONE 2015, 10, e0131689. [Google Scholar]
- Liu, Z.J.; Semenza, G.L.; Zhang, H.F. Hypoxia-inducible factor 1 and breast cancer metastasis. Biomed. Biotechnol. 2015, 16, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Aiwei, Y.; Kieber-Emons, T. Adipocyte hypoxia causes epithelial to mesenchymal transition—Related gene expression and estrogen receptor-negative phenotype in breast cancer cells. Oncol. Rep. 2015, 33, 2689–2694. [Google Scholar]
- Ho, M.Y.; Tang, S.J.; Chuang, M.J.; Cha, T.L.; Li, J.Y.; Sun, G.H.; Sun, K.H. TNF-α induces epithelial-mesenchymal transition of renal cell carcinoma cells via a GSK3β-dependent mechanism. Mol. Cancer Res. 2012, 10, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- Li, C.W.; Xia, W.; Huo, L.; Lim, S.O.; Wu, Y.; Hsu, J.L.; Chao, C.H.; Yamaguchi, H.; Yang, N.K.; Ding, Q.; et al. Epithelial-mesenchymal transition induced by TNF-α requires NF-κB-mediated transcriptional upregulation of Twist 1. Cancer Res. 2012, 72, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.; Ferracin, M.; Liu, C.G.; Veronese, A.; Spizzo, R.; Sabbioni, S.; Magri, E.; Pedriali, M.; Fabbri, M.; Campiglio, M.; et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005, 65, 7065–7070. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, L.V.; Martin, E.C.; Segar, H.C.; Miller, D.F.; Buechlein, A.; Rusch, D.B.; Nephew, K.P.; Burow, M.E.; Collins-Burow, B.M. Dual regulation by micro RNA-200b-3b and microRNA-200b-5p in the inhibition of epithelial-to-mesenchymal transition in triple-negative breast cancer. Oncotarget 2015, 6, 16639–16652. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Gibbons, D.L.; Kurie, J.M. The role of epithelial-mesenchymal transition programming in invasion and metastasis: A clinical perspective. Cancer Manag. Res. 2013, 5, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Guttilla, I.; White, B. ERα, microRNAs, and the epithelial-mesenchymal transition in breast cancer. Trends Endocrinol. Metab. 2012, 3, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Xie, F.; Bao, X.; Chen, W.; Xu, Q. miR-300 inhibits epithelial to mesenchymal transition and metastasis by targeting Twist in human epithelial cancer. Mol. Cancer 2014, 13, 121. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Dong, C.; Zhou, B.P. Epigenetic regulation of EMT: The snail story. Curr. Pharm. Des. 2014, 20, 1698–1705. [Google Scholar] [CrossRef] [PubMed]
- Sigurdsson, V.; Gudjonsson, T. Endothelial induced EMT in breast epithelial cells with stem cells properties. PLoS ONE 2011, 6, e23833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wendt, M.K.; Schiemann, W.P. Mechanisms of epithelial-mesenchymal transition by TGF-β. Future Oncol. 2009, 5, 1145–1168. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Wienberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Groger, C.; Grubinger, M.; Waldhör, T.; Vierlinger, K.; Mikulits, W. Meta-analysis of gene expression signatures defining the epithelial to mesenchymal transition during cancer progression. PLoS ONE 2012, 7, e51136. [Google Scholar] [CrossRef] [PubMed]
- Srishti, K.; Susinjam, B. Breast cancer stem cells, EMT and therapeutic targets. Biochem. Biophys. Res. Commun. 2014, 453, 112–116. [Google Scholar]
- Eaves, C. Cancer stem cells: Here, there, everywhere? Nature 2008, 456, 581–582. [Google Scholar] [CrossRef] [PubMed]
- Alkatout, I.; Klapper, W. Transcription factors associated with epithelial-mesenchymal transition and cancer stem cells in the tumor center and margin of invasive breast cancer. Exp. Mol. Pathol. 2013, 94, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Li, J.P.; Yang, Y.X.; Liu, Q.L.; Zhou, Z.W.; Pan, S.T.; He, Z.X.; Zhang, X.; Yang, T.; Pan, S.Y.; Duan, W.; et al. The pan-inhibitor of Aurora kinases danusertib induces apoptosis and autophagy and suppresses epithelial-to-mesenchymal transition in human breast cancer cells. Drug Des. Dev. Ther. 2015, 9, 1027–1062. [Google Scholar]
- Yen, W.C.; Fischer, M.M.; Axelrod, F.; Bond, C.; Cain, J.; Cancilla, B.; Henner, W.R.; Meisner, R.; Sato, A.; Shah, J.; et al. Targeting Notch signaling with a Notch2/Notch3 antagonist (Tarextumab) inhibits tumor growth and decreases tumor-initiating cell frequency. Clin. Cancer Res. 2015, 21, 2084–2094. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Mernaugh, R.; Yi, F.; Blum, D.; Carbone, D.P.; Dang, T.P. Targeting specific regions of the Notch3 ligand-binding domain induces apoptosis and inhibits tumor growth in lung cancer. Cancer Res. 2010, 70, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Liu, D.; Zhao, Y.; He, J.; Kang, H.; Dai, Z.; Wang, X.; Zhang, S.; Zan, Y. Sinomenine inhibits breast cancer cell invasion and migration by suppressing NF-κB activation mediated by IL-4/miR-324-5p/CUEDC2 axis. Biochem. Biophys. Res. Commun. 2015, 464, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Yan, W.; Dai, Z.; Gao, X.; Ma, Y.; Xu, Q.; Jiang, J.; Zhang, S. Baicalein suppresses metastasis of breast cancer cells by inhibiting EMT via downregulation of SATB1 and Wnt/β-catenin pathway. Drugs Des. Dev. Ther. 2016. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Liu, M.; Wang, Y.; Chen, X.; Xu, J.; Sun, Y.; Zhao, L.; Qu, H.; Fan, Y.; Wu, C. Antagonism of miR-21 reverses epithelial-mesenchymal transition and cancer stem cell phenotype through AKT/ERK1/2 inactivation by targeting PTEN. PLoS ONE 2012, 7, e39520. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Felipe Lima, J.; Nofech-Mozes, S.; Bayani, J.; Bartlett, J.M.S. EMT in Breast Carcinoma—A Review. J. Clin. Med. 2016, 5, 65. https://doi.org/10.3390/jcm5070065
Felipe Lima J, Nofech-Mozes S, Bayani J, Bartlett JMS. EMT in Breast Carcinoma—A Review. Journal of Clinical Medicine. 2016; 5(7):65. https://doi.org/10.3390/jcm5070065
Chicago/Turabian StyleFelipe Lima, Joema, Sharon Nofech-Mozes, Jane Bayani, and John M. S. Bartlett. 2016. "EMT in Breast Carcinoma—A Review" Journal of Clinical Medicine 5, no. 7: 65. https://doi.org/10.3390/jcm5070065