
Epithelial-to-Mesenchymal Transition and Cancer Invasiveness: What Can We Learn from Cholangiocarcinoma?


Abstract
:1. Introduction
2. EMT Involvement in Cancer Cell Dissemination
- (1)
- Detachment from the highly-organized epithelial layer; this requires reducing cell-cell contacts and rearranging the cytoskeletal architecture, in favor of a motile phenotype;
- (2)
- Impairment of the integrity of the basement membrane through active proteolysis, and then invasion of the surrounding stroma as strands or cords. Once in the tumor stroma, cells can efficiently cross-talk with multiple mesenchymal and inflammatory cell types, which in turn support their invasiveness;
- (3)
- Dissemination at distance through the lymphatic and/or hematogenous circulation, taking advantage of the leaky neovasculature arising in the tumor microenvironment; and
- (4)
3. Evidence for EMT in Human Carcinomas
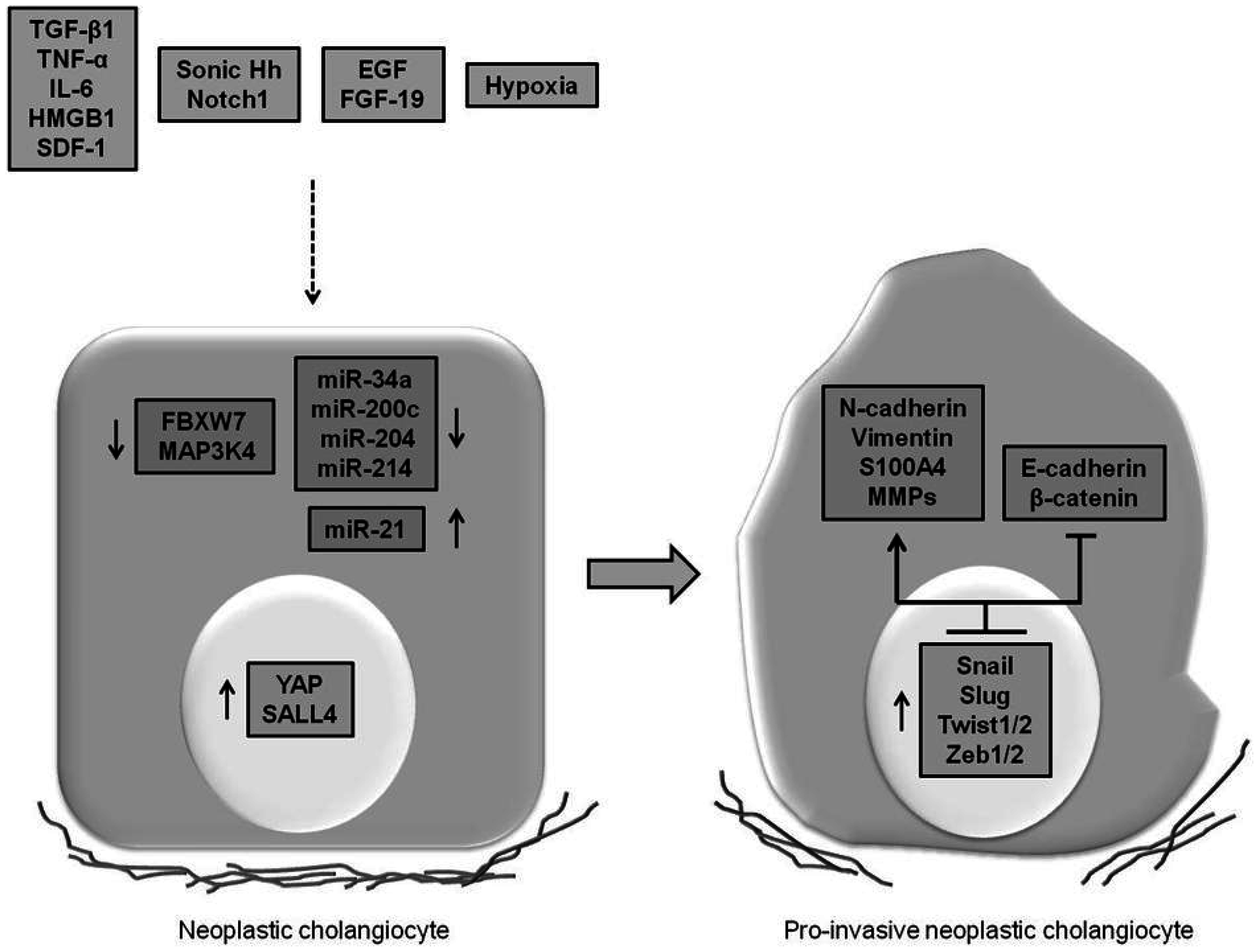
4. Expression of EMT Features in CCA and Underlying Mechanisms Involved
4.1. Cytokines, Growth Factors and Morphogens Promoting EMT

{kind=link}
| EMT Inducer | References |
|---|---|
| Inflammatory cyto/chemokines | |
| TGF-β1 | [55,56,57,58] |
| TNF-α | [59] |
| IL-6 | [60] |
| HMGB1 | [61] |
| SDF-1 | [50] |
| Growth factors | |
| EGF | [62,63] |
| FGF-19 | [64] |
| Morphogens | |
| Notch1/Sox9 | [65,66,67] |
| Sonic Hh | [68] |
4.2. miRNAs Promoting EMT
4.3. Oncogenes and Tumor Suppressor Genes Regulating EMT
4.4. Disease Mechanisms Inducting the “Transitional” Phenotype
5. EMT and CAFs Generation: Insights from CCA
6. CCA as Model to Redefine the Concept of EMT in Cancer Invasiveness
7. Conclusions

Acknowledgments
Conflicts of Interest
References
- Gatto, M.; Bragazzi, M.C.; Semeraro, R.; Napoli, C.; Gentile, R.; Torrice, A.; Gaudio, E.; Alvaro, D. Cholangiocarcinoma: Update and future perspectives. Dig. Liver Dis. 2010, 42, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Davidson, B.R.; Goldin, R.D.; Heaton, N.; Karani, J.; Pereira, S.P.; Rosenberg, W.M.; Tait, P.; Taylor-Robinson, S.D.; Thillainayagam, A.V.; et al. Guidelines for the diagnosis and treatment of cholangiocarcinoma: An update. Gut 2012, 61, 1657–1669. [Google Scholar] [CrossRef] [PubMed]
- Fabris, L.; Alvaro, D. The prognosis of perihilar cholangiocarcinoma after radical treatments. Hepatology 2012, 56, 800–802. [Google Scholar] [CrossRef] [PubMed]
- Zabron, A.; Edwards, R.J.; Khan, S.A. The challenge of cholangiocarcinoma: Dissecting the molecular mechanisms of an insidious cancer. Dis. Model. Mech. 2013, 6, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.G.; Vignjevic, D.M. Modes of cancer cell invasion and the role of the microenvironment. Curr. Opin. Cell Biol. 2015, 36, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Fabris, L.; Cadamuro, M.; Moserle, L.; Dziura, J.; Cong, X.; Sambado, L.; Nardo, G.; Sonzogni, A.; Colledan, M.; Furlanetto, A.; et al. Nuclear expression of S100A4 calcium-binding protein increases cholangiocarcinoma invasiveness and metastatisation. Hepatology 2011, 54, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Gurzu, S.; Turdean, S.; Kovecsi, A.; Contac, A.O.; Jung, I. Epithelial-mesenchymal, mesenchymal-epithelial, and endothelial-mesenchymal transitions in malignant tumors: An update. World J. Clin. Cases 2015, 3, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Acloque, H.; Adams, M.S.; Fishwick, K.; Bronner-Fraser, M.; Nieto, M.A. Epithelial-mesenchymal transitions: The importance of changing cell state in development and disease. J. Clin. Investig. 2009, 119, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Fabris, L.; Strazzabosco, M. Epithelial-mesenchymal interactions in biliary diseases. Semin. Liver Dis. 2011, 31, 11–32. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.; Guilford, P.; Thiery, J.P. Early events in cell adhesion and polarity during epithelial-mesenchymal transition. J. Cell Sci. 2012, 125, 4417–4422. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, Y.; Lamouille, S.; Derynck, R. TGF-β signaling and epithelial-mesenchymal transition in cancer progression. Curr. Opin. Oncol. 2013, 25, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Parker Kerrigan, B.C.; Yang, D.; Hu, L.; Shmulevich, I.; Sood, A.K.; Xue, F.; Zhang, W. Post-transcriptional regulatory network of epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions. J. Hematol. Oncol. 2014, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, S.; Langhans, S.A. Crosstalk of Oncogenic Signaling Pathways during Epithelial-Mesenchymal Transition. Front. Oncol. 2014, 4, 358. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, B.; Zhou, Q.; Wu, Y.; Shang, D.; Guo, Y.; Song, Z.; Zheng, Q.; Xiong, J. Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial-mesenchymal transition. Carcinogenesis 2013, 34, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; He, Y.Y. Autophagy deficiency stabilizes TWIST1 to promote epithelial-mesenchymal transition. Autophagy 2014, 10, 1864–1865. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Portillo, F.; Cano, A. Transcriptional regulation of cadherins during development and carcinogenesis. Int. J. Dev. Biol. 2004, 48, 365–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.M.; Dedhar, S.; Kalluri, R.; Thompson, E.W. The epithelial-mesenchymal transition: New insights in signaling, development, and disease. J. Cell Biol. 2006, 172, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, C.; Gradilone, A.; Naso, G.; Vincenzi, B.; Petracca, A.; Nicolazzo, C.; Palazzo, A.; Saltarelli, R.; Spremberg, F.; Cortesi, E.; et al. Epithelial-mesenchymal transition and stemness features in circulating tumor cells from breast cancer patients. Breast Cancer Res. Treat. 2011, 130, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Gradilone, A.; Raimondi, C.; Nicolazzo, C.; Petracca, A.; Gandini, O.; Vincenzi, B.; Naso, G.; Aglianò, A.M.; Cortesi, E.; Gazzaniga, P. Circulating tumour cells lacking cytokeratin in breast cancer: The importance of being mesenchymal. J. Cell Mol. Med. 2011, 15, 1066–1070. [Google Scholar] [CrossRef] [PubMed]
- Sarrió, D.; Rodriguez-Pinilla, S.M.; Hardisson, D.; Cano, A.; Moreno-Bueno, G.; Palacios, J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008, 68, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, X.; Shang, M.; Zhang, Y.; Xia, B.; Niu, M.; Liu, Y.; Pang, D. Dysregulated expression of Slug, vimentin, and E-cadherin correlates with poor clinical outcome in patients with basal-like breast cancer. J. Surg. Oncol. 2013, 107, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Fuchs, B.C.; Fujii, T.; Shimoyama, Y.; Sugimoto, H.; Nomoto, S.; Takeda, S.; Tanabe, K.K.; Kodera, Y.; Nakao, A. Epithelial-to-mesenchymal transition predicts prognosis of pancreatic cancer. Surgery 2013, 154, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Murai, T.; Yamada, S.; Fuchs, B.C.; Fujii, T.; Nakayama, G.; Sugimoto, H.; Koike, M.; Fujiwara, M.; Tanabe, K.K.; Kodera, Y. Epithelial-to-mesenchymal transition predicts prognosis in clinical gastric cancer. J. Surg. Oncol. 2014, 109, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.J.; Wan, X.B.; Yang, Z.L.; Fu, X.H.; Huang, Y.; Chen, D.K.; Song, S.X.; Liu, Q.; Xiao, H.Y.; Wang, L.; Wang, J.P. Snail promotes lymph node metastasis and Twist enhances tumor deposit formation through epithelial-mesenchymal transition in colorectal cancer. Hum. Pathol. 2013, 44, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wu, H.; Zhang, M.; Ding, L.; Meng, F.; Fan, X. Expression of the epithelial-mesenchymal transition-related proteins and their clinical significance in lung adenocarcinoma. Diagn. Pathol. 2013, 8, 89. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.J.; Wang, H.Y.; Zhang, H.T.; Su, J.M.; Zhu, J.; Wang, H.B.; Zhou, W.Y.; Zhang, H.; Zhao, M.C.; Zhang, L.; et al. Transforming growth factor-β1 promotes lung adenocarcinoma invasion and metastasis by epithelial-to-mesenchymal transition. Mol. Cell Biochem. 2011, 355, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Park, S.M.; Kim, I.K.; Nam, I.K.; Baek, K.E.; Im, M.J.; Yoo, J.M.; Park, S.H.; Ryu, K.J.; Han, H.T.; et al. RhoGDI2 promotes epithelial-mesenchymal transition via induction of Snail in gastric cancer cells. Oncotarget 2014, 5, 1554–1564. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, G.; Zhang, H.; Zhang, F.; Zhou, B.; Ning, F.; Wang, H.S.; Cai, S.H.; Du, J. Acquisition of epithelial-mesenchymal transition phenotype and cancer stem cell-like properties in cisplatin-resistant lung cancer cells through AKT/β-catenin/Snail signaling pathway. Eur. J. Pharmacol. 2014, 723, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.S.; Yang, X.S.; Xia, M.; Jiang, H.Y.; Hou, J.Q. Silencing of twist expression by RNA interference suppresses epithelial-mesenchymal transition, invasion, and metastasis of ovarian cancer. Asian Pac. J. Cancer Prev. 2012, 13, 4435–4439. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, A.; Agyin, J.K.; Wang, L.; Jiang, H.Y.; Hou, J.Q. Inhibition of pulmonary and skeletal metastasis by a transforming growth factor-β type I receptor kinase inhibitor. Cancer Res. 2006, 66, 6714–6721. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, R.; Itoh, S.; Gui, T.; Gai, Z.; Oikawa, K.; Kawai, M.; Tani, M.; Yamaue, H.; Muragaki, Y. SNAIL induces epithelial-to-mesenchymal transition in a human pancreatic cancer cell line (BxPC3) and promotes distant metastasis and invasiveness in vivo. Exp. Mol. Pathol. 2010, 89, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Cadamuro, M.; Nardo, G.; Indraccolo, S.; Dall’olmo, L.; Sambado, L.; Moserle, L.; Franceschet, I.; Colledan, M.; Massani, M.; Stecca, T.; et al. Platelet-derived growth factor-D and Rho GTPases regulate recruitment of cancer-associated fibroblasts in cholangiocarcinoma. Hepatology 2013, 58, 1042–1053. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Wang, X.; Wang, Z.; Dai, L.; Zhang, G.; Yan, Q.; Zhou, W. Clinicopathological and prognostic significance of epithelial mesenchymal transition-related protein expression in intrahepatic cholangiocarcinoma. Onco. Targets Ther. 2012, 5, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Nitta, T.; Mitsuhashi, T.; Hatanaka, Y.; Miyamoto, M.; Oba, K.; Tsuchikawa, T.; Suzuki, Y.; Hatanaka, K.C.; Hirano, S.; Matsuno, Y. Prognostic significance of epithelial-mesenchymal transition-related markers in extrahepatic cholangiocarcinoma: Comprehensive immunohistochemical study using a tissue microarray. Br. J. Cancer 2014, 111, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.Y.; Zhang, C.; Cai, J.B.; Shi, G.M.; Ke, A.W.; Dong, Z.R.; Zhang, P.F.; Fan, J.; Peng, B.G.; Zhou, J. Comprehensive multiple molecular profile of epithelial mesenchymal transition in intrahepatic cholangiocarcinoma patients. PLoS ONE 2014, 9, e96860. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.J.; Choi, J.H. Epithelial-mesenchymal transition phenotypes are associated with patient survival in intrahepatic cholangiocarcinoma. J. Clin. Pathol. 2014, 67, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Techasen, A.; Loilome, W.; Namwat, N.; Khuntikeo, N.; Puapairoj, A.; Jearanaikoon, P.; Saya, H.; Yongvanit, P. Loss of E-cadherin promotes migration and invasion of cholangiocarcinoma cells and serves as a potential marker of metastasis. Tumour. Biol. 2014, 35, 8645–8652. [Google Scholar] [CrossRef] [PubMed]
- Fabris, L.; Cadamuro, M.; Sambado, L.; Beretta, I.; Spirli, C.; Indraccolo, S.; Strazzabosco, M. Selective reduction in S100A4 nuclear expression by low-dose paclitaxel halts invasiveness of human cholangiocarcinoma cells through a RHO-A/CDC42-dependent mechanism [abstract]. In Proceedings of the AASLD 63rd Annual Meeting, Boston, MA, USA, 9–13 November 2012.
- Cadamuro, M.; Morton, S.D.; Strazzabosco, M.; Fabris, L. Unveiling the role of tumor reactive stroma in cholangiocarcinoma: An opportunity for new therapeutic strategies. Transl. Gastrointest. Cancer 2013, 2, 130–144. [Google Scholar]
- Cirri, P.; Chiarugi, P. Cancer associated fibroblasts: The dark side of the coin. Am. J. Cancer Res. 2011, 1, 482–497. [Google Scholar] [PubMed]
- Techasen, A.; Loilome, W.; Namwat, N.; Dokduang, H.; Jongthawin, J.; Yongvanit, P. Cytokines released from activated human macrophages induce epithelial mesenchymal transition markers of cholangiocarcinoma cells. Asian Pac. J. Cancer Prev. 2012, 13, 115–118. [Google Scholar] [PubMed]
- Thanee, M.; Loilome, W.; Techasen, A.; Namwat, N.; Boonmars, T.; Pairojkul, C.; Yongvanit, P. Quantitative changes in tumor-associated M2 macrophages characterize cholangiocarcinoma and their association with metastasis. Asian Pac. J. Cancer Prev. 2015, 16, 3043–3050. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Tajima, H.; Nakanuma, S.; Sakai, S.; Makino, I.; Kinoshita, J.; Hayashi, H.; Nakamura, K.; Oyama, K.; Nakagawara, H.; et al. Angiotensin II enhances epithelial-to-mesenchymal transition through the interaction between activated hepatic stellate cells and the stromal cell-derived factor-1/CXCR4 axis in intrahepatic cholangiocarcinoma. Int. J. Oncol. 2012, 41, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Xiao, C.H.; Tan, L.D.; Wang, Q.S.; Li, X.Q.; Feng, Y.M. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br. J. Cancer 2014, 110, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, J.; Xie, K.; Zhang, T.; Lei, Y.; Chen, Y.; Zhang, L.; Huang, K.; Wang, K.; Wu, H.; et al. FGFR4 promotes stroma-induced epithelial-to-mesenchymal transition in colorectal cancer. Cancer Res. 2013, 73, 5926–5935. [Google Scholar] [CrossRef] [PubMed]
- Sirica, A.E.; Campbell, D.J.; Dumur, C.I. Cancer-associated fibroblasts in intrahepatic cholangiocarcinoma. Curr. Opin. Gastroenterol. 2011, 27, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Zheng, Y.; Li, L.; Zhai, W.; Li, R.; Liang, Z.; Zhao, L. Adrenomedullin promotes intrahepatic cholangiocellular carcinoma metastasis and invasion by inducing epithelial-mesenchymal transition. Oncol. Rep. 2015, 34, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Harada, K.; Itatsu, K.; Ikeda, H.; Kakuda, Y.; Shimomura, S.; Shan Ren, X.; Yoneda, N.; Sasaki, M.; Nakanuma, Y. Epithelial-mesenchymal transition induced by transforming growth factor-β1/Snail activation aggravates invasive growth of cholangiocarcinoma. Am. J. Pathol. 2010, 177, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Shimura, T.; Suzuki, H.; Tsutsumi, S.; Wada, W.; Yajima, T.; Kobayahi, T.; Kubo, N.; Kuwano, H. E/N-cadherin switch mediates cancer progression via TGF-β-induced epithelial-to-mesenchymal transition in extrahepatic cholangiocarcinoma. Br. J. Cancer 2011, 105, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Hirose, A.; Tajima, H.; Ohta, T.; Tsukada, T.; Okamoto, K.; Nakanuma, S.; Sakai, S.; Kinoshita, J.; Makino, I.; Furukawa, H.; et al. Low-dose paclitaxel inhibits the induction of epidermal-mesenchymal transition in the human cholangiocarcinoma CCKS-1 cell line. Oncol. Lett. 2013, 6, 915–920. [Google Scholar] [PubMed]
- Duangkumpha, K.; Techasen, A.; Loilome, W.; Namwat, N.; Thanan, R.; Khuntikeo, N.; Yongvanit, P. BMP-7 blocks the effects of TGF-β-induced EMT in cholangiocarcinoma. Tumour. Biol. 2014, 35, 9667–9676. [Google Scholar] [CrossRef] [PubMed]
- Techasen, A.; Namwat, N.; Loilome, W.; Bungkanjana, P.; Khuntikeo, N.; Puapairoj, A.; Jearanaikoon, P.; Saya, H.; Yongvanit, P. Tumor necrosis factor-α (TNF-α) stimulates the epithelial-mesenchymal transition regulator Snail in cholangiocarcinoma. Med. Oncol. 2012, 29, 3083–3091. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.X.; Jiang, X.M.; Wang, Z.D.; Li, C.L.; Cui, Y.F. Enhanced expression of suppresser of cytokine signaling 3 inhibits the IL-6-induced epithelial-to-mesenchymal transition and cholangiocarcinoma cell metastasis. Med. Oncol. 2015, 32, 105. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.F.; Ge, F.J.; Han, B.; Yang, X.Q.; Su, H.; Zhao, A.C.; Zhao, M.H.; Yang, Y.B.; Yang, J. High-mobility group box 1 expression and lymph node metastasis in intrahepatic cholangiocarcinoma. World J. Gastroenterol. 2015, 21, 3256–3265. [Google Scholar] [PubMed]
- Lee, M.J.; Yu, G.R.; Yoo, H.J.; Kim, J.H.; Yoon, B.I.; Choi, Y.K.; Kim, D.G. ANXA8 down-regulation by EGF-FOXO4 signaling is involved in cell scattering and tumor metastasis of cholangiocarcinoma. Gastroenterology 2009, 137, 1138–1150. [Google Scholar] [CrossRef] [PubMed]
- Clapéron, A.; Mergey, M.; Nguyen Ho-Bouldoires, T.H.; Vignjevic, D.; Wendum, D.; Chrétien, Y.; Merabtene, F.; Frazao, A.; Paradis, V.; et al. EGF/EGFR axis contributes to the progression of cholangiocarcinoma through the induction of an epithelial-mesenchymal transition. J. Hepatol. 2014, 61, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.F.; Yang, X.Q.; Lu, X.F.; Guo, S.; Liu, Y.; Iqbal, M.; Ning, S.L.; Yang, H.; Suo, N.; Chen, Y.X. Fibroblast growth factor receptor 4 promotes progression and correlates to poor prognosis in cholangiocarcinoma. Biochem. Biophys. Res. Commun. 2014, 446, 54–60. [Google Scholar] [CrossRef] [PubMed]
- El Khatib, M.; Bozko, P.; Palagani, V.; Malek, N.P.; Wilkens, L.; Plentz, R.R. Activation of Notch signaling is required for cholangiocarcinoma progression and is enhanced by inactivation of p53 in vivo. PLoS ONE 2013, 8, e77433. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Wang, Y.; Peng, B.; Liang, L.; Li, J. The roles of Notch1 expression in the migration of intrahepatic cholangiocarcinoma. BMC Cancer 2013, 13, 244. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, H.; Kuroki, T.; Kitasato, A.; Adachi, T.; Tanaka, T.; Hirabaru, M.; Hirayama, T.; Kuroshima, N.; Hidaka, M.; Soyama, A.; et al. Sox9 expression in carcinogenesis and its clinical significance in intrahepatic cholangiocarcinoma. Dig. Liver Dis. 2015. [Google Scholar] [CrossRef] [PubMed]
- El Khatib, M.; Kalnytska, A.; Palagani, V.; Kossatz, U.; Manns, M.P.; Malek, N.P.; Wilkens, L.; Plentz, R.R. Inhibition of hedgehog signaling attenuates carcinogenesis in vitro and increases necrosis of cholangiocellular carcinoma. Hepatology 2013, 57, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Kiesslich, T.; Pichler, M.; Neureiter, D. Epigenetic control of epithelial-mesenchymal-transition in human cancer. Mol. Clin. Oncol. 2013, 1, 3–11. [Google Scholar] [PubMed]
- Peinado, H.; Ballestar, E.; Esteller, M.; Cano, A. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol. Cell Biol. 2004, 24, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.O.; Gu, J.M.; Kim, M.S.; Kim, H.S.; Park, Y.N.; Park, C.K.; Cho, J.W.; Park, Y.M.; Jung, G. Epigenetic changes induced by reactive oxygen species in hepatocellular carcinoma: Methylation of the E-cadherin promoter. Gastroenterology 2008, 135, 2128–2140. [Google Scholar] [CrossRef] [PubMed]
- Kurashige, J.; Mima, K.; Sawada, G.; Takahashi, Y.; Eguchi, H.; Sugimachi, K.; Mori, M.; Yanagihara, K.; Yashiro, M.; Hirakawa, K.; et al. Epigenetic modulation and repression of miR-200b by cancer-associated fibroblasts contribute to cancer invasion and peritoneal dissemination in gastric cancer. Carcinogenesis 2015, 36, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Deng, Q.; Pan, Y.; Peng, M.; Wang, X.; Song, L.; Xiao, M.; Wang, Z. Cancer-associated fibroblasts enhance the migration ability of ovarian cancer cells by increasing EZH2 expression. Int. J. Mol. Med. 2014, 33, 91–96. [Google Scholar] [PubMed]
- Lin, H.J.; Zuo, T.; Lin, C.H.; Kuo, C.T.; Liyanarachchi, S.; Sun, S.; Shen, R.; Deatherage, D.E.; Potter, D.; Asamoto, L.; et al. Breast cancer-associated fibroblasts confer AKT1-mediated epigenetic silencing of Cystatin M in epithelial cells. Cancer Res. 2008, 68, 10257–10266. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Pan, X.; Cobb, G.P.; Anderson, T.A. microRNAs as oncogenes and tumor suppressors. Dev. Biol. 2007, 302. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Garrido, P.; García-Fernández de Barrena, M.; Hijona, E.; Carracedo, M.; Marín, J.J.; Bujanda, L.; Banales, J.M. MicroRNAs in biliary diseases. World J. Gastroenterol. 2012, 18, 6189–6196. [Google Scholar] [CrossRef] [PubMed]
- Piontek, K.; Selaru, F.M. MicroRNAs in the biology and diagnosis of cholangiocarcinoma. Semin. Liver Dis. 2015, 35, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Jin, Z.Y.; Liu, C.H.; Xie, F.; Lin, X.S.; Huang, Q. microRNA-21 regulates biological behavior by inducing EMT in human cholangiocarcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 4684–4694. [Google Scholar] [PubMed]
- Li, B.; Han, Q.; Zhu, Y.; Yu, Y.; Wang, J.; Jiang, X. Down-regulation of miR-214 contributes to intrahepatic cholangiocarcinoma metastasis by targeting Twist. FEBS J. 2012, 279, 2393–2398. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.H.; Wei, Y.P.; Shen, N.J.; Wang, Z.C.; Kan, T.; Yu, W.L.; Yi, B.; Zhang, Y.J. miR-204 inhibits epithelial to mesenchymal transition by targeting slug in intrahepatic cholangiocarcinoma cells. Cell Physiol. Biochem. 2013, 32, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Oishi, N.; Kumar, M.R.; Roessler, S.; Ji, J.; Forgues, M.; Budhu, A.; Zhao, X.; Andersen, J.B.; Ye, Q.H.; Jia, H.L.; et al. Transcriptomic profiling reveals hepatic stem-like gene signatures and interplay of miR-200c and epithelial-mesenchymal transition in intrahepatic cholangiocarcinoma. Hepatology 2012, 56, 1792–1803. [Google Scholar] [CrossRef] [PubMed]
- Qiao, P.; Li, G.; Bi, W.; Yang, L.; Yao, L.; Wu, D. microRNA-34a inhibits epithelial mesenchymal transition in human cholangiocarcinoma by targeting Smad4 through transforming growth factor-β/Smad pathway. BMC Cancer 2015, 15, 469. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Zhu, L.; Huang, F.; Nie, W.; Huang, W.; Xu, H.; Zheng, S.; Yi, Z.; Wan, T. SALL4 is a novel therapeutic target in intrahepatic cholangiocarcinoma. Oncotarget 2015, 6, 27416–27426. [Google Scholar] [CrossRef] [PubMed]
- Pei, T.; Li, Y.; Wang, J.; Wang, H.; Liang, Y.; Shi, H.; Sun, B.; Yin, D.; Sun, J.; Song, R.; et al. YAP is a critical oncogene in human cholangiocarcinoma. Oncotarget 2015, 6, 17206–17220. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chai, L.; Fowles, T.C.; Alipio, Z.; Xu, D.; Fink, L.M.; Ward, D.C.; Ma, Y. Genome-wide analysis reveals Sall4 to be a major regulator of pluripotency in murine-embryonic stem cells. Proc. Natl. Acad. Sci. USA 2008, 105, 19756–19761. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lu, X.; Liu, Z.; Chen, L.; Xu, Y.; Wang, Y.; Wei, G.; Chen, Y. FBXW7 suppresses epithelial-mesenchymal transition, stemness and metastatic potential of cholangiocarcinoma cells. Oncotarget 2015, 6, 6310–6325. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.X.; Gao, Q.; Shi, J.Y.; Wang, Z.C.; Zhang, Y.; Gao, P.T.; Wang, X.Y.; Shi, Y.H.; Ke, A.W.; Shi, G.M.; et al. Mitogen-activated protein kinase kinase kinase 4 Deficiency in Intrahepatic Cholangiocarcinoma Leads to Invasive Growth and Epithelial-Mesenchymal Transition. Hepatology 2015. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.G.; Lee, S.H.; Kim, J.S.; Park, J.; Cho, Y.L.; Kim, K.S.; Jo, D.Y.; Song, I.C.; Kim, N.; Yun, H.J.; et al. Loss of NDRG2 promotes epithelial-mesenchymal transition of gallbladder carcinoma cells through MMP-19-mediated Slug expression. J. Hepatol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Li, D.; Cheng, L.; Wu, H.; Gao, Z.; Liu, Z.; Jiang, W.; Gao, Y.H.; Tian, F.; Zhao, L.; et al. Epithelial-mesenchymal transition induced by hepatitis C virus core protein in cholangiocarcinoma. Ann. Surg. Oncol. 2010, 17, 1937–1944. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Del Carmen Iglesias-de la Cruz, M.; Olmeda, D.; Csiszar, K.; Fong, K.S.; Vega, S.; Nieto, M.A.; Cano, A.; Portillo, F. A molecular role for lysyl oxidase-like 2 enzyme in snail regulation and tumor progression. EMBO J. 2005, 24, 3446–3458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenific, C.M.; Debnath, J. Cellular and metabolic functions for autophagy in cancer cells. Trends Cell Biol. 2015, 25, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Thongchot, S.; Yongvanit, P.; Loilome, W.; Seubwai, W.; Phunicom, K.; Tassaneeyakul, W.; Pairojkul, C.; Promkotra, W.; Techasen, A.; Namwat, N. High expression of HIF-1α, BNIP3 and PI3KC3: Hypoxia-induced autophagy predicts cholangiocarcinoma survival and metastasis. Asian Pac. J. Cancer Prev. 2014, 15, 5873–5878. [Google Scholar] [CrossRef] [PubMed]
- Nitta, T.; Sato, Y.; Ren, X.S.; Harada, K.; Sasaki, M.; Hirano, S.; Nakanuma, Y. Autophagy may promote carcinoma cell invasion and correlate with poor prognosis in cholangiocarcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 4913–4921. [Google Scholar] [PubMed]
- Anderberg, C.; Pietras, K. On the origin of cancer-associated fibroblasts. Cell Cycle 2009, 8, 1461–1462. [Google Scholar] [CrossRef] [PubMed]
- Tarin, D.; Thompson, E.W.; Newgreen, D.F. The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res. 2005, 65, 5996–6000. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, G. The epithelial-mesenchymal transition: Fact or fiction in cancer? Hepatology 2009, 50, 1344–1346. [Google Scholar] [CrossRef] [PubMed]
- May, C.D.; Sphyris, N.; Evans, K.W.; Werden, S.J.; Guo, W.; Mani, S.A. Epithelial-mesenchymal transition and cancer stem cells: A dangerously dynamic duo in breast cancer progression. Breast Cancer Res. 2011, 13, 202. [Google Scholar] [CrossRef] [PubMed]
- Shuang, Z.Y.; Wu, W.C.; Xu, J.; Lin, G.; Liu, Y.C.; Lao, X.M.; Zheng, L.; Li, S. Transforming growth factor-β1-induced epithelial-mesenchymal transition generates ALDH-positive cells with stem cell properties in cholangiocarcinoma. Cancer Lett. 2014, 354, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; You, P.; Li, W.L.; Tao, X.R.; Zhu, H.Y.; Yao, Y.C.; Yu, H.Y.; Han, Q.W.; Yu, B.; Liu, F.X.; et al. The existence of multipotent stem cells with epithelial-mesenchymal transition features in the human liver bud. Int. J. Biochem. Cell Biol. 2010, 42, 2047–2055. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, J.; Abu-Kaoud, N.; Al Thani, H.; Rafii, A. Epithelial to Mesenchymal Transition in a Clinical Perspective. J. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brivio, S.; Cadamuro, M.; Fabris, L.; Strazzabosco, M. Epithelial-to-Mesenchymal Transition and Cancer Invasiveness: What Can We Learn from Cholangiocarcinoma? J. Clin. Med. 2015, 4, 2028-2041. https://doi.org/10.3390/jcm4121958
Brivio S, Cadamuro M, Fabris L, Strazzabosco M. Epithelial-to-Mesenchymal Transition and Cancer Invasiveness: What Can We Learn from Cholangiocarcinoma? Journal of Clinical Medicine. 2015; 4(12):2028-2041. https://doi.org/10.3390/jcm4121958
Chicago/Turabian StyleBrivio, Simone, Massimiliano Cadamuro, Luca Fabris, and Mario Strazzabosco. 2015. "Epithelial-to-Mesenchymal Transition and Cancer Invasiveness: What Can We Learn from Cholangiocarcinoma?" Journal of Clinical Medicine 4, no. 12: 2028-2041. https://doi.org/10.3390/jcm4121958