
Clinical Profile of Patients with Idiopathic Pulmonary Fibrosis in Real Life

 and
and
Abstract
:1. Introduction
2. Materials and Methods
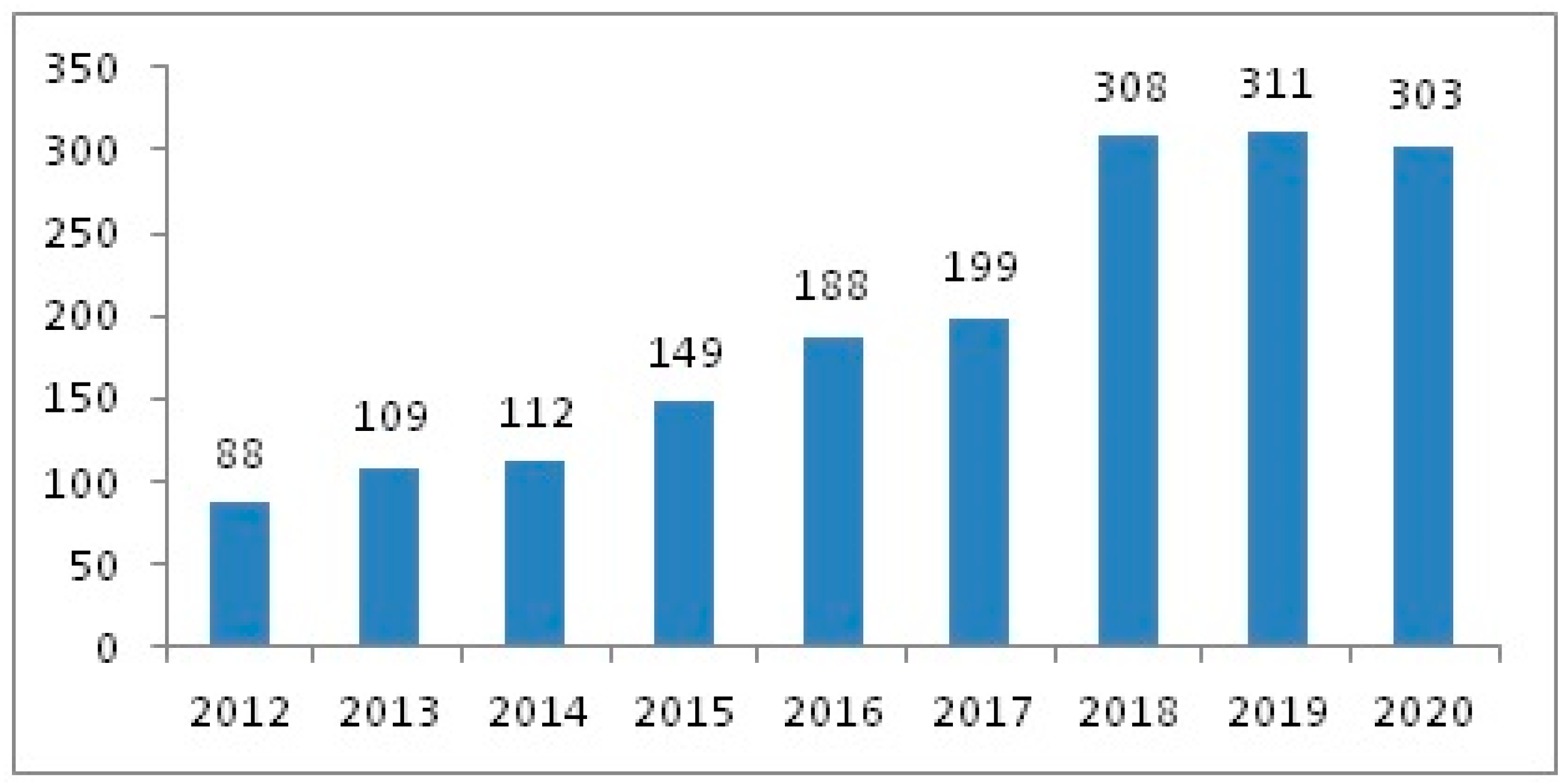
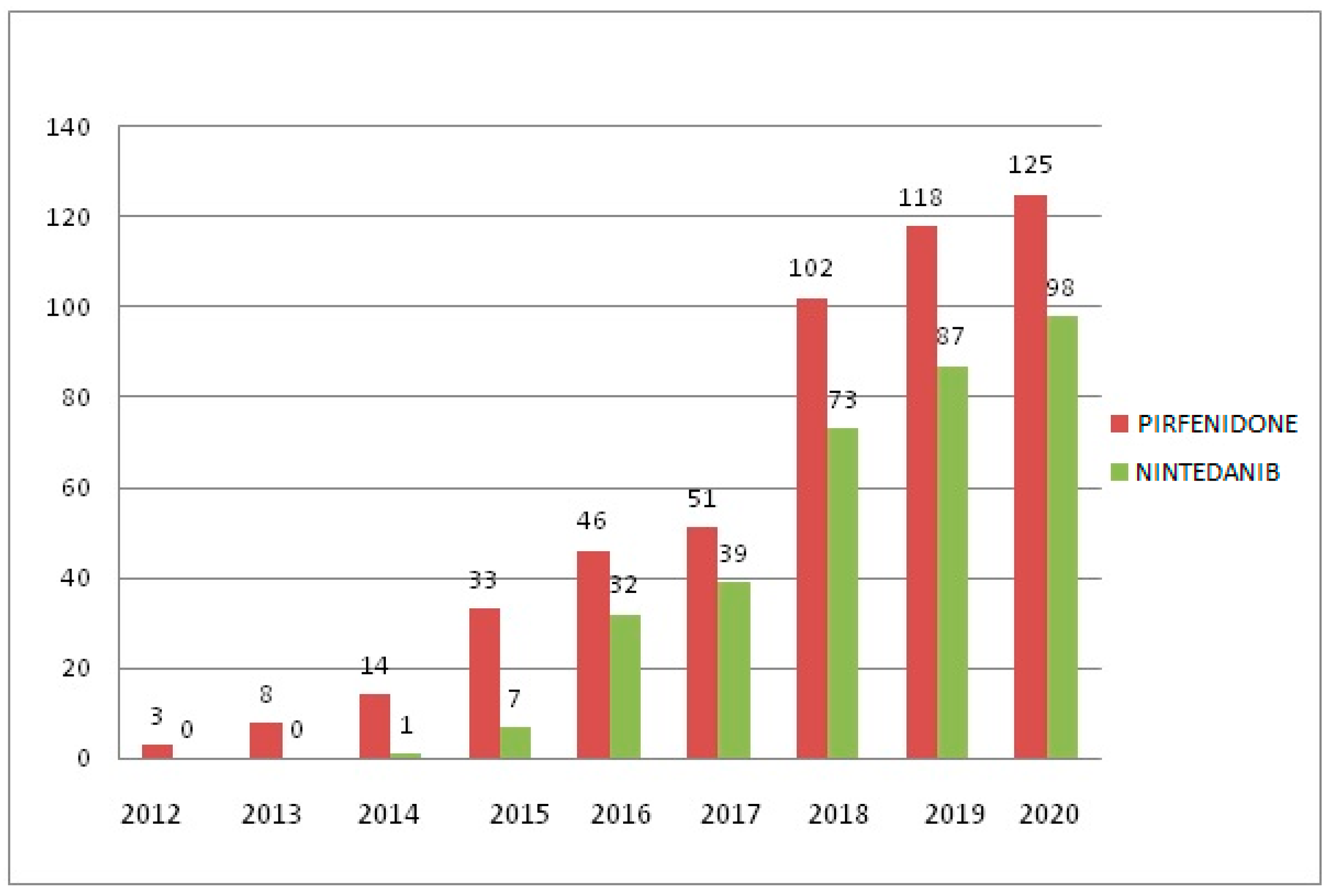
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Oliveira, D.S.; Araújo Filho, J.D.A.; Paiva, A.F.L.; Ikari, E.S.; Chate, R.C.; Nomura, C.H. Idiopathic interstitial pneumonias: Review of the latest American Thoracic So-ciety/European Respiratory Society classification. Radiol. Bras. 2018, 51, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Cottin, V.; Hirani, N.A.; Hotchkin, D.L.; Nambiar, A.M.; Ogura, T.; Otaola, M.; Skowasch, D.; Park, J.S.; Poonyagariyagorn, H.K.; Wuyts, W.; et al. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur. Respir. Rev. 2018, 27, 180076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E., Jr.; Lynch, D.A.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.H.; Selman, M.; Wells, A.U.; et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef] [PubMed]
- El-Chemaly, S.; Ziegler, S.G.; Wilson, K.; Gahl, W.A.; Moss, J.; Gochuico, B.R. Familial pulmonary fibrosis: Natural history of preclinical disease. Am. J. Respir. Crit. Care Med. 2010, 181, A2980. [Google Scholar]
- García-Sancho, C.; Buendía-Roldán, I.; Fernández-Plata, M.R.; Navarro, C.; Pérez-Padilla, R.; Vargas, M.H.; Loyd, J.E.; Selman, M. Familial pul-monary fibrosis is the strongest risk factor for idiopathic pulmonary fibrosis. Respir. Med. 2011, 105, 1902–1907. [Google Scholar] [CrossRef] [Green Version]
- Xaubet, A.; Molina, M.; Ancochea, J. Nueva clasificación de las neumonías intersti-ciales idiopáticas. Med. Respir. 2014, 7, 21–28. [Google Scholar]
- Xaubet, A.; Ancochea, J.; Bollo, E.; Fernández-Fabrellas, E.; Franquet, T.; Molina-Molina, M.; Montero, M.A.; Serrano-Mollar, A. Guidelines for the Diagnosis and Treatment of Idiopathic Pulmonary Fibrosis. Arch. Bronconeumol. Engl. Ed. 2013, 49, 343–353. [Google Scholar] [CrossRef] [Green Version]
- Xaubet, A.; Ancochea, J.; Morell, F.; Rodriguez-Arias, J.M.; Villena, V.; Blanquer, R.; Montero, C.; Sueiro, A.; Disdier, C.; Vendrell, M. Report on the incidence of intersticial lung diseades in Spain. Sarcoidosis Vasc. Diffuse Lung Dis. 2004, 21, 64–70. [Google Scholar]
- Demedts, M.; Wells, A.U.; Anto, J.M.; Costabel, U.; Hubbard, R.; Cullinan, P.; Slabbynck, H.; Rizzato, G.; Poletti, V.; Verbeken, E.K.; et al. Inter-stitial lung diseases: An epidemiological overview. Eur. Respir. J. 2001, 18, 2S–16S. [Google Scholar]
- Pérez, E.R.F.; Daniels, C.E.; Sauver, J.S.; Hartman, T.E.; Bartholmai, B.J.; Eunhee, S.Y.; Ryu, J.H.; Schroeder, D.R. Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis: A population based study. Chest 2010, 137, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Raghu, G.; Weycker, D.; Edelsberg, J.; Bradford, W.Z.; Oster, G. Incidence and Prevalence of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2006, 174, 810–816. [Google Scholar] [CrossRef] [Green Version]
- Gribbin, J.; Hubbard, R.B.; Le Jeune, I.; Smith, C.J.P.; West, J.; Tata, L.J. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax 2006, 61, 980–985. [Google Scholar] [CrossRef] [Green Version]
- King, T.E., Jr.; Tooze, J.A.; Schwarz, M.I.; Brown, K.R.; Cherniack, R.M. Predicting Survival in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2001, 164, 1171–1181. [Google Scholar] [CrossRef]
- Martinez, F.J.; Safrin, S.; Weycker, D.; Starko, K.M.; Bradford, W.Z.; King, T.E., Jr.; Flaherty, K.R.; Schwartz, D.A.; Noble, P.W.; Raghu, G.; et al. The clinical course of patients with id-iopathic pulmonary fibrosis. Ann. Intern. Med. 2005, 142, 963–967. [Google Scholar] [CrossRef]
- Maher, T.M.; Wells, A.U.; Laurent, G.J. Idiopathic pulmonary fibrosis: Multiple causes and multiple mechanisms? Eur. Respir. J. 2007, 835–839. [Google Scholar] [CrossRef] [Green Version]
- Ancochea, J.; Gómez, J.; Vilar, J.; Xaubet, A. Consenso para el diagnóstico de las neumonías intersticiales idiopáticas. Arch. Bronconeumol. 2010, 46, 2–21. [Google Scholar]
- Cavazza, A.; Rossi, G.; Carbonelli, C.; Spaggiari, L.; Paci, M.; Roggeri, A. The role of histology in idiopathic pulmonary fibrosis: An update. Respir. Med. 2010, 104, S11–S22. [Google Scholar] [CrossRef] [Green Version]
- Ichikado, K. High-resolution computed tomography findings of acute respira-tory distress syndrome, acute interstitial pneumonia, and acute exacerbation of idio-pathic pulmonary fibrosi. Semin. Ultrasound CT MRI 2014, 35, 39–46. [Google Scholar] [CrossRef]
- Jabob, J.; Hansell, D.M. HRCT of fibrosing lung disease. Respirology 2015, 125–131. [Google Scholar]
- Corte, T.J.; Wort, S.J.; Wells, A.U. Pulmonary hypertension in idiopathic pulmonary fibrosis: A review. Sarcoidosis Vasc. Diffus. Lung Dis. Off. J. WASOG 2009, 26, 7–19. [Google Scholar]
- Cottin, V.; Nunes, H.; Brillet, P.; Delaval, P.; Devouassoux, G.; Tillie-Leblond, I.; Israel-Biet, D.; Valeyre, D.; Cordier, J.F. Combined pulmonary fibrosis and emphy-sema: A distinct underrecognized entity. Eur. Respir. J. 2005, 26, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Navaratnam, V.; Fleming, K.M.; West, J.; Smith, C.J.P.; Jenkins, G.; Fogarty, A.; Hubbard, R.B. The rising incidence of idiopathic pulmonary fibrosis in the UK. Thorax 2011, 66, 462–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xaubet, A.; Molina-Molina, M.; Acosta, O.; Bollo, E.; Castillo, D.; Fernández-Fabrellas, E.; Rodríguez-Portal, J.A.; Valenzuela, C.; Ancochea, J. Guidelines for the medical treatment of idiopathic pulmonary fibrosis. Arch. Bronconeumol. 2017, 53, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J.; et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two ran-domised trials. Lancet 2011, 377, 1760–1769. [Google Scholar] [CrossRef] [PubMed]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A Phase 3 Trial of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richeldi, L.; Du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [Green Version]
- Kolonics-Farkas, A.M.; Šterclová, M.; Mogulkoc, N.; Kus, J.; Hájková, M.; Müller, V.; Jovanovic, D.; Tekavec-Trkanjec, J.; Littnerová, S.; Hejduk, K.; et al. Anticoagulant Use and Bleeding Risk in Central European patients with Idiopathic Pulmonary Fibrosis (IPF) Treated with Antifibrotic Therapy: Real-World Data from EMPIRE. Drug Saf. 2020, 43, 971–980. [Google Scholar] [CrossRef]
- Lee, J.S.; Ryu, J.H.; Elicker, B.M.; Lydell, C.P.; Jones, K.D.; Wolters, P.J.; King, T.E., Jr.; Collard, H.R. Gastroesophageal Reflux Therapy Is Associated with Longer Survival in Patients with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 1390–1394. [Google Scholar] [CrossRef] [Green Version]
- Xaubet, A.; Molina-Molina, M.; Ancochea, J. Fibrosis pulmonar relacionada con el tabaco. Med. Respir. 2015, 8, 39–46. [Google Scholar]
- Canales, L.; Menke, S.; Marchesseau, S.; D’Agostino, A.; del Rio-Bermudez, C.; Taberna, M.; Tello, J. Assessing the Performance of Clinical Natural Language Processing Systems: Development of an Evaluation Methodology. JMIR Public Health Surveill. 2021, 9, e20492. [Google Scholar] [CrossRef]
- Izquierdo, J.L.; Almonacid, C.; González, Y.; Del Rio-Bermudez, C.; Ancochea, J.; Cárdenas, R.; Lumbreras, S.; Soriano, J.B. The impact of COVID-19 on patients with asthma: A Big data analysis. Eur. Respir. J. 2021, 57, 2003142. [Google Scholar] [CrossRef]
- Benson, T. Principles of Health Interoperability HL7 and SNOMED; Springer: London, UK, 2012. [Google Scholar]
- Baeza-Yates, R.A.; Ribeiro-Neto, B. Modern Information Retrieval; Addison-Wesley Longman: Boston, MA, USA, 1999. [Google Scholar]
- Izquierdo, J.L.; Morena, D.; González, Y.; Paredero, J.M.; Pérez, B.; Graziani, D.; Gutiérrez, M.; Rodríguez, J.M. Manejo clínico de la EPOC en situación de vida real. Análisis a partir de big data. Arch. Bronconeumol. 2020, 57, 94–100. [Google Scholar] [CrossRef]
- Cottin, V.; Le Pavec, J.; Prévot, G.; Mal, H.; Humbert, M.; Simonneau, G.; Cordier, J.F. Pulmonary hypertension in patients with combined pulmonary fibrosis and emphysema syndrome. Eur. Respir. J. 2010, 35, 105–111. [Google Scholar] [CrossRef]
- Patel, N.M.; Lederer, D.J.; Borczuk, A.C.; Kawut, S.M. Pulmonary hypertension in idiopathic pulmonary fibrosis. Chest 2007, 132, 998–1006. [Google Scholar] [CrossRef]
- Lancaster, L.H.; Mason, W.R.; Parnell, J.A.; Rice, T.W.; Loyd, J.E.; Milstone, A.P.; Collard, H.R.; Malow, B.A. Obstructive sleep apnea is common in idiopathic pulmonary fibrosis. Chest 2009, 136, 772–778. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
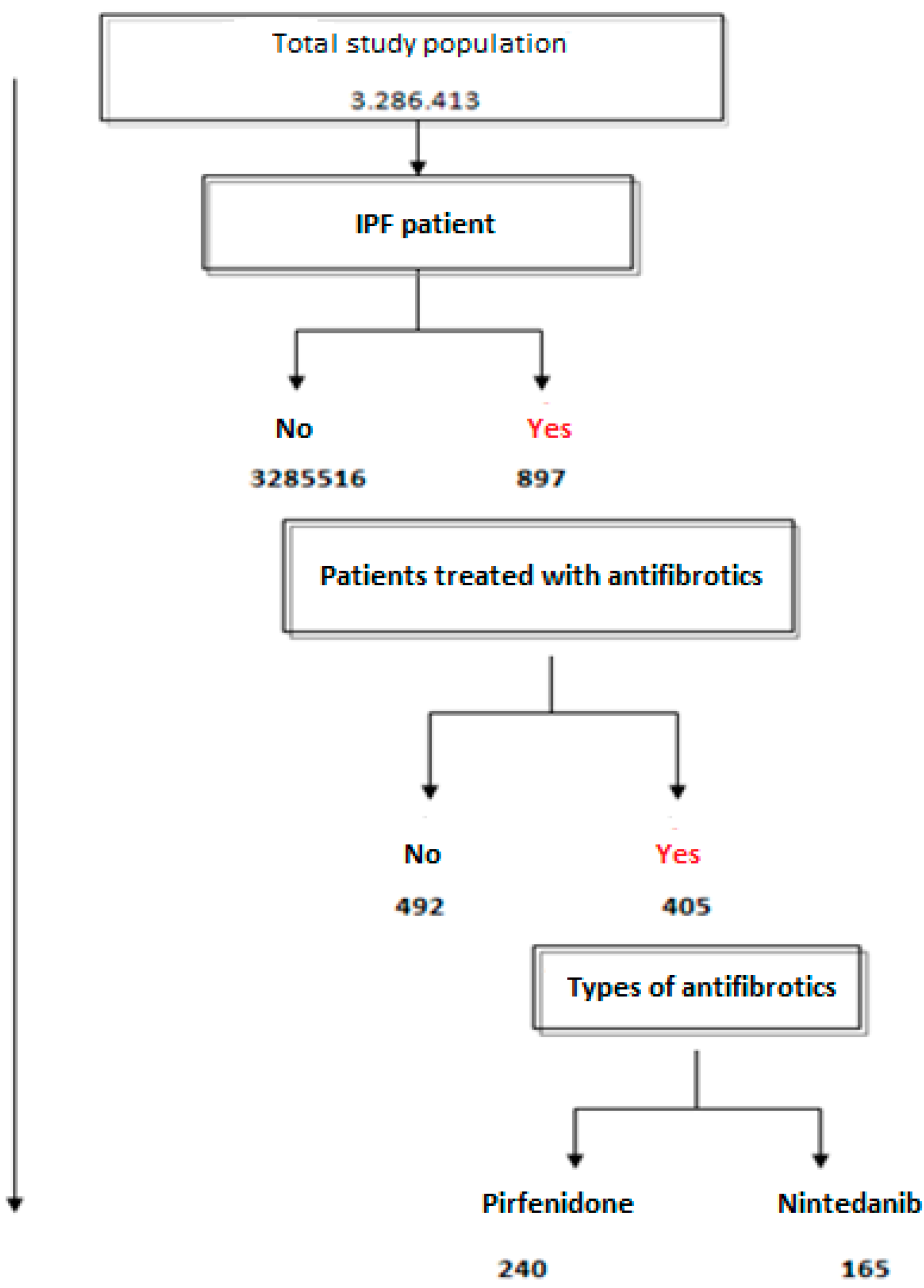
| Inclusion criteria |
|---|
| Patients with clinical diagnosis of idiopathic pulmonary fibrosis. The selected concept also includes idiopathic pulmonary fibrosis (acute fatal form), idiopathic pulmonary fibrosis, acute interstitial pneumonia and Hamman Rich syndrome |
| Exclusion criteria |
| Patients with a specific diagnosis other than idiopathic pulmonary fibrosis, including sarcoidosis, hypersensitivity pneumonitis, pulmonary edema, pneumonia, pulmonary embolism, pneumothorax, rib fractures, aspiration, pleural effusion or any other associated respiratory or non-respiratory disorder |
| IPF Patients | % | General Population | % | p Value/OR (95%CI) | |
|---|---|---|---|---|---|
| N | 897 | 1,410,880 | |||
| Mean (SD) age, years | 74.2 (15.3) | 68.3 (12.2) | <0.001 | ||
| Male sex | 581 | 64.8 | 657,470 | 46.6 | 2.1 (1.8–2.4) |
| Arterial hypertension | 634 | 70.7 | 511,527 | 36.3 | 4.2 (3.7–4.9) |
| Dyslipidemia | 504 | 56.2 | 350,926 | 24.9 | 3.9 (3.4–4.4) |
| Diabetes mellitus | 363 | 40.5 | 241,004 | 17.1 | 3.3 (2.9–3.8) |
| Congestive heart failure | 384 | 42.8 | 68,566 | 4.9 | 14.7 (12.8–16.7) |
| Hiatal hernia | 201 | 22.4 | 81,452 | 5.8 | 4.7 (4.0–5.5) |
| Obesity | 182 | 20.3 | 130,460 | 9.6 | 2.5 (2.1–2.9) |
| Pulmonary hypertension | 250 | 27.9 | 34,014 | 2.4 | 15.6 (13.5–18.1) |
| Atrial fibrillation | 209 | 23.3 | 81,207 | 5.8 | 4.9 (4.3–5.8) |
| Ischemic cardiopathy | 200 | 22.3 | 47,669 | 3.4 | 8.2 (7.1–9.6) |
| Gastroesophageal reflux disease | 112 | 12.5 | 46,153 | 3.3 | 4.2 (3.5–5.1) |
| Pulmonary emphysema | 144 | 16.1 | 14,776 | 1.1 | 10.8 (8.7–13.4) |
| Sleep apnea syndrome | 123 | 13.7 | 38,722 | 2.7 | 5.6 (4.7–6.8) |
| SEX (Males, %) | OR(95%CI) | Mean Age (Years, 95%CI) | p Value | ||
|---|---|---|---|---|---|
| Chest CT | Yes (763) | 66.2% | 1.5 (1.1–2.1) | 73.8 (95%CI 73–74.5) | p < 0.001 |
| No (134) | 57.5% | 76.2 (95%CI 73.7–78.7) | |||
| Simple bronchoscopy | Yes (304) | 64.8% | 1.1 (0.8–1.4) | 70.1 (95%CI 68.8–71.3) | p < 0.001 |
| No (593) | 63.8% | 75.8 (95%CI 74.9–76.7) | |||
| Surgical pulmonary biopsy | Yes (127) | 66.9% | 1.1 (0.8–1.7) | 66 (95%CI 64.3–67.8) | p < 0.001 |
| No (770) | 64.2% | 75.3 (95%CI 74.5–76) |
| Comorbidities | Patients Treated | % | Patients Not Treated | % | OR (95%CI) |
|---|---|---|---|---|---|
| Arterial hypertension | 235 | 58.0 | 399 | 81.1 | 0.3 (0.2–0.4) |
| Dyslipidemia | 215 | 53.1 | 289 | 58.7 | 0.8 (0.6–1.0) |
| Diabetes mellitus | 143 | 35.3 | 220 | 44.7 | 0.7 (0.5–0.9) |
| Obesity | 84 | 20.7 | 98 | 19.9 | 1.1 (0.8–1.5) |
| Pulmonary hypertension | 107 | 26.4 | 143 | 29.1 | 0.6 (0.4–0.8) |
| Heart failure | 109 | 26.9 | 275 | 55.9 | 0.3 (0.2–0.4) |
| Mitral insufficiency | 77 | 19.0 | 135 | 27.4 | 0.7 (0.5–0.9) |
| Ischemic cardiopathy | 78 | 19.3 | 122 | 24.8 | 0.7 (0.5–0.9) |
| Emphysema | 78 | 19.6 | 66 | 13.4 | 0.9 (0.6–1.2) |
| Atrial fibrillation | 49 | 12.1 | 160 | 32.5 | 0.3 (0.2–0.4) |
| Sleep apnea syndrome | 63 | 15.6 | 60 | 12.2 | 1.3 (0.9–1.9) |
| Adverse Events | Nintedanib | % | OR (95%CI) | Pirfenidone | % | OR (95%CI) |
|---|---|---|---|---|---|---|
| Diarrhea | 48 | 29.1 | 5.6 (3.6–8.7) | 46 | 19.2 | 2.2 (4.5–3.3) |
| Nausea | 20 | 12.1 | 2.8 (1.5–4.8) | 25 | 10.4 | 1.1 (0.7–1.8) |
| Loss of appetite | 28 | 17.0 | 5.1 (2.9–8.9) | 27 | 11.3 | 1.2 (0.7–1.9) |
| Dizziness | 37 | 22.4 | 5.2 (3.1–8.4) | 48 | 20.0 | 2.2 (1.5–3.2) |
| Headache | 15 | 9.1 | 2.8 (1.5–5.5) | 21 | 8.8 | 1.6 (0.9–2.7) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morena, D.; Fernández, J.; Campos, C.; Castillo, M.; López, G.; Benavent, M.; Izquierdo, J.L. Clinical Profile of Patients with Idiopathic Pulmonary Fibrosis in Real Life. J. Clin. Med. 2023, 12, 1669. https://doi.org/10.3390/jcm12041669
Morena D, Fernández J, Campos C, Castillo M, López G, Benavent M, Izquierdo JL. Clinical Profile of Patients with Idiopathic Pulmonary Fibrosis in Real Life. Journal of Clinical Medicine. 2023; 12(4):1669. https://doi.org/10.3390/jcm12041669
Chicago/Turabian StyleMorena, Diego, Jesús Fernández, Carolina Campos, María Castillo, Guillermo López, María Benavent, and José Luis Izquierdo. 2023. "Clinical Profile of Patients with Idiopathic Pulmonary Fibrosis in Real Life" Journal of Clinical Medicine 12, no. 4: 1669. https://doi.org/10.3390/jcm12041669