
MicroRNA Regulation of Epithelial to Mesenchymal Transition


{kind=link}
{kind=link}
Abstract
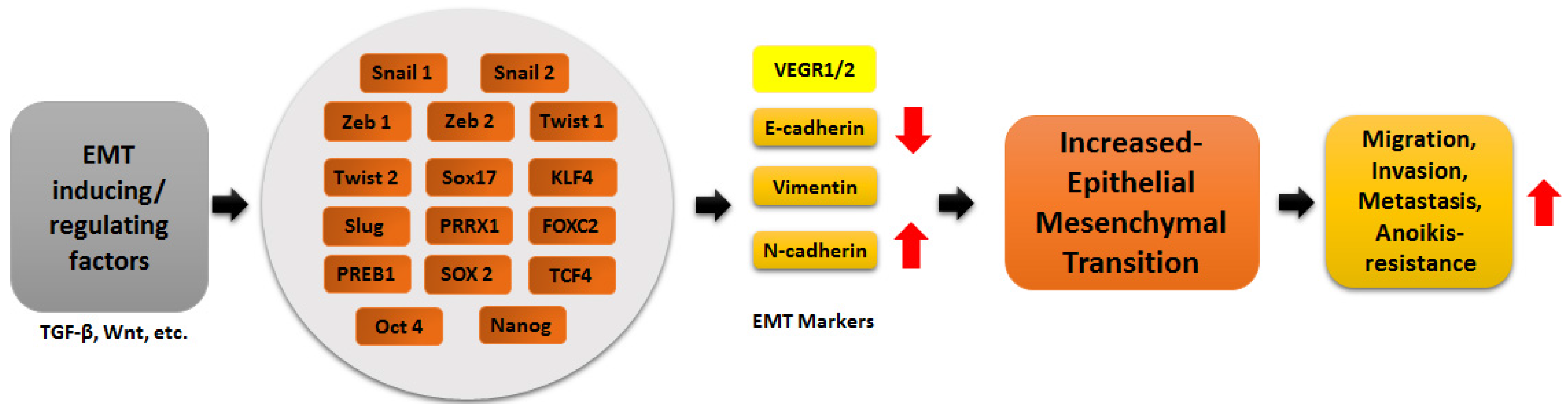
:1. Epithelial to Mesenchymal Transition (EMT)

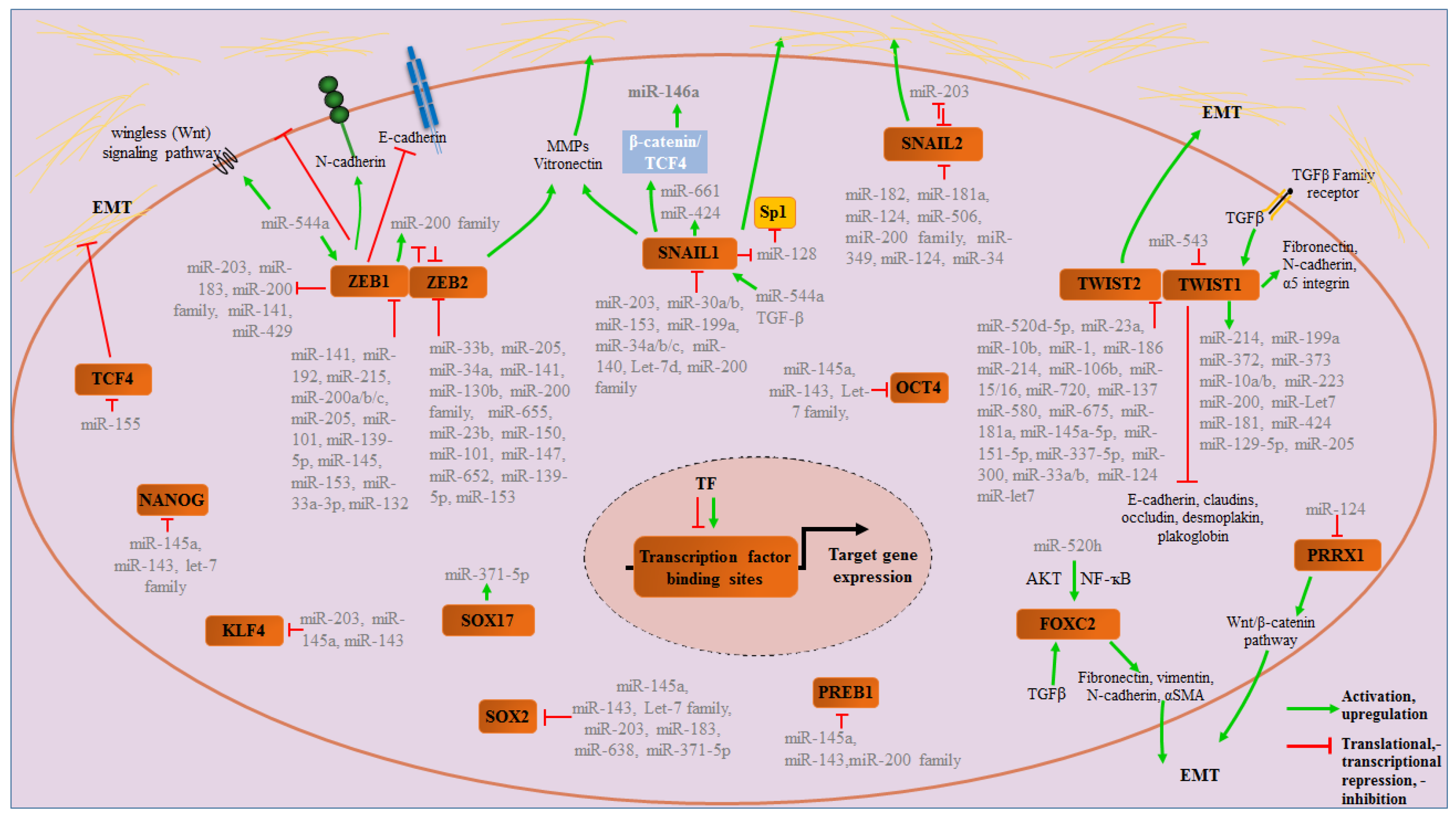
2. MicroRNAs
3. Transcription Factors in EMT Regulation
3.1. The ZEB Family

3.2. The Snail Family (Snai1, Snai2 and Snai3)
3.3. The Twist Family (TWIST1 and TWIST2)
3.4. Pluripotency Transcription Factors
3.5. Other Transcription Factors
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Cavey, M.; Lecuit, T. Molecular bases of cell-cell junctions stability and dynamics. Cold Spring Harb. Perspect. Biol. 2009, 1, a002998. [Google Scholar]
- Zaravinos, A. The regulatory role of micrornas in emt and cancer. J. Oncol. 2015, 2015, 865816. [Google Scholar]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar]
- Liu, W.; Vivian, C.J.; Brinker, A.E.; Hampton, K.R.; Lianidou, E.; Welch, D.R. Microenvironmental influences on metastasis suppressor expression and function during a metastatic cell’s journey. Cancer Microenviron. 2014, 7, 117–131. [Google Scholar]
- De Craene, B.; Berx, G. Regulatory networks defining emt during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar]
- Bedi, U.; Mishra, V.K.; Wasilewski, D.; Scheel, C.; Johnsen, S.A. Epigenetic plasticity: A central regulator of epithelial-to-mesenchymal transition in cancer. Oncotarget 2014, 5, 2016–2029. [Google Scholar] [CrossRef] [PubMed]
- Demirkan, B. The roles of epithelial-to-mesenchymal transition (emt) and mesenchymal-to-epithelial transition (met) in breast cancer bone metastasis: Potential targets for prevention and treatment. J. Clin. Med. 2013, 2, 264–282. [Google Scholar] [CrossRef] [PubMed]
- Corallino, S.; Malabarba, M.G.; Zobel, M.; Di Fiore, P.P.; Scita, G. Epithelial-to-mesenchymal plasticity harnesses endocytic circuitries. Front. Oncol. 2015, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.S.; Yang, M.H. Epithelial-mesenchymal transition-related factors in solid tumor and hematological malignancy. J. Chin. Med. Assoc. 2015, 78, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Cannito, S.; Novo, E.; di Bonzo, L.V.; Busletta, C.; Colombatto, S.; Parola, M. Epithelial-mesenchymal transition: From molecular mechanisms, redox regulation to implications in human health and disease. Antioxid. Redox Signal 2010, 12, 1383–1430. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [PubMed]
- Nieto, M.A. Epithelial plasticity: A common theme in embryonic and cancer cells. Science 2013, 342, 1234850. [Google Scholar] [PubMed]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Ben-Jacob, E.; Onuchic, J.N.; Levine, H. Implications of the hybrid epithelial/mesenchymal phenotype in metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef] [PubMed]
- Revenu, C.; Gilmour, D. Emt 2.0: Shaping epithelia through collective migration. Curr. Opin. Genet. Dev. 2009, 19, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Theveneau, E.; Marchant, L.; Kuriyama, S.; Gull, M.; Moepps, B.; Parsons, M.; Mayor, R. Collective chemotaxis requires contact-dependent cell polarity. Dev. Cell 2010, 19, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Ninov, N.; Chiarelli, D.A.; Martin-Blanco, E. Extrinsic and intrinsic mechanisms directing epithelial cell sheet replacement during drosophila metamorphosis. Development 2007, 134, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, Y.; Yoshinaga, K.; Kitao, H.; Ando, K.; Kimura, Y.; Saeki, H.; Oki, E.; Morita, M.; Kakeji, Y.; Hirahashi, M.; et al. Podoplanin is expressed at the invasive front of esophageal squamous cell carcinomas and is involved in collective cell invasion. Cancer Sci. 2013, 104, 1718–1725. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Liu, Q.; Chen, J.; Chen, J.; Chen, F.; He, C.; Huang, D.; Wu, W.; Lin, L.; Huang, W.; et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell 2014, 25, 605–620. [Google Scholar] [CrossRef] [PubMed]
- Sarrio, D.; Rodriguez-Pinilla, S.M.; Hardisson, D.; Cano, A.; Moreno-Bueno, G.; Palacios, J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008, 68, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. Emt: When epithelial cells decide to become mesenchymal-like cells. J. Clin. Investig. 2009, 119, 1417–1419. [Google Scholar] [CrossRef] [PubMed]
- Klymkowsky, M.W.; Savagner, P. Epithelial-mesenchymal transition: A cancer researcher’s conceptual friend and foe. Am. J. Pathol. 2009, 174, 1588–1593. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Christofori, G. Emt, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Barrallo-Gimeno, A.; Nieto, M.A. The snail genes as inducers of cell movement and survival: Implications in development and cancer. Development 2005, 132, 3151–3161. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Weinberg, R.A. Epithelial-mesenchymal plasticity: A central regulator of cancer progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Mudduluru, G.; Abba, M.; Batliner, J.; Patil, N.; Scharp, M.; Lunavat, T.R.; Leupold, J.H.; Oleksiuk, O.; Juraeva, D.; Thiele, W.; et al. A systematic approach to defining the microrna landscape in metastasis. Cancer Res. 2015, 75, 3010–3019. [Google Scholar] [CrossRef] [PubMed]
- Aigner, K.; Dampier, B.; Descovich, L.; Mikula, M.; Sultan, A.; Schreiber, M.; Mikulits, W.; Brabletz, T.; Strand, D.; Obrist, P.; et al. The transcription factor zeb1 (deltaef1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene 2007, 26, 6979–6988. [Google Scholar] [CrossRef] [PubMed]
- Bolos, V.; Peinado, H.; Perez-Moreno, M.A.; Fraga, M.F.; Esteller, M.; Cano, A. The transcription factor slug represses e-cadherin expression and induces epithelial to mesenchymal transitions: A comparison with snail and e47 repressors. J. Cell. Sci. 2003, 116, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Grooteclaes, M.L.; Frisch, S.M. Evidence for a function of ctbp in epithelial gene regulation and anoikis. Oncogene 2000, 19, 3823–3828. [Google Scholar] [CrossRef] [PubMed]
- Hajra, K.M.; Fearon, E.R. Cadherin and catenin alterations in human cancer. Genes Chromosom. Cancer 2002, 34, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Hajra, K.M.; Chen, D.Y.; Fearon, E.R. The slug zinc-finger protein represses e-cadherin in breast cancer. Cancer Res. 2002, 62, 1613–1618. [Google Scholar] [PubMed]
- Huber, M.A.; Kraut, N.; Beug, H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell. Biol. 2005, 17, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Sancho, E.; Franci, C.; Dominguez, D.; Monfar, M.; Baulida, J.; Garcia De Herreros, A. The transcription factor snail is a repressor of e-cadherin gene expression in epithelial tumour cells. Nat. Cell. Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Perez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing e-cadherin expression. Nat. Cell. Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, zeb and bhlh factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, J.; Horiuchi, A.; Kikuchi, N.; Hayashi, A.; Osada, R.; Ohira, S.; Shiozawa, T.; Konishi, I. Changes in the expression of e-cadherin repressors, snail, slug, sip1, and twist, in the development and progression of ovarian carcinoma: The important role of snail in ovarian tumorigenesis and progression. Med. Mol. Morphol. 2009, 42, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Byles, V.; Zhu, L.; Lovaas, J.D.; Chmilewski, L.K.; Wang, J.; Faller, D.V.; Dai, Y. Sirt1 induces emt by cooperating with emt transcription factors and enhances prostate cancer cell migration and metastasis. Oncogene 2012, 31, 4619–4629. [Google Scholar] [CrossRef] [PubMed]
- Dave, N.; Guaita-Esteruelas, S.; Gutarra, S.; Frias, A.; Beltran, M.; Peiro, S.; de Herreros, A.G. Functional cooperation between snail1 and twist in the regulation of zeb1 expression during epithelial to mesenchymal transition. J. Biol. Chem. 2011, 286, 12024–12032. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Portillo, F.; Cano, A. Transcriptional regulation of cadherins during development and carcinogenesis. Int. J. Dev. Biol. 2004, 48, 365–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheelock, M.J.; Shintani, Y.; Maeda, M.; Fukumoto, Y.; Johnson, K.R. Cadherin switching. J. Cell. Sci. 2008, 121, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Schickel, R.; Boyerinas, B.; Park, S.M.; Peter, M.E. Micrornas: Key players in the immune system, differentiation, tumorigenesis and cell death. Oncogene 2008, 27, 5959–5974. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, M.; Croce, C.M. Micrornas: Master regulators as potential therapeutics in cancer. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 25–43. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.G.; Sanchez-Elsner, T.; Stephanou, A.; Cragg, M.S.; Townsend, P.A. Regulating the genome surveillance system: Mirnas and the p53 super family. Apoptosis 2010, 15, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Baehrecke, E.H. Mirnas: Micro managers of programmed cell death. Curr. Biol. 2003, 13, R473–R475. [Google Scholar] [CrossRef]
- Takeishi, Y. Biomarkers in heart failure. Int. Heart J. 2014, 55, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Stellato, C. Posttranscriptional gene regulation: Novel pathways for glucocorticoids’ anti-inflammatory action. Transl. Med. UniSa. 2012, 3, 67–73. [Google Scholar] [PubMed]
- States, J.C.; Srivastava, S.; Chen, Y.; Barchowsky, A. Arsenic and cardiovascular disease. Toxicol. Sci. 2009, 107, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Sinkovics, J.G. Molecular biology of oncogenic inflammatory processes. I. Non-oncogenic and oncogenic pathogens, intrinsic inflammatory reactions without pathogens, and microrna/DNA interactions (review). Int. J. Oncol. 2012, 40, 305–349. [Google Scholar] [CrossRef] [PubMed]
- Ranjha, R.; Paul, J. Micro-rnas in inflammatory diseases and as a link between inflammation and cancer. Inflamm. Res. 2013, 62, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Malemud, C.J. The discovery of novel experimental therapies for inflammatory arthritis. Mediators Inflamm. 2009, 2009, 698769. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Andreeva, T.V.; Grigorenko, A.P.; Rogaev, E.I. Studying micro rna function and dysfunction in alzheimer’s disease. Front Genet. 2012, 3, 327. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Eom, S.; Park, D.; Kim, H.; Jeoung, D. The hyaluronic acid-hdac3-mirna network in allergic inflammation. Front Immunol. 2015, 6, 210. [Google Scholar] [CrossRef] [PubMed]
- Hamar, P. Role of regulatory micro rnas in type 2 diabetes mellitus-related inflammation. Nucleic Acid Ther. 2012, 22, 289–294. [Google Scholar] [PubMed]
- Deb, R.; Kumar, A.; Chakraborty, S.; Verma, A.K.; Tiwari, R.; Dhama, K.; Singh, U.; Kumar, S. Trends in diagnosis and control of bovine mastitis: A review. Pak. J. Biol. Sci. 2013, 16, 1653–1661. [Google Scholar] [CrossRef] [PubMed]
- Ather, M.H.; Siddiqui, T. The genetics of neuroendocrine prostate cancers: A review of current and emerging candidates. Appl. Clin. Genet. 2012, 5, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.; O’Connell, R.M. Noncoding rnas and chronic inflammation: Micro-managing the fire within. Bioessays 2015, 37, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Oleksiuk, O.; Abba, M.; Tezcan, K.C.; Schaufler, W.; Bestvater, F.; Patil, N.; Birk, U.; Hafner, M.; Altevogt, P.; Cremer, C.; et al. Single-molecule localization microscopy allows for the analysis of cancer metastasis-specific mirna distribution on the nanoscale. Oncotarget 2015. [Google Scholar]
- Utikal, J.; Abba, M.; Novak, D.; Moniuszko, M.; Allgayer, H. Function and significance of micrornas in benign and malignant human stem cells. Semin. Cancer Biol. 2015, 35, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Abba, M.; Patil, N.; Allgayer, H. Micrornas in the regulation of mmps and metastasis. Cancers (Basel) 2014, 6, 625–645. [Google Scholar] [CrossRef] [PubMed]
- Abba, M.; Mudduluru, G.; Allgayer, H. Micrornas in cancer: Small molecules, big chances. Anticancer Agents Med. Chem. 2012, 12, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Mobley, A.K.; Braeuer, R.R.; Kamiya, T.; Shoshan, E.; Bar-Eli, M. Driving transcriptional regulators in melanoma metastasis. Cancer Metastasis Rev. 2012, 31, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of nf-kappab signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Boldin, M.P.; Baltimore, D. Micrornas, new effectors and regulators of nf-kappab. Immunol. Rev. 2012, 246, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.M.; Cao, B.; Zhou, X.; Lu, H. New insights into p53 functions through its target micrornas. J. Mol. Cell. Biol. 2014, 6, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Rokavec, M.; Li, H.; Jiang, L.; Hermeking, H. The p53/mir-34 axis in development and disease. J. Mol. Cell. Biol. 2014, 6, 214–230. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.J.; Carrasco, R.D. Crosstalk between microrna30a/b/c/d/e-5p and the canonical wnt pathway: Implications for multiple myeloma therapy. Cancer Res. 2014, 74, 5351–5358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ma, L. Microrna control of epithelial-mesenchymal transition and metastasis. Cancer Metastasis Rev. 2012, 31, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Romero-Cordoba, S.L.; Salido-Guadarrama, I.; Rodriguez-Dorantes, M.; Hidalgo-Miranda, A. Mirna biogenesis: Biological impact in the development of cancer. Cancer Biol. Ther. 2014, 15, 1444–1455. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Tang, H. Micrornas regulate the epithelial to mesenchymal transition (emt) in cancer progression. Microrna 2014, 3, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Zhang, Y.; Deng, M.; Ye, R.; Zhao, S.; Wang, Y.; Li, J.; Zhao, Z. Microrna control of epithelial-mesenchymal transition in cancer stem cells. Int. J. Cancer. 2014, 135, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Wang, Z.; Fillmore, R.; Xi, Y. Mir-200, a new star mirna in human cancer. Cancer Lett. 2014, 344, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Rusek, A.M.; Abba, M.; Eljaszewicz, A.; Moniuszko, M.; Niklinski, J.; Allgayer, H. Microrna modulators of epigenetic regulation, the tumor microenvironment and the immune system in lung cancer. Mol. Cancer 2015, 14, 34. [Google Scholar] [CrossRef] [PubMed]
- Mimeault, M.; Batra, S.K. Molecular biomarkers of cancer stem/progenitor cells associated with progression, metastases, and treatment resistance of aggressive cancers. Cancer Epidemiol. Biomarkers Prev. 2014, 23, 234–254. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Kang, Y. Multilayer control of the emt master regulators. Oncogene 2014, 33, 1755–1763. [Google Scholar] [CrossRef] [PubMed]
- Tania, M.; Khan, M.A.; Fu, J. Epithelial to mesenchymal transition inducing transcription factors and metastatic cancer. Tumour Biol. 2014, 35, 7335–7342. [Google Scholar] [CrossRef] [PubMed]
- Son, H.; Moon, A. Epithelial-mesenchymal transition and cell invasion. Toxicol. Res. 2010, 26, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Vandewalle, C.; Van Roy, F.; Berx, G. The role of the zeb family of transcription factors in development and disease. Cell. Mol. Life Sci. 2009, 66, 773–787. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Tillo, E.; Liu, Y.; de Barrios, O.; Siles, L.; Fanlo, L.; Cuatrecasas, M.; Darling, D.S.; Dean, D.C.; Castells, A.; Postigo, A. Emt-activating transcription factors in cancer: Beyond emt and tumor invasiveness. Cell. Mol. Life Sci. 2012, 69, 3429–3456. [Google Scholar] [CrossRef] [PubMed]
- Hill, L.; Browne, G.; Tulchinsky, E. Zeb/mir-200 feedback loop: At the crossroads of signal transduction in cancer. Int. J. Cancer 2013, 132, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Lai-Cheong, J.E.; Arita, K.; McGrath, J.A. Genetic diseases of junctions. J. Investig. Dermatol. 2007, 127, 2713–2725. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Heid, H.W.; Schafer, S.; Nuber, U.A.; Zimbelmann, R.; Franke, W.W. Desmosomes and cytoskeletal architecture in epithelial differentiation: Cell type-specific plaque components and intermediate filament anchorage. Eur. J. Cell. Biol. 1994, 65, 229–245. [Google Scholar] [PubMed]
- Tepass, U. Claudin complexities at the apical junctional complex. Nat. Cell. Biol. 2003, 5, 595–597. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. Epithelial-mesenchymal transition in gastric cancer (review). Int. J. Oncol. 2005, 27, 1677–1683. [Google Scholar] [PubMed]
- McConkey, D.J.; Choi, W.; Marquis, L.; Martin, F.; Williams, M.B.; Shah, J.; Svatek, R.; Das, A.; Adam, L.; Kamat, A.; et al. Role of epithelial-to-mesenchymal transition (emt) in drug sensitivity and metastasis in bladder cancer. Cancer Metastasis Rev. 2009, 28, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Yasumi, M.; Sakisaka, T.; Hoshino, T.; Kimura, T.; Sakamoto, Y.; Yamanaka, T.; Ohno, S.; Takai, Y. Direct binding of lgl2 to lgn during mitosis and its requirement for normal cell division. J. Biol. Chem. 2005, 280, 6761–6765. [Google Scholar] [CrossRef] [PubMed]
- Musch, A.; Cohen, D.; Yeaman, C.; Nelson, W.J.; Rodriguez-Boulan, E.; Brennwald, P.J. Mammalian homolog of drosophila tumor suppressor lethal (2) giant larvae interacts with basolateral exocytic machinery in madin-darby canine kidney cells. Mol. Biol. Cell 2002, 13, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Elsum, I.A.; Martin, C.; Humbert, P.O. Scribble regulates an emt polarity pathway through modulation of mapk-erk signaling to mediate junction formation. J. Cell. Sci. 2013, 126, 3990–3999. [Google Scholar] [CrossRef] [PubMed]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between zeb1 and members of the mir-200 family promotes emt and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The mir-200 family and mir-205 regulate epithelial to mesenchymal transition by targeting zeb1 and sip1. Nat. Cell. Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Korpal, M.; Lee, E.S.; Hu, G.; Kang, Y. The mir-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of e-cadherin transcriptional repressors zeb1 and zeb2. J. Biol. Chem. 2008, 283, 14910–14914. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The mir-200 family determines the epithelial phenotype of cancer cells by targeting the e-cadherin repressors zeb1 and zeb2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Veronese, A.; Pichiorri, F.; Lee, T.J.; Jeon, Y.J.; Volinia, S.; Pineau, P.; Marchio, A.; Palatini, J.; Suh, S.S.; et al. P53 regulates epithelial-mesenchymal transition through micrornas targeting zeb1 and zeb2. J. Exp.Med. 2011, 208, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Karaayvaz, M.; Jia, N.; Kaneuchi, M.; Hamada, J.; Watari, H.; Sudo, S.; Ju, J.; Sakuragi, N. Mutant p53 gain-of-function induces epithelial-mesenchymal transition through modulation of the mir-130b-zeb1 axis. Oncogene 2013, 32, 3286–3295. [Google Scholar] [CrossRef] [PubMed]
- Qiu, G.; Lin, Y.; Zhang, H.; Wu, D. Mir-139-5p inhibits epithelial-mesenchymal transition, migration and invasion of hepatocellular carcinoma cells by targeting zeb1 and zeb2. Biochem. Biophys. Res. Commun. 2015, 463, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Park, M.K.; Park, J.H.; Lee, H.J.; Shin, D.H.; Kang, Y.; Lee, C.H.; Kong, G. Loss of the polycomb protein mel-18 enhances the epithelial-mesenchymal transition by zeb1 and zeb2 expression through the downregulation of mir-205 in breast cancer. Oncogene 2014, 33, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, K.; Isomoto, H.; Yamaguchi, N.; Inoue, N.; Machida, H.; Nakayama, T.; Hayashi, T.; Kunizaki, M.; Hidaka, S.; Nagayasu, T.; et al. Mirna-205 modulates cellular invasion and migration via regulating zinc finger e-box binding homeobox 2 expression in esophageal squamous cell carcinoma cells. J. Transl. Med. 2011, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Majid, S.; Dar, A.A.; Saini, S.; Deng, G.; Chang, I.; Greene, K.; Tanaka, Y.; Dahiya, R.; Yamamura, S. Microrna-23b functions as a tumor suppressor by regulating zeb1 in bladder cancer. PLoS ONE 2013, 8, e67686. [Google Scholar] [CrossRef] [PubMed]
- Yokobori, T.; Suzuki, S.; Tanaka, N.; Inose, T.; Sohda, M.; Sano, A.; Sakai, M.; Nakajima, M.; Miyazaki, T.; Kato, H.; et al. Mir-150 is associated with poor prognosis in esophageal squamous cell carcinoma via targeting the emt inducer zeb1. Cancer Sci. 2013, 104, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Yang, Z.; Ye, W.; Xu, H.; Hua, X. Microrna-150 predicts a favorable prognosis in patients with epithelial ovarian cancer, and inhibits cell invasion and metastasis by suppressing transcriptional repressor zeb1. PLoS ONE 2014, 9, e103965. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.G.; McCarthy, S.; Gruidl, M.; Timme, C.; Yeatman, T.J. Microrna-147 induces a mesenchymal-to-epithelial transition (met) and reverses egfr inhibitor resistance. PLoS ONE 2014, 9, e84597. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Li, M.; An, J.; Zhao, B.; Zhong, W.; Gu, Q.; Cao, L.; Yang, H.; Hu, C. Microrna-33b inhibits lung adenocarcinoma cell growth, invasion, and epithelial-mesenchymal transition by suppressing wnt/beta-catenin/zeb1 signaling. Int. J. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Yanaka, Y.; Muramatsu, T.; Uetake, H.; Kozaki, K.I.; Inazawa, J. Mir-544a induces epithelial-mesenchymal transition through the activation of wnt signaling pathway in gastric cancer. Carcinogenesis 2015. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Hara, T.; Choi, Y.; Subramanian, M.; Francis, P.; Bilke, S.; Walker, R.L.; Pineda, M.; Zhu, Y.; Yang, Y.; et al. A p21-zeb1 complex inhibits epithelial-mesenchymal transition through the microrna 183-96-182 cluster. Mol. Cell. Biol. 2014, 34, 533–550. [Google Scholar] [CrossRef] [PubMed]
- White, E.J.; Brewer, G.; Wilson, G.M. Post-transcriptional control of gene expression by auf1: Mechanisms, physiological targets, and regulation. Biochim. Biophys. Acta. 2013, 1829, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Al-Khalaf, H.H.; Aboussekhra, A. Microrna-141 and microrna-146b-5p inhibit the prometastatic mesenchymal characteristics through the rna-binding protein auf1 targeting the transcription factor zeb1 and the protein kinase akt. J. Biol. Chem. 2014, 289, 31433–31447. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, Y.; Zheng, X.; Tu, X.; Li, H.; Chen, J.; Zang, Y.; Zhang, J. Loss of microrna-101 promotes epithelial to mesenchymal transition in hepatocytes. J. Cell. Physiol. 2015, 230, 2706–2717. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Xie, M.; Shi, Y.; Luo, B.; Gong, G.; Li, J.; Wang, J.; Zhao, W.; Zi, Y.; Wu, X.; et al. Microrna-153 functions as a tumor suppressor by targeting set7 and zeb2 in ovarian cancer cells. Oncol. Rep. 2015, 34, 111–120. [Google Scholar] [PubMed]
- Huang, N.; Wu, Z.; Lin, L.; Zhou, M.; Wang, L.; Ma, H.; Xia, J.; Bin, J.; Liao, Y.; Liao, W. Mir-338-3p inhibits epithelial-mesenchymal transition in gastric cancer cells by targeting zeb2 and macc1/met/akt signaling. Oncotarget 2015, 6, 15222–15234. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Qiu, K.; Li, M.; Liang, Y. Double-negative feedback loop between long non-coding rna tug1 and mir-145 promotes epithelial to mesenchymal transition and radioresistance in human bladder cancer cells. FEBS Lett. 2015, 589, 3175–3181. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Wang, M.; Guo, W.; Huang, S.; Wang, Z.; Zhao, X.; Du, H.; Song, L.; Peng, X. Double-negative feedback loop between zeb2 and mir-145 regulates epithelial-mesenchymal transition and stem cell properties in prostate cancer cells. Cell Tissue Res. 2014, 358, 763–778. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.B.; Luo, H.P.; Shi, Q.; Hao, Z.N.; Ding, Y.; Wang, Q.S.; Li, S.B.; Xiao, G.C.; Tong, S.L. Mir-132 inhibits colorectal cancer invasion and metastasis via directly targeting zeb2. World J. Gastroenterol. 2014, 20, 6515–6522. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Li, Y.; Fang, N.; Liu, B.; Zu, L.; Chang, R.; Li, X.; Zhou, Q. Mir-132 suppresses the migration and invasion of lung cancer cells via targeting the emt regulator zeb2. PLoS ONE 2014, 9, e91827. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, C.; Chen, Z.; Jin, Y.; Wang, Y.; Kolokythas, A.; Dai, Y.; Zhou, X. Microrna-138 suppresses epithelial-mesenchymal transition in squamous cell carcinoma cell lines. Biochem. J. 2011, 440, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Stinson, S.; Lackner, M.R.; Adai, A.T.; Yu, N.; Kim, H.J.; O’Brien, C.; Spoerke, J.; Jhunjhunwala, S.; Boyd, Z.; Januario, T.; et al. Mir-221/222 targeting of trichorhinophalangeal 1 (trps1) promotes epithelial-to-mesenchymal transition in breast cancer. Sci. Signal 2011, 4, pt5. [Google Scholar] [PubMed]
- Stinson, S.; Lackner, M.R.; Adai, A.T.; Yu, N.; Kim, H.J.; O’Brien, C.; Spoerke, J.; Jhunjhunwala, S.; Boyd, Z.; Januario, T.; et al. Trps1 targeting by mir-221/222 promotes the epithelial-to-mesenchymal transition in breast cancer. Sci. Signal 2011, 4, ra41. [Google Scholar] [PubMed]
- Wang, Y.; Zhou, B.P. Epithelial-mesenchymal transition---A hallmark of breast cancer metastasis. Cancer Hallm. 2013, 1, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B.P. The role of snail in emt and tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Dong, C.; Zhou, B.P. Epigenetic regulation of emt: The snail story. Curr. Pharm. Des. 2014, 20, 1698–1705. [Google Scholar] [CrossRef] [PubMed]
- Baulida, J.; Garcia de Herreros, A. Snail1-driven plasticity of epithelial and mesenchymal cells sustains cancer malignancy. Biochim. Biophys. Acta. 2015, 1856, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.; Ayyanathan, K. Snail/gfi-1 (snag) family zinc finger proteins in transcription regulation, chromatin dynamics, cell signaling, development, and disease. Cytokine Growth Factor. Rev. 2013, 24, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, H.; Murayama, T.; Yokode, M.; Mori, S.; Sano, H.; Ozaki, H.; Yokota, Y.; Nishikawa, S.; Kita, T. A novel snail-related transcription factor smuc regulates basic helix-loop-helix transcription factor activities via specific e-box motifs. Nucleic Acids Res. 2000, 28, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Bueno, G.; Cubillo, E.; Sarrio, D.; Peinado, H.; Rodriguez-Pinilla, S.M.; Villa, S.; Bolos, V.; Jorda, M.; Fabra, A.; Portillo, F.; et al. Genetic profiling of epithelial cells expressing e-cadherin repressors reveals a distinct role for snail, slug, and e47 factors in epithelial-mesenchymal transition. Cancer Res. 2006, 66, 9543–9556. [Google Scholar] [CrossRef] [PubMed]
- Olmeda, D.; Montes, A.; Moreno-Bueno, G.; Flores, J.M.; Portillo, F.; Cano, A. Snai1 and snai2 collaborate on tumor growth and metastasis properties of mouse skin carcinoma cell lines. Oncogene 2008, 27, 4690–4701. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Lopez, A.; Moreno-Bueno, G.; Cano, A. Role of microrna in epithelial to mesenchymal transition and metastasis and clinical perspectives. Cancer Manag. Res. 2014, 6, 205–216. [Google Scholar] [PubMed]
- Diaz-Lopez, A.; Diaz-Martin, J.; Moreno-Bueno, G.; Cuevas, E.P.; Santos, V.; Olmeda, D.; Portillo, F.; Palacios, J.; Cano, A. Zeb1 and snail1 engage mir-200f transcriptional and epigenetic regulation during emt. J. Int. Cancer 2015, 136, E62–E73. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Martin, J.; Diaz-Lopez, A.; Moreno-Bueno, G.; Castilla, M.A.; Rosa-Rosa, J.M.; Cano, A.; Palacios, J. A core microrna signature associated with inducers of the epithelial-to-mesenchymal transition. J. Pathol. 2014, 232, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Kumarswamy, R.; Mudduluru, G.; Ceppi, P.; Muppala, S.; Kozlowski, M.; Niklinski, J.; Papotti, M.; Allgayer, H. Microrna-30a inhibits epithelial-to-mesenchymal transition by targeting snai1 and is downregulated in non-small cell lung cancer. J. Int. Cancer 2012, 130, 2044–2053. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Tu, K.; Liu, Q. Effects of microrna-30a on migration, invasion and prognosis of hepatocellular carcinoma. FEBS Lett. 2014, 588, 3089–3097. [Google Scholar] [CrossRef] [PubMed]
- Kourtidis, A.; Ngok, S.P.; Pulimeno, P.; Feathers, R.W.; Carpio, L.R.; Baker, T.R.; Carr, J.M.; Yan, I.K.; Borges, S.; Perez, E.A.; et al. Distinct e-cadherin-based complexes regulate cell behaviour through mirna processing or src and p120 catenin activity. Nat. Cell. Biol. 2015, 17, 1145–1157. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.H.; Kim, H.S.; Li, X.Y.; Lee, I.; Choi, H.S.; Kang, S.E.; Cha, S.Y.; Ryu, J.K.; Yoon, D.; Fearon, E.R.; et al. A p53/mirna-34 axis regulates snail1-dependent cancer cell epithelial-mesenchymal transition. J. Cell Biol. 2011, 195, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Sun, Q.; Zhang, J.; Yu, J.; Chen, W.; Zhang, Z. Downregulation of mir-153 contributes to epithelial-mesenchymal transition and tumor metastasis in human epithelial cancer. Carcinogenesis 2013, 34, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Sun, J.; Bai, Z.; Li, H.; He, S.; Chen, R.; Che, X. Microrna-153 acts as a prognostic marker in gastric cancer and its role in cell migration and invasion. Onco. Targets. Ther. 2015, 8, 357–364. [Google Scholar] [PubMed]
- Bai, Z.; Sun, J.; Wang, X.; Wang, H.; Pei, H.; Zhang, Z. Microrna-153 is a prognostic marker and inhibits cell migration and invasion by targeting snai1 in human pancreatic ductal adenocarcinoma. Oncol. Rep. 2015, 34, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Mizutani, K.; Minami, A.; Nobutani, K.; Kurita, S.; Nagino, M.; Shimono, Y.; Takai, Y. Suppression of the tgf-beta1-induced protein expression of snai1 and n-cadherin by mir-199a. Genes Cells 2014, 19, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Sun, Y.; Hu, L.; Zheng, H.; Ji, P.; Pecot, C.V.; Zhao, Y.; Reynolds, S.; Cheng, H.; Rupaimoole, R.; et al. Integrated analyses identify a master microrna regulatory network for the mesenchymal subtype in serous ovarian cancer. Cancer Cell 2013, 23, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Hu, L.; Zheng, H.; Bagnoli, M.; Guo, Y.; Rupaimoole, R.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Ji, P.; Chen, K.; et al. Mir-506 inhibits multiple targets in the epithelial-to-mesenchymal transition network and is associated with good prognosis in epithelial ovarian cancer. J. Pathol. 2015, 235, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Arora, H.; Qureshi, R.; Park, W.Y. Mir-506 regulates epithelial mesenchymal transition in breast cancer cell lines. PLoS ONE 2013, 8, e64273. [Google Scholar] [CrossRef] [PubMed]
- Roy-Chaudhuri, B.; Valdmanis, P.N.; Zhang, Y.; Wang, Q.; Luo, Q.J.; Kay, M.A. Regulation of microrna-mediated gene silencing by microrna precursors. Nat. Struct. Mol. Biol. 2014, 21, 825–832. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Zhou, X.; Li, S.; Jin, Y.; Chen, Z.; Chen, D.; Cai, Y.; Liu, Z.; Zhao, T.; Wang, A. Microrna-181a suppresses salivary adenoid cystic carcinoma metastasis by targeting mapk-snai2 pathway. Biochim. Biophys. Acta. 2013, 1830, 5258–5266. [Google Scholar] [CrossRef] [PubMed]
- Moes, M.; Le Bechec, A.; Crespo, I.; Laurini, C.; Halavatyi, A.; Vetter, G.; Del Sol, A.; Friederich, E. A novel network integrating a mirna-203/snai1 feedback loop which regulates epithelial to mesenchymal transition. PLoS ONE 2012, 7, e35440. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Park, S.I.; McCauley, L.K.; Wang, C.Y. Signaling between transforming growth factor beta (tgf-beta) and transcription factor snai2 represses expression of microrna mir-203 to promote epithelial-mesenchymal transition and tumor metastasis. J. Bio. Chem. 2013, 288, 10241–10253. [Google Scholar] [CrossRef] [PubMed]
- Hamamori, Y.; Sartorelli, V.; Ogryzko, V.; Puri, P.L.; Wu, H.Y.; Wang, J.Y.; Nakatani, Y.; Kedes, L. Regulation of histone acetyltransferases p300 and pcaf by the bhlh protein twist and adenoviral oncoprotein e1a. Cell 1999, 96, 405–413. [Google Scholar] [CrossRef]
- Castanon, I.; Von Stetina, S.; Kass, J.; Baylies, M.K. Dimerization partners determine the activity of the twist bhlh protein during drosophila mesoderm development. Development 2001, 128, 3145–3159. [Google Scholar] [PubMed]
- Firulli, B.A.; Krawchuk, D.; Centonze, V.E.; Vargesson, N.; Virshup, D.M.; Conway, S.J.; Cserjesi, P.; Laufer, E.; Firulli, A.B. Altered twist1 and hand2 dimerization is associated with saethre-chotzen syndrome and limb abnormalities. Nat. Genet. 2005, 37, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Connerney, J.; Andreeva, V.; Leshem, Y.; Muentener, C.; Mercado, M.A.; Spicer, D.B. Twist1 dimer selection regulates cranial suture patterning and fusion. Dev. Dyn. 2006, 235, 1345–1357. [Google Scholar] [CrossRef] [PubMed]
- Connerney, J.; Andreeva, V.; Leshem, Y.; Mercado, M.A.; Dowell, K.; Yang, X.; Lindner, V.; Friesel, R.E.; Spicer, D.B. Twist1 homodimers enhance fgf responsiveness of the cranial sutures and promote suture closure. Dev. Biol. 2008, 318, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Yuan, J.; Li, K. Emt transcription factors: Implication in osteosarcoma. Med. Oncol. 2013, 30, 697. [Google Scholar] [CrossRef] [PubMed]
- Tseng, J.C.; Chen, H.F.; Wu, K.J. A twist tale of cancer metastasis and tumor angiogenesis. Histol. Histopathol. 2015, 30, 1283–1294. [Google Scholar] [PubMed]
- Puisieux, A.; Valsesia-Wittmann, S.; Ansieau, S. A twist for survival and cancer progression. Br. J. Cancer 2006, 94, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Norozi, F.; Ahmadzadeh, A.; Shahjahani, M.; Shahrabi, S.; Saki, N. Twist as a new prognostic marker in hematological malignancies. Clin. Transl. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Chen, H.C.; Zhang, D.; Fu, J. Twist: A molecular target in cancer therapeutics. Tumour Biol. 2013, 34, 2497–2506. [Google Scholar] [CrossRef] [PubMed]
- Karreth, F.; Tuveson, D.A. Twist induces an epithelial-mesenchymal transition to facilitate tumor metastasis. Cancer Biol. Ther. 2004, 3, 1058–1059. [Google Scholar] [CrossRef] [PubMed]
- Hung, J.J.; Yang, M.H.; Hsu, H.S.; Hsu, W.H.; Liu, J.S.; Wu, K.J. Prognostic significance of hypoxia-inducible factor-1alpha, twist1 and snail expression in resectable non-small cell lung cancer. Thorax 2009, 64, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, L.Y.; Wong, G.; Earle, C.; Krueger, K.; Spevak, C.C. Hyaluronan-cd44 interaction promotes c-src-mediated twist signaling, microrna-10b expression, and rhoa/rhoc up-regulation, leading to rho-kinase-associated cytoskeleton activation and breast tumor cell invasion. J. Biol. Chem. 2010, 285, 36721–36735. [Google Scholar] [CrossRef] [PubMed]
- Haque, I.; Banerjee, S.; Mehta, S.; De, A.; Majumder, M.; Mayo, M.S.; Kambhampati, S.; Campbell, D.R.; Banerjee, S.K. Cysteine-rich 61-connective tissue growth factor-nephroblastoma-overexpressed 5 (ccn5)/wnt-1-induced signaling protein-2 (wisp-2) regulates microrna-10b via hypoxia-inducible factor-1alpha-twist signaling networks in human breast cancer cells. J. Biol. Chem. 2011, 286, 43475–43485. [Google Scholar] [CrossRef] [PubMed]
- Loayza-Puch, F.; Yoshida, Y.; Matsuzaki, T.; Takahashi, C.; Kitayama, H.; Noda, M. Hypoxia and ras-signaling pathways converge on, and cooperatively downregulate, the reck tumor-suppressor protein through micrornas. Oncogene 2010, 29, 2638–2648. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.; Chen, R.; Alvero, A.B.; Fu, H.H.; Holmberg, J.; Glackin, C.; Rutherford, T.; Mor, G. Twisting stemness, inflammation and proliferation of epithelial ovarian cancer cells through mir199a2/214. Oncogene 2010, 29, 3545–3553. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Y.; Zhang, H.; Liu, X.; Gong, T.; Li, M.; Sun, L.; Ji, G.; Shi, Y.; Han, Z.; et al. Mirna-223 promotes gastric cancer invasion and metastasis by targeting tumor suppressor epb41l3. Mol. Cancer. Res. 2011, 9, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Glaser, S.S.; Francis, H.; DeMorrow, S.; Han, Y.; Passarini, J.D.; Stokes, A.; Cleary, J.P.; Liu, X.; Venter, J.; et al. Functional analysis of micrornas in human hepatocellular cancer stem cells. J. Cell. Mol. Med. 2012, 16, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, J.; Huang, H.; Hou, J.; Zhang, B.; Wang, A. Mir-181a-twist1 pathway in the chemoresistance of tongue squamous cell carcinoma. Biochem. Biophys. Res. Commun. 2013, 441, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Drasin, D.J.; Guarnieri, A.L.; Neelakantan, D.; Kim, J.; Cabrera, J.H.; Wang, C.A.; Zaberezhnyy, V.; Gasparini, P.; Cascione, L.; Huebner, K.; et al. Twist1-induced mir-424 reversibly drives mesenchymal programming while inhibiting tumor initiation. Cancer Res. 2015, 75, 1908–1921. [Google Scholar] [CrossRef] [PubMed]
- Wiklund, E.D.; Bramsen, J.B.; Hulf, T.; Dyrskjot, L.; Ramanathan, R.; Hansen, T.B.; Villadsen, S.B.; Gao, S.; Ostenfeld, M.S.; Borre, M.; et al. Coordinated epigenetic repression of the mir-200 family and mir-205 in invasive bladder cancer. Int. J. Cancer. 2011, 128, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Hsu, C.C.; Chang, C.H.; Tsai, L.L.; Chang, Y.C.; Lu, S.W.; Yu, C.H.; Huang, H.S.; Wang, J.J.; Tsai, C.H.; et al. Let-7d functions as novel regulator of epithelial-mesenchymal transition and chemoresistant property in oral cancer. Oncol. Rep. 2011, 26, 1003–1010. [Google Scholar] [PubMed]
- Haga, C.L.; Phinney, D.G. Micrornas in the imprinted dlk1-dio3 region repress the epithelial-to-mesenchymal transition by targeting the twist1 protein signaling network. J. Biol. Chem. 2012, 287, 42695–42707. [Google Scholar] [CrossRef] [PubMed]
- Nairismagi, M.L.; Vislovukh, A.; Meng, Q.; Kratassiouk, G.; Beldiman, C.; Petretich, M.; Groisman, R.; Fuchtbauer, E.M.; Harel-Bellan, A.; Groisman, I. Translational control of twist1 expression in mcf-10a cell lines recapitulating breast cancer progression. Oncogene 2012, 31, 4960–4966. [Google Scholar] [CrossRef] [PubMed]
- Nairismagi, M.L.; Fuchtbauer, A.; Labouriau, R.; Bramsen, J.B.; Fuchtbauer, E.M. The proto-oncogene twist1 is regulated by micrornas. PLoS ONE 2013, 8, e66070. [Google Scholar] [CrossRef] [PubMed]
- Li, L.Z.; Zhang, C.Z.; Liu, L.L.; Yi, C.; Lu, S.X.; Zhou, X.; Zhang, Z.J.; Peng, Y.H.; Yang, Y.Z.; Yun, J.P. Mir-720 inhibits tumor invasion and migration in breast cancer by targeting twist1. Carcinogenesis 2014, 35, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, J.M.; Elahi, A.; Clark, C.W.; Wang, J.; Humphries, L.A.; Centeno, B.; Bloom, G.; Fuchs, B.C.; Yeatman, T.; Shibata, D. Mir-675 mediates downregulation of twist1 and rb in afp-secreting hepatocellular carcinoma. Ann. Surg. Oncol. 2013, 20, S625–S635. [Google Scholar] [CrossRef] [PubMed]
- Tsukerman, P.; Yamin, R.; Seidel, E.; Khawaled, S.; Schmiedel, D.; Bar-Mag, T.; Mandelboim, O. Mir-520d-5p directly targets twist1 and downregulates the metastamir mir-10b. Oncotarget 2014, 5, 12141–12150. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cui, J.; Liao, G.; Zhang, Y.; Ye, K.; Lu, T.; Qi, J.; Wan, G. Mir-137 regulates epithelial-mesenchymal transition in gastrointestinal stromal tumor. Tumour Biol. 2014, 35, 9131–9138. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Huang, Z.; Wu, S.; Zang, X.; Liu, M.; Shi, J. Mir-33a is up-regulated in chemoresistant osteosarcoma and promotes osteosarcoma cell resistance to cisplatin by down-regulating twist. J. Exp. Clin. Cancer Res. 2014, 33, 12. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yang, J.; Li, J.; Shen, X.; Le, Y.; Zhou, C.; Wang, S.; Zhang, S.; Xu, D.; Gong, Z. Mircorna-33a inhibits epithelial-to-mesenchymal transition and metastasis and could be a prognostic marker in non-small cell lung cancer. Sci. Rep. 2015, 5, 13677. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Shen, H.; Yin, X.; Long, L.; Xie, C.; Liu, Y.; Hui, L.; Lin, X.; Fang, Y.; Cao, Y.; et al. Mir-186 regulation of twist1 and ovarian cancer sensitivity to cisplatin. Oncogene 2015. [Google Scholar]
- Chang, Y.S.; Chen, W.Y.; Yin, J.J.; Sheppard-Tillman, H.; Huang, J.; Liu, Y.N. Egf receptor promotes prostate cancer bone metastasis by downregulating mir-1 and activating twist1. Cancer Res. 2015, 75, 3077–3086. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Morel, A.P.; Lievre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef] [PubMed]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The emt-activator zeb1 promotes tumorigenicity by repressing stemness-inhibiting micrornas. Nat. Cell. Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Schepers, G.E.; Teasdale, R.D.; Koopman, P. Twenty pairs of sox: Extent, homology, and nomenclature of the mouse and human sox transcription factor gene families. Dev. Cell 2002, 3, 167–170. [Google Scholar] [CrossRef]
- Stolt, C.C.; Wegner, M. Soxe function in vertebrate nervous system development. Int. J. Biochem. Cell. Biol 2010, 42, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Guth, S.I.; Bosl, M.R.; Sock, E.; Wegner, M. Evolutionary conserved sequence elements with embryonic enhancer activity in the vicinity of the mammalian sox8 gene. Int. J. Biochem. Cell. Biol. 2010, 42, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Wegner, M. All purpose sox: The many roles of sox proteins in gene expression. Int. J. Biochem. Cell. Biol. 2010, 42, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Sakai, D.; Suzuki, T.; Osumi, N.; Wakamatsu, Y. Cooperative action of sox9, snail2 and pka signaling in early neural crest development. Development 2006, 133, 1323–1333. [Google Scholar] [CrossRef] [PubMed]
- Suske, G.; Bruford, E.; Philipsen, S. Mammalian sp/klf transcription factors: Bring in the family. Genomics 2005, 85, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.; Fleetwood, J.; Eaton, S.; Crossley, M.; Bao, S. Kruppel-like transcription factors: A functional family. Int. J. Biochem. Cell. Biol. 2008, 40, 1996–2001. [Google Scholar] [CrossRef] [PubMed]
- Chambers, I.; Colby, D.; Robertson, M.; Nichols, J.; Lee, S.; Tweedie, S.; Smith, A. Functional expression cloning of nanog, a pluripotency sustaining factor in embryonic stem cells. Cell 2003, 113, 643–655. [Google Scholar] [CrossRef]
- Sureban, S.M.; May, R.; Qu, D.; Weygant, N.; Chandrakesan, P.; Ali, N.; Lightfoot, S.A.; Pantazis, P.; Rao, C.V.; Postier, R.G.; et al. Dclk1 regulates pluripotency and angiogenic factors via microrna-dependent mechanisms in pancreatic cancer. PLoS One 2013, 8, e73940. [Google Scholar] [CrossRef] [PubMed]
- Sureban, S.M.; May, R.; Weygant, N.; Qu, D.; Chandrakesan, P.; Bannerman-Menson, E.; Ali, N.; Pantazis, P.; Westphalen, C.B.; Wang, T.C.; et al. Xmd8-92 inhibits pancreatic tumor xenograft growth via a dclk1-dependent mechanism. Cancer Lett. 2014, 351, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Wu, Y.; Liu, B.; Wang, P.; Chen, Y. Downregulation of mir-638 promotes invasion and proliferation by regulating sox2 and induces emt in nsclc. FEBS Lett. 2014, 588, 2238–2245. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.C.; Chan, L.S.; Ip, J.C.; Lo, C.; Yip, T.T.; Ngan, R.K.; Wong, R.N.; Lo, K.W.; Ng, W.T.; Lee, A.W.; et al. Therapeutic targeting of cbp/beta-catenin signaling reduces cancer stem-like population and synergistically suppresses growth of ebv-positive nasopharyngeal carcinoma cells with cisplatin. Sci. Rep. 2015, 5, 9979. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lv, Z.; He, G.; Wang, J.; Zhang, X.; Lu, G.; Ren, X.; Wang, F.; Zhu, X.; Ding, Y.; et al. The sox17/mir-371-5p/sox2 axis inhibits emt, stem cell properties and metastasis in colorectal cancer. Oncotarget 2015, 6, 9099–9112. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.D.; Diao, L.T.; Xu, H.; Xiao, Z.D.; Li, J.H.; Zhou, H.; Qu, L.H. Beta-catenin/lef1 transactivates the microrna-371-373 cluster that modulates the wnt/beta-catenin-signaling pathway. Oncogene 2012, 31, 2968–2978. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.; Hermeking, H. Znf281/zbp-99: A new player in epithelial-mesenchymal transition, stemness, and cancer. J. Mol. Med. (Berl.) 2014, 92, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Ocana, O.H.; Corcoles, R.; Fabra, A.; Moreno-Bueno, G.; Acloque, H.; Vega, S.; Barrallo-Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer prrx1. Cancer Cell 2012, 22, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zheng, L.; Huang, J.; Gao, F.; Lin, X.; He, L.; Li, D.; Li, Z.; Ding, Y.; Chen, L. Mir-124 radiosensitizes human colorectal cancer cells by targeting prrx1. PLoS ONE 2014, 9, e93917. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Zhuang, X.; Ju, S.; Zhang, S.; Jiang, H.; Mu, J.; Zhang, L.; Miller, D.; Grizzle, W.; Zhang, H.G. Mir-155 promotes macroscopic tumor formation yet inhibits tumor dissemination from mammary fat pads to the lung by preventing emt. Oncogene 2011, 30, 3440–3453. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abba, M.L.; Patil, N.; Leupold, J.H.; Allgayer, H. MicroRNA Regulation of Epithelial to Mesenchymal Transition. J. Clin. Med. 2016, 5, 8. https://doi.org/10.3390/jcm5010008
Abba ML, Patil N, Leupold JH, Allgayer H. MicroRNA Regulation of Epithelial to Mesenchymal Transition. Journal of Clinical Medicine. 2016; 5(1):8. https://doi.org/10.3390/jcm5010008
Chicago/Turabian StyleAbba, Mohammed L., Nitin Patil, Jörg Hendrik Leupold, and Heike Allgayer. 2016. "MicroRNA Regulation of Epithelial to Mesenchymal Transition" Journal of Clinical Medicine 5, no. 1: 8. https://doi.org/10.3390/jcm5010008