
Clinical and Genetic Screening for Hypertrophic Cardiomyopathy in Paediatric Relatives: Changing Paradigms in Clinical Practice

Abstract
:
1. Introduction
2. Aetiology of Childhood HCM
3. Screening during Childhood and Adolescence for Those with an Affected First Degree Relative
3.1. The Role of Predictive Genetic Testing in Children in Familial Screening for HCM
3.2. Challenges and ‘the Unknowns’ in Familial Screening in Childhood for HCM
4. Conclusions and Future Directions
Funding
Conflicts of Interest
References
- Arola, A.; Jokinen, E.; Ruuskanen, O.; Saraste, M.; Pesonen, E.; Kuusela, A.L.; Tikanoja, T.; Paavilainen, T.; Simell, O. Epidemiology of idiopathic cardiomyopathies in children and adolescents: A nationwide study in Finland. Am. J. Epidemiol. 1997, 146, 385–393. [Google Scholar] [CrossRef] [Green Version]
- Lipshultz, S.E.; Sleeper, L.A.; Towbin, J.A.; Lowe, A.M.; Orav, E.J.; Cox, G.F.; Lurie, P.R.; McCoy, K.L.; McDonald, M.A.; Messere, J.E.; et al. The Incidence of Pediatric Cardiomyopathy in Two Regions of the United States. N. Engl. J. Med. 2003, 348, 1647–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nugent, A.W.; Daubeney, P.E.; Chondros, P.; Carlin, J.B.; Cheung, M.; Wilkinson, L.C.; Davis, A.M.; Kahler, S.G.; Chow, C.W.; Wilkinson, J.L.; et al. The epidemiology of childhood cardiomyopathy in Australia. N. Engl. J. Med. 2003, 348, 1639–1646. [Google Scholar] [CrossRef] [PubMed]
- Norrish, G.; Field, E.; Mcleod, K.; Ilina, M.; Stuart, G.; Bhole, V.; Uzun, O.; Brown, E.; Daubeney, P.E.F.; Lota, A.; et al. Clinical presentation and survival of childhood hypertrophic cardiomyopathy: A retrospective study in United Kingdom. Eur. Heart J. 2018, 40, 986–993. [Google Scholar] [CrossRef]
- Alexander, P.M.A.; Nugent, A.W.; Daubeney, P.E.F.; Lee, K.J.; Sleeper, L.A.; Schuster, T.; Turner, C.; Davis, A.M.; Semsarian, C.; Colan, S.D.; et al. Long-Term Outcomes of Hypertrophic Cardiomyopathy Diagnosed During Childhood. Circulation 2018, 138, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Norrish, G.; Kolt, G.; Cervi, E.; Field, E.; Dady, K.; Ziółkowska, L.; Olivotto, I.; Favilli, S.; Passantino, S.; Limongelli, G.; et al. Clinical presentation and long-term outcomes of infantile hypertrophic cardiomyopathy: A European multicentre study. ESC Heart Fail. 2021, 8, 5057–5067. [Google Scholar] [CrossRef]
- Norrish, G.; Cleary, A.; Field, E.; Cervi, E.; Boleti, O.; Ziółkowska, L.; Olivotto, I.; Khraiche, D.; Limongelli, G.; Anastasakis, A.; et al. Clinical Features and Natural History of Preadolescent Nonsyndromic Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2022, 79, 1986–1997. [Google Scholar] [CrossRef]
- Marston, N.A.-O.; Han, L.A.-O.; Olivotto, I.A.-O.; Day, S.A.-O.; Ashley, E.A.; Michels, M.A.-O.; Pereira, A.C.; Ingles, J.A.-O.; Semsarian, C.; Jacoby, D.A.-O.X.; et al. Clinical characteristics and outcomes in childhood-onset hypertrophic cardiomyopathy. Eur. Heart J. 2021, 42, 1988–1996. [Google Scholar] [CrossRef]
- Ho, C.Y.; Day, S.M.; Ashley, E.A.; Michels, M.; Pereira, A.C.; Jacoby, D.; Cirino, A.L.; Fox, J.C.; Lakdawala, N.K.; Ware, J.S.; et al. Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy. Circulation 2018, 138, 1387–1398. [Google Scholar] [CrossRef]
- van Capelle, C.I.; Poelman, E.; Frohn-Mulder, I.M.; Koopman, L.P.; van den Hout, J.M.P.; Régal, L.; Cools, B.; Helbing, W.A.; van der Ploeg, A.T. Cardiac outcome in classic infantile Pompe disease after 13 years of treatment with recombinant human acid alpha-glucosidase. Int. J. Cardiol. 2018, 269, 104–110. [Google Scholar] [CrossRef] [Green Version]
- Andelfinger, G.; Marquis, C.; Raboisson, M.J.; Théoret, Y.; Waldmüller, S.; Wiegand, G.; Gelb, B.D.; Zenker, M.; Delrue, M.A.; Hofbeck, M. Hypertrophic Cardiomyopathy in Noonan Syndrome Treated by MEK-Inhibition. J. Am. Coll. Cardiol. 2019, 73, 2237–2239. [Google Scholar] [CrossRef]
- Gross, A.M.; Frone, M.; Gripp, K.W.; Gelb, B.D.; Schoyer, L.; Schill, L.; Stronach, B.; Biesecker, L.G.; Esposito, D.; Hernandez, E.R.; et al. Advancing RAS/RASopathy therapies: An NCI-sponsored intramural and extramural collaboration for the study of RASopathies. Am. J. Med. Genet. Part A 2020, 182, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Maron, M.S.; Masri, A.; Choudhury, L.; Olivotto, I.; Saberi, S.; Wang, A.; Garcia-Pavia, P.; Lakdawala, N.K.; Nagueh, S.F.; Rader, F.; et al. Phase 2 Study of Aficamten in Patients With Obstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2023, 81, 34–45. [Google Scholar] [CrossRef]
- Spertus, J.A.; Fine, J.T.; Elliott, P.; Ho, C.Y.; Olivotto, I.; Saberi, S.; Li, W.; Dolan, C.; Reaney, M.; Sehnert, A.J.; et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): Health status analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2021, 397, 2467–2475. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.Y.; Owens, A.; Geske, J.B.; Wolski, K.; Naidu, S.S.; Smedira, N.G.; Cremer, P.C.; Schaff, H.; McErlean, E.; Sewell, C.; et al. Myosin Inhibition in Patients With Obstructive Hypertrophic Cardiomyopathy Referred for Septal Reduction Therapy. J. Am. Coll. Cardiol. 2022, 80, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Green, E.M.; Wakimoto, H.; Anderson, R.L.; Evanchik, M.J.; Gorham, J.M.; Harrison, B.C.; Henze, M.; Kawas, R.; Oslob, J.D.; Rodriguez, H.M.; et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 2016, 351, 617–621. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Marti-Gutierrez, N.; Park, S.-W.; Wu, J.; Lee, Y.; Suzuki, K.; Koski, A.; Ji, D.; Hayama, T.; Ahmed, R.; et al. Correction of a pathogenic gene mutation in human embryos. Nature 2017, 548, 413–419. [Google Scholar] [CrossRef]
- Dainis, A.; Zaleta-Rivera, K.; Ribeiro, A.; Chang, A.C.H.; Shang, C.; Lan, F.; Burridge, P.W.; Liu, W.R.; Wu, J.C.; Chang, A.C.Y.; et al. Silencing of MYH7 ameliorates disease phenotypes in human iPSC-cardiomyocytes. Physiol. Genom. 2020, 52, 293–303. [Google Scholar] [CrossRef]
- Chai, A.A.-O.; Cui, M.; Chemello, F.A.-O.X.; Li, H.A.-O.; Chen, K.; Tan, W.; Atmanli, A.; McAnally, J.R.; Zhang, Y.A.-O.; Xu, L.A.-O.; et al. Base editing correction of hypertrophic cardiomyopathy in human cardiomyocytes and humanized mice. Nat. Med. 2023, 29, 401–411. [Google Scholar] [CrossRef]
- Monda, E.; Rubino, M.; Lioncino, M.; Di Fraia, F.; Pacileo, R.; Verrillo, F.; Cirillo, A.; Caiazza, M.; Fusco, A.; Esposito, A.; et al. Hypertrophic Cardiomyopathy in Children: Pathophysiology, Diagnosis, and Treatment of Non-sarcomeric Causes. Front. Pediatr. 2021, 9, 632293. [Google Scholar] [CrossRef]
- Ware, S.M.; Bhatnagar, S.; Dexheimer, P.J.; Wilkinson, J.D.; Sridhar, A.; Fan, X.; Shen, Y.; Tariq, M.; Schubert, J.A.; Colan, S.D.; et al. The genetic architecture of pediatric cardiomyopathy. Am. J. Hum. Genet. 2022, 109, 282–298. [Google Scholar] [CrossRef] [PubMed]
- Kaski, J.P.; Syrris, P.; Esteban, M.T.T.; Jenkins, S.; Pantazis, A.; Deanfield, J.E.; McKenna, W.J.; Elliott, P.M. Prevalence of Sarcomere Protein Gene Mutations in Preadolescent Children With Hypertrophic Cardiomyopathy. Circ. Cardiovasc. Genet. 2009, 2, 436–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdmann, J.; Moretti, A. The Genetic Landscape of Cardiomyopathies; Springer International Publishing AG: Cham, Switzerland, 2019; Volume 7, pp. 45–91. [Google Scholar]
- Wilde, A.A.M.; Semsarian, C.; Márquez, M.F.; Sepehri Shamloo, A.; Ackerman, M.J.; Ashley, E.A.; Sternick Eduardo, B.; Barajas-Martinez, H.; Behr, E.R.; Bezzina, C.R.; et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the state of genetic testing for cardiac diseases. J. Arrhythmia 2022, 38, 491–553. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Mas, R.; Gutiérrez-Fernández, A.; Gómez, J.; Coto, E.; Astudillo, A.; Puente, D.A.; Reguero, J.R.; Álvarez, V.; Morís, C.; León, D.; et al. Mutations in filamin C cause a new form of familial hypertrophic cardiomyopathy. Nat. Commun. 2014, 5, 5326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelb, B.D.; Roberts, A.E.; Tartaglia, M. Cardiomyopathies in Noonan syndrome and the other RASopathies. Prog. Pediatr. Cardiol. 2015, 39, 13–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porto, A.G.; Brun, F.; Severini, G.M.; Losurdo, P.; Fabris, E.; Taylor, M.R.G.; Mestroni, L.; Sinagra, G. Clinical Spectrum of PRKAG2 Syndrome. Circ. Arrhythmia Electrophysiol. 2016, 9, e003121. [Google Scholar] [CrossRef] [Green Version]
- Arad, M.; Maron, B.J.; Gorham, J.M.; Johnson, W.H.; Saul, J.P.; Perez-Atayde, A.R.; Spirito, P.; Wright, G.B.; Kanter, R.J.; Seidman, J.G.; et al. Glycogen Storage Diseases Presenting as Hypertrophic Cardiomyopathy. N. Engl. J. Med. 2005, 352, 362–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finsterer, J. Barth syndrome: Mechanisms and management. Appl. Clin. Genet. 2019, 12, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [CrossRef] [PubMed]
- Gersh, B.J.; Maron, B.J.; Bonow, R.O.; Dearani, J.A.; Fifer, M.A.; Link, M.S.; Naidu, S.S.; Nishimura, R.A.; Ommen, S.R.; Rakowski, H.; et al. 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy. Circulation 2011, 124, e783–e831. [Google Scholar] [CrossRef] [Green Version]
- Maron, B.J. Clinical Course and Management of Hypertrophic Cardiomyopathy. N. Engl. J. Med. 2018, 379, 655–668. [Google Scholar] [CrossRef]
- Maron, B.J.; Rowin, E.J.; Casey, S.A.; Maron, M.S. How Hypertrophic Cardiomyopathy Became a Contemporary Treatable Genetic Disease With Low Mortality: Shaped by 50 Years of Clinical Research and Practice. JAMA Cardiol. 2016, 1, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Maron, B.J.; Spirito, P.; Wesley, Y.; Arce, J. Development and Progression of Left Ventricular Hypertrophy in Children with Hypertrophic Cardiomyopathy. N. Engl. J. Med. 1986, 315, 610–614. [Google Scholar] [CrossRef]
- Jensen, M.K.; Havndrup, O.; Christiansen, M.; Andersen, P.S.; Diness, B.; Axelsson, A.; Skovby, F.; KØBer, L.; Bundgaard, H. Penetrance of Hypertrophic Cardiomyopathy in Children and Adolescents: A 12-Year Follow-up Study of Clinical Screening and Predictive Genetic Testing. Circulation 2013, 127, 48–54. [Google Scholar] [CrossRef] [Green Version]
- Norrish, G.; Jager, J.; Field, E.; Quinn, E.; Fell, H.; Lord, E.; Cicerchia, M.N.; Ochoa, J.P.; Cervi, E.; Elliott, P.M.; et al. Yield of Clinical Screening for Hypertrophic Cardiomyopathy in Child First-Degree Relatives. Circulation 2019, 140, 184–192. [Google Scholar] [CrossRef]
- Lafreniere-Roula, M.; Bolkier, Y.; Zahavich, L.; Mathew, J.; George, K.; Wilson, J.; Stephenson, E.A.; Benson, L.N.; Manlhiot, C.; Mital, S. Family screening for hypertrophic cardiomyopathy: Is it time to change practice guidelines? Eur. Heart J. 2019, 40, 3672–3681. [Google Scholar] [CrossRef] [Green Version]
- Page, S.P.; Kounas, S.; Syrris, P.; Christiansen, M.; Frank-Hansen, R.; Andersen, P.S.; Elliott, P.M.; McKenna, W.J. Cardiac myosin binding protein-C mutations in families with hypertrophic cardiomyopathy: Disease expression in relation to age, gender, and long term outcome. Circ. Cardiovasc. Genet. 2012, 1, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Field, E.A.-O.; Norrish, G.; Acquaah, V.; Dady, K.; Cicerchia, M.N.; Ochoa, J.P.; Syrris, P.; McLeod, K.; McGowan, R.; Fell, H.; et al. Cardiac myosin binding protein-C variants in paediatric-onset hypertrophic cardiomyopathy: Natural history and clinical outcomes. J. Med. Genet. 2022, 59, 768–775. [Google Scholar] [CrossRef]
- Spanaki, A.; O’Curry, S.; Winter-Beatty, J.; Mead-Regan, S.; Hawkins, K.; English, J.; Head, C.; Ridout, D.; Tome-Esteban, M.T.; Elliott, P.; et al. Psychosocial adjustment and quality of life in children undergoing screening in a specialist paediatric hypertrophic cardiomyopathy clinic. Cardiol. Young 2016, 26, 961–967. [Google Scholar] [CrossRef]
- Ommen, S.R.; Mital, S.; Burke, M.A.; Day, S.M.; Deswal, A.; Elliott, P.; Evanovich, L.L.; Hung, J.; Joglar, J.A.; Kantor, P.; et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy. Circulation 2020, 142, e558–e631. [Google Scholar] [CrossRef]
- Smedsrud, M.K.; Chivulescu, M.; Forså, M.I.; Castrini, I.; Aabel, E.W.; Rootwelt-Norberg, C.; Bogsrud, M.P.; Edvardsen, T.; Hasselberg, N.E.; Früh, A.; et al. Highly malignant disease in childhood-onset arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2022, 43, 4694–4703. [Google Scholar] [CrossRef]
- Kaski, J.P. Arrhythmogenic cardiomyopathies in children: Seek and you shall find. Eur. Heart J. 2022, 43, 4704–4706. [Google Scholar] [CrossRef]
- Lorenzini, M.; Norrish, G.; Field, E.; Ochoa, J.P.; Cicerchia, M.; Akhtar, M.M.; Syrris, P.; Lopes, L.R.; Kaski, J.P.; Elliott, P.M. Penetrance of Hypertrophic Cardiomyopathy in Sarcomere Protein Mutation Carriers. J. Am. Coll. Cardiol. 2020, 76, 550–559. [Google Scholar] [CrossRef]
- Arbour, L.; Canadian Paediatric Society; Bioethics Committee. Guidelines for genetic testing of healthy children. Paediatr. Child Health 2003, 8, 42–45. [Google Scholar] [CrossRef] [Green Version]
- NHS Commissioning Board. Clinical Commissioning Policy: Pre-Implantation Genetic Diagnosis (PGD); NHS Commissioning Board: Leeds, UK, 2013. [Google Scholar]
- Wakefield, C.E.; Hanlon, L.V.; Tucker, K.M.; Patenaude, A.F.; Signorelli, C.; McLoone, J.K.; Cohn, R.J. The psychological impact of genetic information on children: A systematic review. Genet. Med. 2016, 18, 755–762. [Google Scholar] [CrossRef] [Green Version]
- Ingles, J.; Burns, C.; Barratt, A.; Semsarian, C. Application of Genetic Testing in Hypertrophic Cardiomyopathy for Preclinical Disease Detection. Circ. Cardiovasc. Genet. 2015, 8, 852–859. [Google Scholar] [CrossRef] [Green Version]
- GradDipGenCouns, J.I.; Yeates, L.; O’Brien, L.; McGaughran, J.; Scuffham, P.A.; Atherton, J.; Semsarian, C. Genetic testing for inherited heart diseases: Longitudinal impact on health-related quality of life. Genet. Med. 2012, 14, 749–752. [Google Scholar] [CrossRef] [Green Version]
- de Feria, A.E.; Kott, A.E.; Becker, J.R. Sarcomere mutation negative hypertrophic cardiomyopathy is associated with ageing and obesity. Open Heart 2021, 8, e001560. [Google Scholar] [CrossRef]
- Ko, C.; Arscott, P.; Concannon, M.; Saberi, S.; Day, S.M.; Yashar, B.M.; Helms, A.S. Genetic testing impacts the utility of prospective familial screening in hypertrophic cardiomyopathy through identification of a nonfamilial subgroup. Genet. Med. 2018, 20, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Canepa, M.; Fumagalli, C.; Tini, G.; Vincent-Tompkins, J.; Day, S.M.; Ashley, E.A.; Mazzarotto, F.; Ware, J.S.; Michels, M.; Jacoby, D.; et al. Temporal Trend of Age at Diagnosis in Hypertrophic Cardiomyopathy: An Analysis of the International Sarcomeric Human Cardiomyopathy Registry. Circ. Heart Fail. 2020, 13, e007230. [Google Scholar] [CrossRef]
- Tadros, R.A.-O.; Francis, C.; Xu, X.; Vermeer, A.M.C.; Harper, A.A.-O.; Huurman, R.A.-O.; Kelu Bisabu, K.A.-O.X.; Walsh, R.A.-O.; Hoorntje, E.T.; Te Rijdt, W.P.; et al. Shared genetic pathways contribute to risk of hypertrophic and dilated cardiomyopathies with opposite directions of effect. Nat. Genet. 2021, 53, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Biddinger, K.J.; Jurgens, S.J.; Maamari, D.; Gaziano, L.; Choi, S.H.; Morrill, V.N.; Halford, J.L.; Khera, A.V.; Lubitz, S.A.; Ellinor, P.T.; et al. Rare and Common Genetic Variation Underlying the Risk of Hypertrophic Cardiomyopathy in a National Biobank. JAMA Cardiol. 2022, 7, 715–722. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
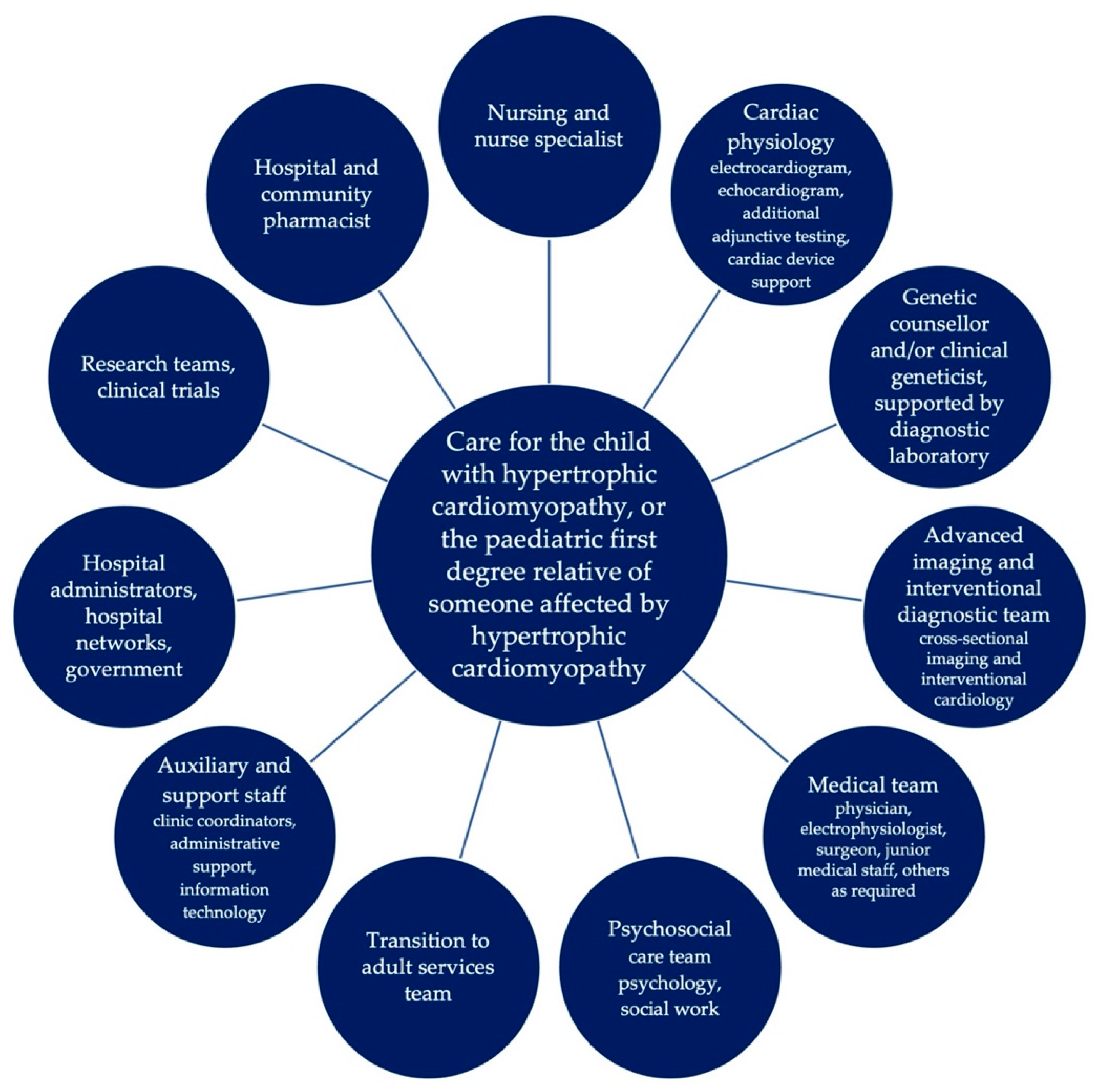{kind=link}
| Category | Example Conditions | Cardiac Features | The Most Common Mode of Inheritance | Example Candidate Genes |
|---|---|---|---|---|
| “Sarcomeric” HCM [9,22,24,25] | “Thick filament” disease “Thin filament” disease Other | Variable hypertrophy, arrhythmic risk, and patterns of hypertrophy with some genotype-phenotype correlation | Autosomal dominant * | MYH7, MYBPC3 TNNI3, TNNT2, TPM1, MYL2, MYL3, ACTC1, TNNC1 CSPR3, FLNC ** Additional noncoding variants influencing phenotype |
| Non-sarcomeric HCM | ||||
| Ras/MAPK disease [26] | Noonan syndrome, Costello syndrome, and cardiofaciocutaneous syndrome | Polyvalvulopathy, biventricular hypertrophy, infancy/early childhood presentation | Autosomal dominant | RAF1, RIT1, PTPN1, HRAS, MEK1, MEK2 |
| Inborn errors of metabolism [27,28] | Glycogen storage disorders (Pompe disease, Danon disease, Cori-Forbes disease, and PRKAG2 syndrome) Lysosomal storage disorders (mucopolysaccharidoses) | Disease specific; specific ECG changes (such as short PR, ventricular pre-excitation), variable hypertrophy, polyvavulopathy in some conditions, early childhood onset in some conditions | Autosomal recessive | GLA LAMP2 PRKAG2 |
| Neuromuscular disease [12,29] | Mitochondrial disorders (Friedreich’s ataxia, Barth syndrome) | Disease-specific Friedreich’s ataxia—concentric left ventricular hypertrophy, higher rate of atrial arrhythmia, lower riskof sudden death. | Autosomal recessive, X-linked | FXN TAZ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lawley, C.M.; Kaski, J.P. Clinical and Genetic Screening for Hypertrophic Cardiomyopathy in Paediatric Relatives: Changing Paradigms in Clinical Practice. J. Clin. Med. 2023, 12, 2788. https://doi.org/10.3390/jcm12082788
Lawley CM, Kaski JP. Clinical and Genetic Screening for Hypertrophic Cardiomyopathy in Paediatric Relatives: Changing Paradigms in Clinical Practice. Journal of Clinical Medicine. 2023; 12(8):2788. https://doi.org/10.3390/jcm12082788
Chicago/Turabian StyleLawley, Claire M., and Juan Pablo Kaski. 2023. "Clinical and Genetic Screening for Hypertrophic Cardiomyopathy in Paediatric Relatives: Changing Paradigms in Clinical Practice" Journal of Clinical Medicine 12, no. 8: 2788. https://doi.org/10.3390/jcm12082788