
New Insights into the Crossroads between EMT and Stemness in the Context of Cancer

Abstract
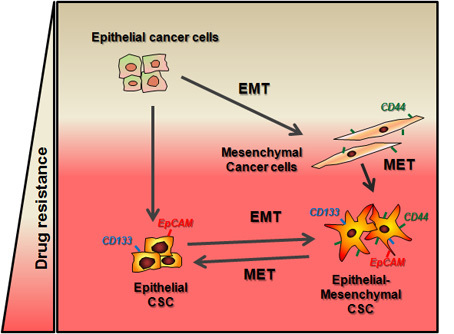
:
{kind=link}
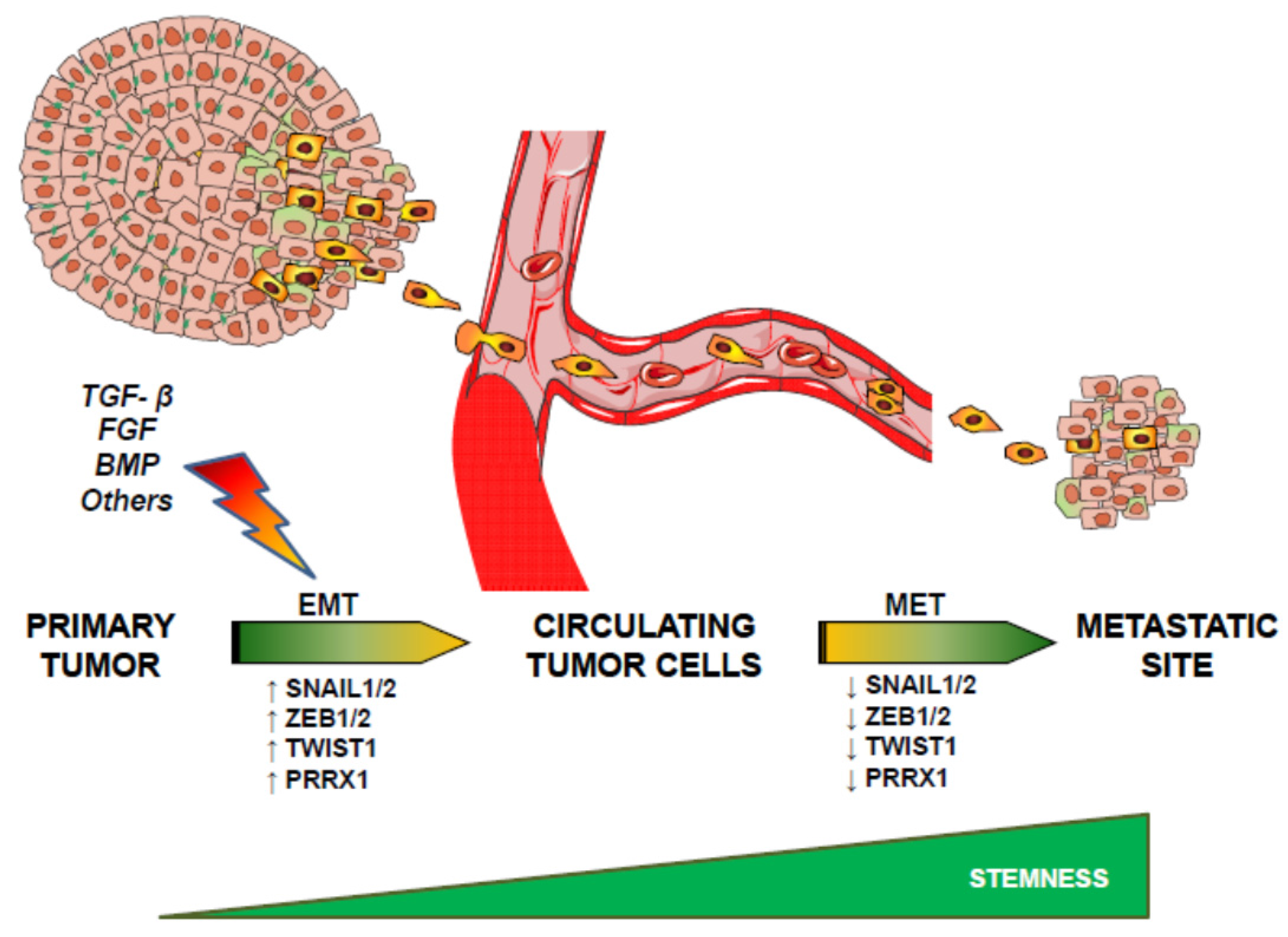{kind=link}
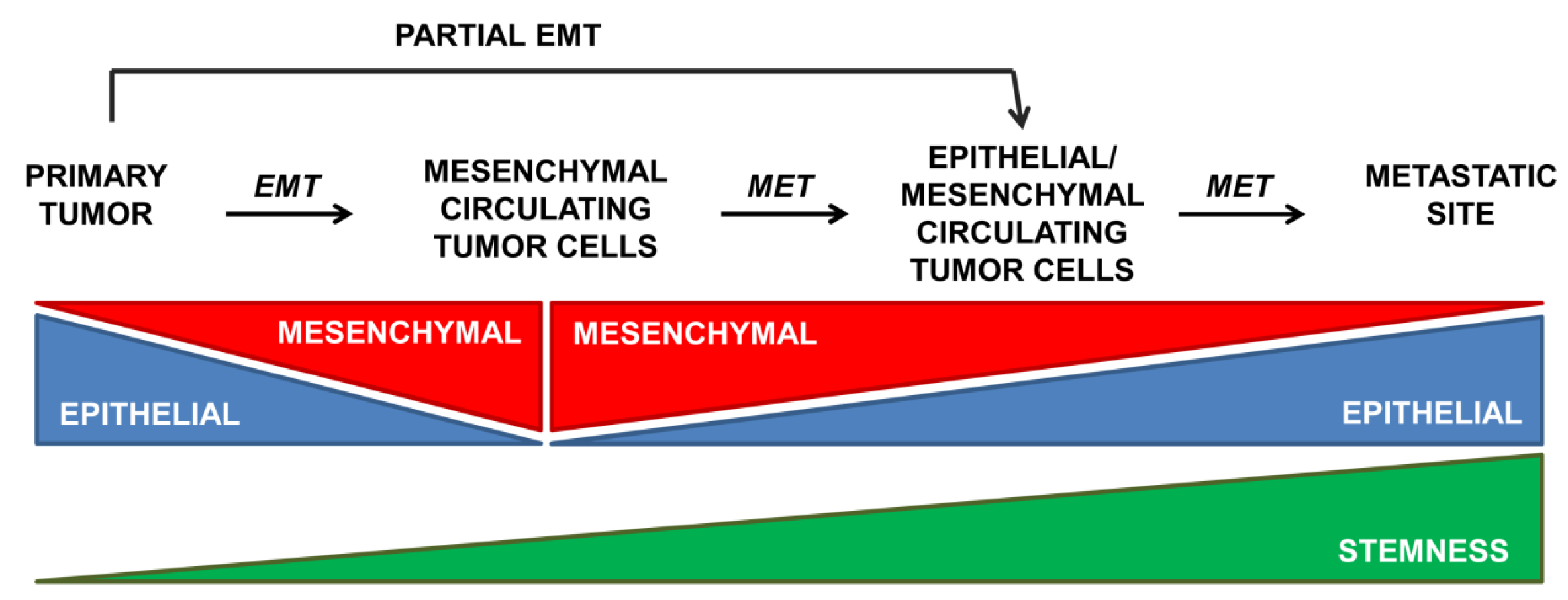{kind=link}
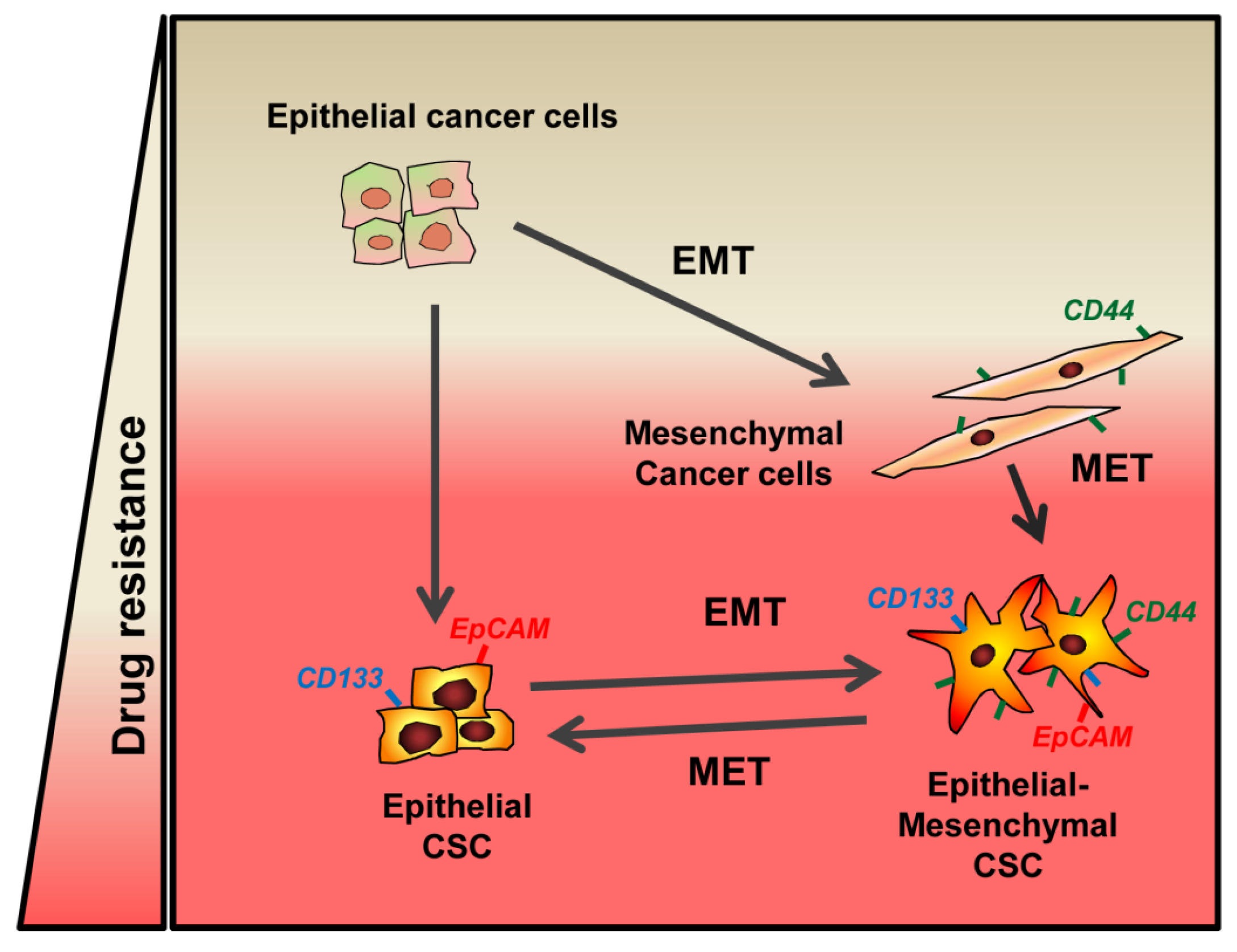{kind=link}
1. Introduction
2. Epithelial-Mesenchymal Transition (EMT) and Stemness: General Overview
3. EMT and Stemness: Coupled, Antagonistic or Independent Processes?
4. Epithelial Plasticity: The EMT Transient State
5. EMT and Stemness in the Crossroads of Chemotherapy Resistance
6. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nieto, M.A. Epithelial plasticity: A common theme in embryonic and cancer cells. Science 2013, 342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, A.M. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial–mesenchymal transitin in tumour progression. Nat. Rev. Cancer 2002, 2, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Medema, J.P. Cancer stem cells: The challenges ahead. Nat. Cell Biol. 2013, 15, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; Hausen, A.Z.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting micrornas. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Ocaña, O.H.; Córcoles, R.; Fabra, A.; Moreno-Bueno, G.; Acloque, H.; Vega, S.; Barrallo-Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer PRRX1. Cancer Cell 2012, 22, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.H.; Donaher, J.L.; Murphy, D.A.; Chau, S.; Yang, J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012, 22, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 23. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, I.; Fernando, J.; Mainez, J.; Sancho, P. TGF-β signaling in cancer treatment. Curr. Pharm. Des. 2014, 20, 2934–2947. [Google Scholar] [CrossRef] [PubMed]
- Caja, L.; Sancho, P.; Bertran, E.; Fabregat, I. Dissecting the effect of targeting the epidermal growth factor receptor on TGF-β-induced-apoptosis in human hepatocellular carcinoma cells. J. Hepatol. 2011, 55, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Drabsch, Y.; Dijke, P.T. TGF-β signalling and its role in cancer progression and metastasis. Cancer Metastasis Rev. 2012, 3–4, 553–568. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Cano, A. The epithelial–mesenchymal transition under control: Global programs to regulate epithelial plasticity. Semin. Cancer Biol. 2012, 22, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Ye, J.; Wu, D.; Wu, P.; Chen, Z.; Chen, J.; Gao, S.; Huang, J. LEIGC long non-coding RNA acts as a tumor suppressor in gastric carcinoma by inhibiting the epithelial-to-mesenchymal transition. BMC Cancer 2014, 14, 932. [Google Scholar] [CrossRef] [PubMed]
- Díaz, V.; Viñas-Castells, R.; Herreros, A.G.D. Regulation of the protein stability of EMT transcription factors. Cell Adhes. Migr. 2014, 8, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Bill, R.; Christofori, G. The relevance of EMT in breast cancer metastasis: Correlation or causality? FEBS Lett. 2015, 589, 1577–1587. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; Barrio, M.G.D.; Portillo, F.; Nieto, M.A. The transcription factor Snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Sancho, E.; Francí, C.; Domínguez, D.; Monfar, M.; Baulida, J.; Herreros, A.G.D. The transcription factor Snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.J.; Moreno-Bueno, G.; Sarrio, D.; Locascio, A.; Cano, A.; Palacios, J.; Nieto, M.A. Correlation of Snail expression with histological grade and lymph node status in breast carcinomas. Oncogene 2002, 21, 3241–3246. [Google Scholar] [CrossRef] [PubMed]
- Olmeda, D.; Moreno-Bueno, G.; Flores, J.M.; Fabra, A.; Portillo, F.; Cano, A. Snai1 is required for tumor growth and lymph node metastasis of human breast carcinoma MDA-MB-231 cells. Cancer Res. 2007, 67, 11721–11731. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Puisieux, A.; Brabletz, T.; Caramel, J. Oncogenic roles of EMT-inducing transcription factors. Nat. Cell Biol. 2014, 16, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, S.; Langhans, S.A. Crosstalk of oncogenic signaling pathways during epithelial-mesenchymal transition. Front. Oncol. 2014, 4, 358. [Google Scholar] [CrossRef] [PubMed]
- Trimboli, A.J.; Fukino, K.; Bruin, A.D.; Wei, G.; Shen, L.; Tanner, S.M.; Creasap, N.; Rosol, T.J.; Robinson, M.L.; Eng, C.; et al. Direct evidence for epithelial-mesenchymal transitions in breast cancer. Cancer Res. 2008, 68, 937–945. [Google Scholar] [CrossRef] [PubMed]
- McGirt, L.Y.; Jia, P.; Baerenwald, D.A.; Duszynski, R.J.; Dahlman, K.B.; Zic, J.A.; Zwerner, J.P.; Hucks, D.; Dave, U.; Zhao, Z.; et al. Whole-genome sequencing reveals oncogenic mutations in mycosis fungoides. Blood 2015, 126, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Barrallo-Gimeno, A.; Nieto, A.M. The Snail genes as inducers of cell movement and survival: Implications in development and cancer. Development 2005, 132, 3151–3161. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Seidel, M.; Wu, W.; Kamizono, S.; Ferrando, A.; Bronson, R.; Iwasaki, H.; Akashi, K.; Morimoto, A.; Hitzler, J.; et al. Slug, a highly conserved zinc finger transcriptional repressor, protects hematopoietic progenitor cells from radiation-induced apoptosis in vivo. Cancer Cell 2002, 2, 279–288. [Google Scholar] [CrossRef]
- Vega, S.; Morales, A.V.; Ocaña, O.H.; Valdés, F.; Fabregat, I.; Nieto, M.A. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004, 18, 1131–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, D.L.; Mainez, J.; Vega, S.; Sancho, P.; Murillo, M.M.; Frutos, C.A.D.; Castillo, G.D.; López-Blau, C.; Fabregat, I.; Nieto, M.A. Snail1 suppresses TGF-β-induced apoptosis and is sufficient to trigger EMT in hepatocytes. J. Cell Sci. 2010, 123, 3467–3477. [Google Scholar] [CrossRef] [PubMed]
- Beck, B.; Lapouge, G.; Rorive, S.; Drogat, B.; Desaedelaere, K.; Delafaille, S.; Dubois, C.; Salmon, I.; Willekens, K.; Marine, J.; et al. Different levels of Twist1 regulate skin tumor initiation, stemness, and progression. Cell Stem Cell 2015, 16, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, A.; Álvarez, A.M.; Pedrosa, J.M.L.; Roncero, C.; Benito, M.; Fabregat, I. Apoptotic response to TGF-β in fetal hepatocytes depends upon their state of differentiation. Exp. Cell Res. 1999, 252, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Valdés, F.; Álvarez, A.M.; Locascio, A.; Vega, S.; Herrera, B.; Fernández, M.; Benito, M.; Nieto, M.A.; Fabregat, I. The epithelial mesenchymal transition confers resistance to the apoptotic effects of transforming growth factor β in fetal rat hepatocytes. Mol. Cancer Res. 2002, 1, 68–78. [Google Scholar] [PubMed]
- Caja, L.; Bertran, E.; Campbell, J.; Fausto, N.; Fabregat, I. The transforming growth factor-β (TGF-β) mediates acquisition of a mesenchymal stem cell-like phenotype in human liver cells. J. Cell. Physiol. 2010, 226, 1214–1223. [Google Scholar] [CrossRef] [PubMed]
- Castillo, G.D.; Alvarez-Barrientos, A.; Carmona-Cuenca, I.; Fernández, M.; Sánchez, A.; Fabregat, I. Isolation and characterization of a putative liver progenitor population after treatment of fetal rat hepatocytes with TGF-β. J. Cell. Physiol. 2008, 215, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Morel, A.; Lièvre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Dang, H.; Ding, W.; Emerson, D.; Rountree, C.B. Snail1 induces epithelial-to-mesenchymal transition and tumor initiating stem cell characteristics. BMC Cancer 2011, 11, 396. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Ding, J.; Chen, C.; Sun, W.; Ning, B.-F.; Wen, W.; Huang, L.; Han, T.; Yang, W.; Wang, C.; et al. Hepatic transforming growth factor β gives rise to tumor-initiating cells and promotes liver cancer development. Hepatology 2012, 56, 2255–2267. [Google Scholar] [CrossRef] [PubMed]
- Fernando, J.; Malfettone, A.; Cepeda, E.B.; Vilarrasa-Blasi, R.; Bertran, E.; Raimondi, G.; Fabra, A.; Alvarez-Barrientos, A.; Fernández-Salguero, P.; Fernández-Rodríguez, C.M.; et al. A mesenchymal-like phenotype and expression of CD44 predict lack of apoptotic response to sorafenib in liver tumor cells. Int. J. Cancer 2015, 136, E161–E172. [Google Scholar] [CrossRef] [PubMed]
- Bertran, E.; Crosas-Molist, E.; Sancho, P.; Caja, L.; Lopez-Luque, J.; Navarro, E.; Egea, G.; Lastra, R.; Serrano, T.; Ramos, E.; et al. Overactivation of the TGF-β pathway confers a mesenchymal-like phenotype and CXCR4 dependent migratory properties to liver tumor cells. Hepatology 2013, 58, 2032–2044. [Google Scholar] [CrossRef] [PubMed]
- Kurrey, N.; Jalgaonkar, S.; Joglekar, A.; Ghanate, A.; Chaskar, P.; Doiphode, R.; Bapat, S. Snail and Slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells 2009, 27, 2059–2068. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Hsu, D.S.S.; Wang, H.W.; Wang, H.J.; Lan, H.Y.; Yang, W.H.; Huang, C.H.; Kao, S.Y.; Tzeng, C.H.; Tai, S.K.; et al. Bmi1 is essential in Twist1-induced epithelial–mesenchymal transition. Nat. Cell Biol. 2010, 12, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Chao, C.H.; Xia, W.; Yang, J.Y.; Xiong, Y.; Li, C.W.; Yu, W.H.; Rehman, S.K.; Hsu, J.L.; Lee, H.H.; et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat. Cell Biol. 2011, 13, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Tschaharganeh, D.F.; Xue, W.; Calvisi, D.F.; Evert, M.; Michurina, T.V.; Dow, L.E.; Banito, A.; Katz, S.F.; Kastenhuber, E.R.; Weissmueller, S.; et al. p53-dependent Nestin regulation links tumor suppression to cellular plasticity in liver cancer. Cell 2014, 158, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Tilló, E.; Barrios, O.D.; Siles, L.; Cuatrecasas, M.; Castells, A.; Postigo, A. Β-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc. Natl. Acad. Sci. USA 2011, 108, 19204–19209. [Google Scholar] [CrossRef] [PubMed]
- Mima, K.; Okabe, H.; Ishimoto, T.; Hayashi, H.; Nakagawa, S.; Kuroki, H.; Watanabe, M.; Beppu, T.; Tamada, M.; Nagano, O.; et al. CD44s regulates the TGF-β–mediated mesenchymal phenotype and is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Res. 2012, 23, 3414–3423. [Google Scholar] [CrossRef] [PubMed]
- Nomura, A.; Banerjee, S.; Chugh, R.; Dudeja, V.; Yamamoto, M.; Vickers, S.M.; Saluja, A.K. CD133 initiates tumors, induces epithelial-mesenchymal transition and increases metastasis in pancreatic cancer. Oncotarget 2015, 6, 8313–8322. [Google Scholar] [CrossRef] [PubMed]
- Preca, B.-T.; Bajdak, K.; Mock, K.; Sundararajan, V.; Pfannstiel, J.; Maurer, J.; Wellner, U.; Hopt, U.T.; Brummer, T.; Brabletz, S.; et al. A self-enforcing CD44s/ZEB1 feedback loop maintains EMT and stemness properties in cancer cells. Int. J. Cancer 2015, 137, 2566–2577. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Liang, J.; Ni, S.; Zhou, T.; Qing, X.; Li, H.; He, W.; Chen, J.; Li, F.; Zhuang, Q.; et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 2010, 7, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Celià-Terrassa, T.; Meca-Cortés, Ó.; Mateo, F.; Paz, A.M.D.; Rubio, N.; Arnal-Estapé, A.; Ell, B.J.; Bermudo, R.; Díaz, A.; Guerra-Rebollo, M.; et al. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J. Clin. Investig. 2012, 122, 1849–1868. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.; Luitel, K.; Kim, M.; Zhang, K.; Longmore, G.; Tran, D. Transient Snail1 expression is necessary for metastatic competence in breast cancer. Cancer Res. 2014, 74, 6330–6340. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Keckesova, Z.; Donaher, J.L.; Shibue, T.; Tischler, V.; Reinhardt, F.; Itzkovitz, S.; Noske, A.; Zürrer-Härdi, U.; Bell, G.; et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 2012, 148, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Luanpitpong, S.; Li, J.; Manke, A.; Brundage, K.; Ellis, E.; McLaughlin, S.; Angsutararux, P.; Chanthra, N.; Voronkova, M.; Chen, Y.; et al. SLUG is required for SOX9 stabilization and functions to promote cancer stem cells and metastasis in human lung carcinoma. Oncogene 2015. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.M.; Panzilius, E.; Bartsch, H.S.; Irmler, M.; Beckers, J.; Kari, V.; Linnemann, J.R.; Dragoi, D.; Hirschi, B.; Kloos, U.J.; et al. Stem-cell-like properties and epithelial plasticity arise as stable traits after transient Twist1 activation. Cell Rep. 2015, 10, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Stankic, M.; Pavlovic, S.; Chin, Y.; Brogi, E.; Padua, D.; Norton, L.; Massagué, J.; Benezra, R. TGF-β-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Rep. 2013, 5, 1228–1242. [Google Scholar] [CrossRef] [PubMed]
- Costa-Silva, B.; Aiello, N.; Ocean, A.; Singh, S.; Zhang, H.; Thakur, B.; Becker, A.; Hoshino, A.; Mark, M.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Costa-Silva, B.; Shen, T.; Rodrigues, G.; Hashimoto, A.; Tesic, M.M.; Molina, H.; Kohsaka, S.; Giannatale, A.D.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Grande, M.T.; Sánchez-Laorden, B.; López-Blau, C.; Frutos, C.A.D.; Boutet, A.; Arévalo, M.; Rowe, R.G.; Weiss, S.J.; López-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovisa, S.; LeBleu, V.S.; Tampe, B.; Sugimoto, H.; Vadnagara, K.; Carstens, J.L.; Wu, C.-C.; Hagos, Y.; Burckhardt, B.C.; Pentcheva-Hoang, T.; et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998–908. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T. EMT and MET in metastasis: Where are the cancer stem cells? Cancer Cell 2012, 22, 699–701. [Google Scholar] [CrossRef] [PubMed]
- Ombrato, L.; Malanchi, I. The EMT universe: Space between cancer cell dissemination and metastasis initiation. Crit. Rev. Oncog. 2014, 19, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Weinberg, R.A. Epithelial–mesenchymal plasticity: A central regulator of cancer progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Jia, D.; Boareto, M.; Mani, S.A.; Pienta, K.J.; Ben-Jacob, E.; Levine, H. Coupling the modules of EMT and stemness: A tunable “stemness window” model. Oncotarget 2015, 6, 25161–25174. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ma, J.; Qian, X.; Wu, Q.; Xia, J.; Miele, L.; Sarkar, F.; Wang, Z. Regulation of EMT by Notch signaling pathway in tumor progression. Curr. Cancer Drug Targets 2013, 13, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Kong, D.; Banerjee, S.; Ahmad, A.; Azmi, A.; Ali, S.; Abbruzzese, J.; Gallick, G.; Sarkar, F. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009, 69, 2400–2407. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ling, M.; Guan, X.; Tsao, S.; Cheung, H.; Lee, D.; Wong, Y. Identification of a novel function of TWIST, a bHLH protein, in the development of acquired taxol resistance in human cancer cells. Oncogene 2004, 23, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; Stephens, M.; Pathak, H.; Rangarajan, A. Transcription factors that mediate epithelial–mesenchymal transition lead to multidrug resistance by upregulating ABC transporters. Cell Death Dis. 2011, 2, e179. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Peng, Y.; Liu, Y.; Xin, H.; Zhan, X.; Tan, W. Twist1 and Snail link Hedgehog signaling to tumor-initiating cell-like properties and acquired chemoresistance independently of ABC transporters. Stem Cells 2015, 33, 1063–1074. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.C.; Choi, H.; Rayes, T.E.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Mishra, L.; Li, S. EMT, CTCs, CSCs in tumor relapse and drug-resistance. Oncotarget 2015, 6, 10697–10711. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Li, R.; Tao, K.; Cao, D.; Ti, Z.; Ding, R.; Cai, L.; Zhang, F.; Dou, K. Characterization of a stem-like population in hepatocellular carcinoma MHCC97 cells. Oncol. Rep. 2010, 23, 827–831. [Google Scholar] [PubMed]
- Liu, T.; Xu, F.; Du, X.; Lai, D.; Liu, T.; Zhao, Y.; Huang, Q.; Jiang, L.; Huang, W.; Cheng, W.; et al. Establishment and characterization of multi-drug resistant, prostate carcinoma-initiating stem like cells from human prostate cancer cell lines 22RV1. Mol. Cell. Biochem. 2010, 340, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Becker, A.; Zimmer, A.; Lu, J.; Buettner, R.; Kirfel, J. SNAI1-mediated epithelial-mesenchymal transition confers chemoresistance and cellular plasticity by regulating genes involved in cell death and stem cell maintenance. PLOS ONE 2015, 8, e66558. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.; Kim, L.; Kong, H.; Low, L.; Iacopetta, B.; Soong, R.; Salto-Tellez, M. CD133 expression predicts for non-response to chemotherapy in colorectal cancer. Mod. Pathol. 2010, 23, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.; Lu, L.; Irvin, D.; Black, K.; Yu, J. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Sarvi, S.; Mackinnon, A.; Avlonitis, N.; Bradley, M.; Rintoul, R.; Rassl, D.; Wang, W.; Forbes, S.; Gregory, C.; Sethi, T. CD133+ cancer stem-like cells in small cell lung cancer are highly tumorigenic and chemoresistant but sensitive to a novel neuropeptide antagonist. Cancer Res. 2014, 74, 1554–1565. [Google Scholar] [CrossRef] [PubMed]
- Bates, R.C.; Edwards, N.S.; Burns, G.F.; Fisher, D.E. A CD44 survival pathway triggers chemoresistance via lyn kinase and phosphoinositide 3-kinase/Akt in colon carcinoma cells. Cancer Res. 2001, 61, 5275–5283. [Google Scholar] [PubMed]
- Bourguignon, L.; Peyrollier, K.; Xia, W.; Gilad, E. Hyaluronan-CD44 interaction activates stem cell marker Nanog, Stat-3-mediated MDR1 gene expression, and ankyrin-regulated multidrug efflux in breast and ovarian tumor cells. J. Biol. Chem. 2008, 283, 17635–17651. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, L.; Shiina, M.; Li, J. Hyaluronan-CD44 interaction promotes oncogenic signaling, microrna functions, chemoresistance, and radiation resistance in cancer stem cells leading to tumor progression. Adv. Cancer Res. 2014, 123, 255–275. [Google Scholar] [PubMed]
- Hashimoto, N.; Tsunedomi, R.; Yoshimura, K.; Watanabe, Y.; Hazama, S.; Oka, M. Cancer stem-like sphere cells induced from de-differentiated hepatocellular carcinoma-derived cell lines possess the resistance to anti-cancer drugs. BMC Cancer 2014, 14, 722. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Ahmad, A.; Azmi, A.S.; Kong, D.; Banerjee, S. Targeting miRNAs involved in cancer stem cell and EMT regulation: An emerging concept in overcoming drug resistance. Drug Resist. Updat. 2010, 13, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Garg, M. Targeting microRNAs in epithelial-to-mesenchymal transition-induced cancer stem cells: Therapeutic approaches in cancer. Expert Opin. Ther. Targets 2015, 19, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zheng, L.; Huang, J.; Gao, F.; Lin, X.; He, L.; Li, D.; Li, Z.; Ding, Y.; Chen, L. MiR-124 radiosensitizes human colorectal cancer cells by targeting PRRX1. PLoS ONE 2014, 9, e93917. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Fang, B.; Zeng, F.; Ma, C.; Pang, H.; Cheng, L.; Shi, Y.; Wang, H.; Yin, B.; Xia, J.; et al. Down-regulation of miR-223 reverses epithelial-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Oncotarget 2015, 6, 1740–1749. [Google Scholar] [CrossRef] [PubMed]
- Bai, W.; Ye, X.; Zhang, M.; Zhu, H.; Xi, W.; Huang, X.; Zhao, J.; Gu, B.; Zheng, G.; Yang, A.; et al. MiR-200c suppresses TGF-β signaling and counteracts trastuzumab resistance and metastasis by targeting ZNF217 and ZEB1 in breast cancer. Int. J. Cancer 2014, 135, 1356–1368. [Google Scholar] [CrossRef] [PubMed]
- Meidhof, S.; Brabletz, S.; Lehmann, W.; Preca, B.; Mock, K.; Ruh, M.; Schüler, J.; Berthold, M.; Weber, A.; Burk, U.; et al. ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol. Med. 2015, 7, 831–847. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Tam, W.L.; Shibue, T.; Kaygusuz, Y.; Reinhardt, F.; Eaton, E.N.; Weinberg, R.A. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 2015, 525, 256–260. [Google Scholar] [CrossRef] [PubMed]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fabregat, I.; Malfettone, A.; Soukupova, J. New Insights into the Crossroads between EMT and Stemness in the Context of Cancer. J. Clin. Med. 2016, 5, 37. https://doi.org/10.3390/jcm5030037
Fabregat I, Malfettone A, Soukupova J. New Insights into the Crossroads between EMT and Stemness in the Context of Cancer. Journal of Clinical Medicine. 2016; 5(3):37. https://doi.org/10.3390/jcm5030037
Chicago/Turabian StyleFabregat, Isabel, Andrea Malfettone, and Jitka Soukupova. 2016. "New Insights into the Crossroads between EMT and Stemness in the Context of Cancer" Journal of Clinical Medicine 5, no. 3: 37. https://doi.org/10.3390/jcm5030037