
Review of the Use of Animal Models of Human Polycystic Kidney Disease for the Evaluation of Experimental Therapeutic Modalities


Abstract
:1. Introduction
2. Spontaneous Model Animals
2.1. cpk Mice
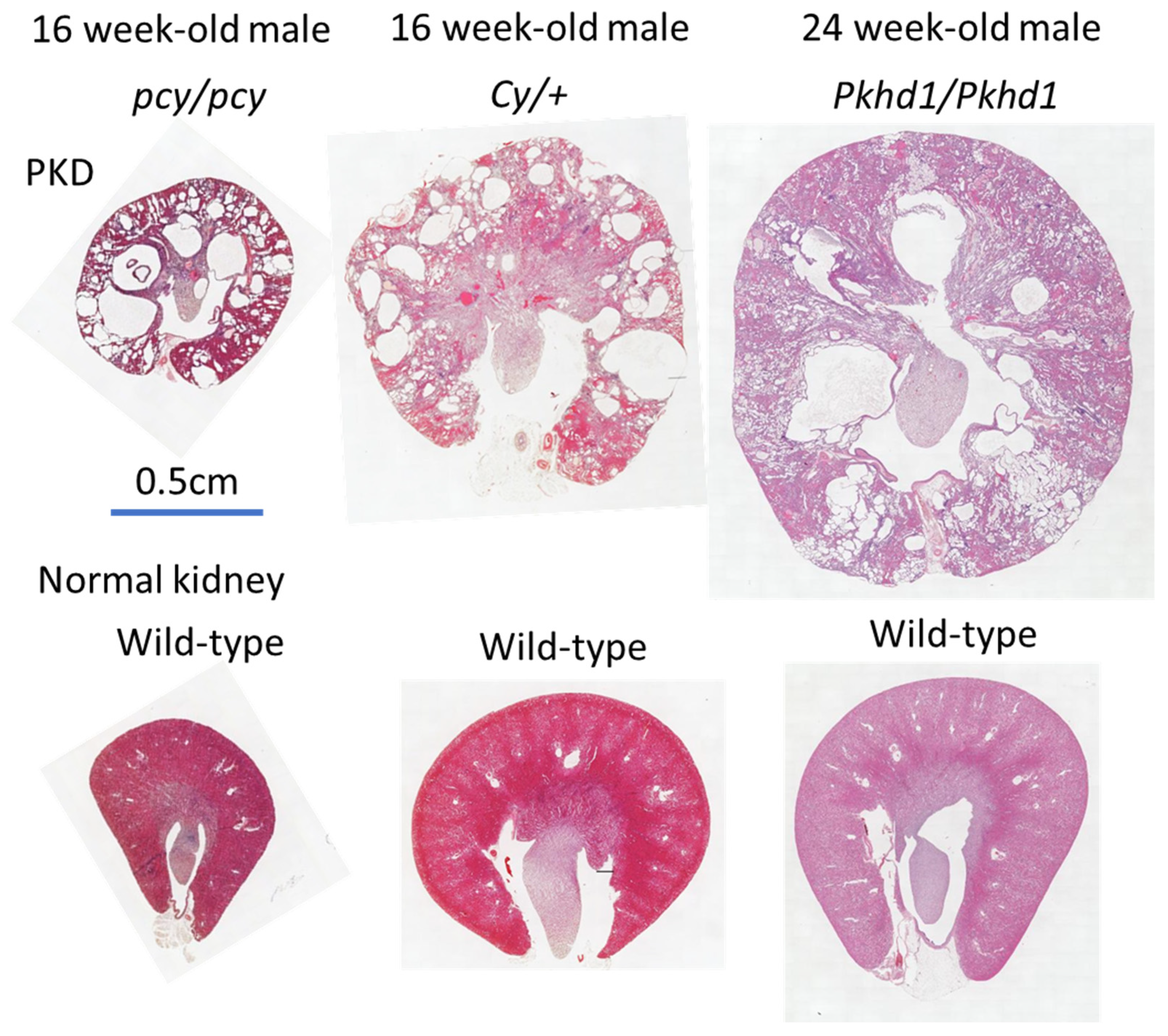
2.2. pcy Mice
2.3. Cy Rats
2.4. PCK Rats
3. Genetically Modified Models
3.1. KO and CKO Mice
3.2. Double-Mutant Mice
3.3. Knock-in Mice
3.4. Genome Editing
4. Discussion
5. Summary and Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sekine, A.; Hidaka, S.; Moriyama, T.; Shikida, Y.; Shimazu, K.; Ishikawa, E.; Uchiyama, K.; Kataoka, H.; Kawano, H.; Kurashige, M.; et al. Cystic kidney diseases that require a differential diagnosis from autosomal dominant polycystic kidney disease (ADPKD). J. Clin. Med. 2022, 11, 6528. [Google Scholar] [CrossRef] [PubMed]
- Mei, C.-L.; Xue, C.; Yu, S.-Q.; Dai, B.; Chen, J.-H.; Li, Y.; Chen, L.-M.; Liu, Z.-S.; Wu, Y.-G.; Hu, Z.; et al. Executive Summary: Clinical Practice Guideline for Autosomal Dominant Polycystic Kidney Disease in China. Kidney Dis. 2020, 6, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Cornec-Le Gall, E.; Alam, A.; Perrone, R.D. Autosomal dominant polycystic kidney disease. Lancet 2019, 393, 919–935. [Google Scholar] [CrossRef] [PubMed]
- Colbert, G.B.; Elrggal, M.E.; Gaur, L.; Lerma, E.V. Update and review of adult polycystic kidney disease. Dis.-A-Mon. 2020, 66, 100887. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Primers 2018, 4, 50. [Google Scholar] [CrossRef]
- Elisakova, V.; Merta, M.; Reiterova, J.; Baxova, A.; Kotlas, J.; Hirschfeldova, K.; Obeidova, L.; Tesar, V.; Stekrova, J. Bilineal inheritance of pathogenic PKD1 and PKD2 variants in a Czech family with autosomal dominant polycystic kidney disease—A case report. BMC Nephrol. 2018, 19, 163. [Google Scholar] [CrossRef] [PubMed]
- Dong, K.; Zhang, C.; Tian, X.; Coman, D.; Hyder, F.; Ma, M.; Somlo, S. Renal plasticity revealed through reversal of polycystic kidney disease in mice. Nat. Genet. 2021, 53, 1649–1663. [Google Scholar] [CrossRef]
- Gall, E.C.-L.; Audrézet, M.-P.; Chen, J.-M.; Hourmant, M.; Morin, M.-P.; Perrichot, R.; Charasse, C.; Whebe, B.; Renaudineau, E.; Jousset, P.; et al. Type of PKD1 Mutation Influences Renal Outcome in ADPKD. J. Am. Soc. Nephrol. 2013, 24, 1006–1013. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, A.R.; Somlo, S. Loss of Cilia Does Not Slow Liver Disease Progression in Mouse Models of Autosomal Recessive Polycystic Kidney Disease. Kidney360 2020, 1, 962–968. [Google Scholar] [CrossRef]
- Goggolidou, P.; Richards, T. The genetics of Autosomal Recessive Polycystic Kidney Disease (ARPKD). Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166348. [Google Scholar] [CrossRef] [PubMed]
- Guay-Woodford, L.M.; Desmond, R.A. Autosomal Recessive Polycystic Kidney Disease: The Clinical Experience in North America. Pediatrics 2003, 111, 1072–1080. [Google Scholar] [CrossRef] [Green Version]
- MacKay, C.E.; Floen, M.; Leo, M.D.; Hasan, R.; Garrud, T.A.C.; Fernández-Peña, C.; Singh, P.; Malik, K.U.; Jaggar, J.H. A plasma membrane-localized polycystin-1/polycystin-2 complex in endothelial cells elicits vasodilation. eLife 2022, 11, e74765. [Google Scholar] [CrossRef]
- Kim, I.; Li, C.; Liang, D.; Chen, X.-Z.; Coffy, R.J.; Ma, J.; Zhao, P.; Wu, G. Polycystin-2 Expression Is Regulated by a PC2-binding Domain in the Intracellular Portion of Fibrocystin. J. Biol. Chem. 2008, 283, 31559–31566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildebrandt, F.; Zhou, W. Nephronophthisis-Associated Ciliopathies. J. Am. Soc. Nephrol. 2007, 18, 1855–1871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, S.A.; Modarage, K.; Goggolidou, P. The Role of Wnt Signalling in Chronic Kidney Disease (CKD). Genes 2020, 11, 496. [Google Scholar] [CrossRef] [PubMed]
- Vogel, P.; Read, R.W.; Hansen, G.M.; Powell, D.R. Histopathology is required to identify and characterize myopathies in high-throughput phenotype screening of genetically engineered mice. Vet. Pathol. 2021, 58, 1158–1171. [Google Scholar] [CrossRef] [PubMed]
- Shakya, M.; Lindberg, I. Mouse Models of Human Proprotein Convertase Insufficiency. Endocr. Rev. 2021, 42, 259–294. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.-L.; Zhang, H.-B.; Li, L.; Yang, Z.-L.; Jiang, L. Characterization of two novel knock-in mouse models of syndromic retinal ciliopathy carrying hypomorphic Sdccag8 mutations. Zool. Res. 2022, 43, 442–456. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Yu, J.; Kim, H.K.; Kim, J.Y.; Kim, M.S.; Cho, Y.G.; Bae, S.; Kang, K.K.; Jung, Y.J. Genome editing of golden SNP-carrying lycopene epsilon-cyclase (LcyE) gene using the CRSPR-Cas9/HDR and geminiviral replicon system in rice. Int. J. Mol. Sci. 2022, 23, 10383. [Google Scholar] [CrossRef]
- Richards, T.; Modarage, K.; Malik, S.A.; Goggolidou, P. The cellular pathways and potential therapeutics of Polycystic Kidney Disease. Biochem. Soc. Trans. 2021, 49, 1171–1188. [Google Scholar] [CrossRef]
- Yang, C.; Harafuji, N.; O’Connor, A.K.; Kesterson, R.A.; Watts, J.A.; Majmundar, A.J.; Braun, D.A.; Lek, M.; Laricchia, K.M.; Fathy, H.M.; et al. Cystin genetic variants cause autosomal recessive polycystic kidney disease associated with altered Myc expression. Sci. Rep. 2021, 11, 18274. [Google Scholar] [CrossRef]
- Swenson-Fields, K.I.; Vivian, C.J.; Salah, S.M.; Peda, J.D.; Davis, B.M.; van Rooijen, N.; Wallace, D.P.; Fields, T.A. Macrophages promote polycystic kidney disease progression. Kidney Int. 2013, 83, 855–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, J.A.; Rezaei, M.; Broderick, C.; Lin, L.; Wang, X.; Hoppe, B.; Cowley, B.D.; Savica, V.; Torres, V.E.; Khan, S.; et al. Crystal deposition triggers tubule dilation that accelerates cystogenesis in polycystic kidney disease. J. Clin. Investig. 2019, 129, 4506–4522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibazaki, S.; Yu, Z.; Nishio, S.; Tian, X.; Thomson, R.B.; Mitobe, M.; Louvi, A.; Velazquez, H.; Ishibe, S.; Cantley, L.G.; et al. Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1. Hum. Mol. Genet. 2008, 17, 1505–1516. [Google Scholar] [CrossRef] [Green Version]
- Nagao, S.; Kugita, M.; Yoshihara, D.; Yamaguchi, T. Animal Models for Human Polycystic Kidney Disease. Exp. Anim. 2012, 61, 477–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, T.; Lysecki, C.; Reid, A.; Nagao, S.; Aukema, H.M. Renal Cyclooxygenase Products are Higher and Lipoxygenase Products are Lower in Early Disease in the pcy Mouse Model of Adolescent Nephronophthisis. Lipids 2013, 49, 39–47. [Google Scholar] [CrossRef]
- Takenaka, T.; Kobori, H.; Inoue, T.; Miyazaki, T.; Suzuki, H.; Nishiyama, A.; Ishii, N.; Hayashi, M. Klotho supplementation ameliorates blood pressure and renal function in DBA/2-pcy mice, a model of polycystic kidney disease. Am. J. Physiol. Physiol. 2020, 318, F557–F564. [Google Scholar] [CrossRef]
- Neudecker, S.; Walz, R.; Menon, K.; Maier, E.; Bihoreau, M.-T.; Obermüller, N.; Kränzlin, B.; Gretz, N.; Hoffmann, S.C. Transgenic Overexpression of Anks6(p.R823W) Causes Polycystic Kidney Disease in Rats. Am. J. Pathol. 2010, 177, 3000–3009. [Google Scholar] [CrossRef]
- Nagao, S.; Morita, M.; Kugita, M.; Yoshihara, D.; Yamaguchi, T.; Kurahashi, H.; Calvet, J.P.; Wallace, D.P. Polycystic kidney disease in Han:SPRD Cy rats is associated with elevated expression and mislocalization of SamCystin. Am. J. Physiol. Physiol. 2010, 299, F1078–F1086. [Google Scholar] [CrossRef] [Green Version]
- Kaspareit-Rittinghausen, J.; Rapp, K.; Deerberg, F.; Wcislo, A.; Messow, C. Hereditary Polycystic Kidney Disease Associated with Osteorenal Syndrome in Rats. Vet. Pathol. 1989, 26, 195–201. [Google Scholar] [CrossRef]
- Bae, K.T.; Kumamoto, K.; Yoshimura, A.; Kugita, M.; Horie, S.; Yamaguchi, T.; Bae, J.T.; Nagao, S. Novel 3D capsule device to restrict kidney volume expansion on polycystic kidney progression: Feasibility study in a rat model. J. Nephrol. 2021, 35, 1033–1040. [Google Scholar] [CrossRef]
- Wang, X.; Wu, Y.; Ward, C.J.; Harris, P.C.; Torres, V.E. Vasopressin Directly Regulates Cyst Growth in Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2007, 19, 102–108. [Google Scholar] [CrossRef] [Green Version]
- Mason, S.B.; Liang, Y.; Sinders, R.M.; Miller, C.A.; Eggleston-Gulyas, T.; Crisler-Roberts, R.; Harris, P.C.; Ii, V.H.G. Disease Stage Characterization of Hepatorenal Fibrocystic Pathology in the PCK Rat Model of ARPKD. Anat. Rec. 2010, 293, 1279–1288. [Google Scholar] [CrossRef]
- Sanzen, T.; Harada, K.; Yasoshima, M.; Kawamura, Y.; Ishibashi, M.; Nakanuma, Y. Polycystic Kidney Rat Is a Novel Animal Model of Caroli’s Disease Associated with Congenital Hepatic Fibrosis. Am. J. Pathol. 2001, 158, 1605–1612. [Google Scholar] [CrossRef]
- Torres, V.E. Role of Vasopressin Antagonists. Clin. J. Am. Soc. Nephrol. 2008, 3, 1212–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sans-Atxer, L.; Joly, D. Tolvaptan in the treatment of autosomal dominant polycystic kidney disease: Patient selection and special considerations. Int. J. Nephrol. Renov. Dis. 2018, 11, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Nagao, S.; Nishii, K.; Katsuyama, M.; Kurahashi, H.; Marunouchi, T.; Takahashi, H.; Wallace, D.P. Increased Water Intake Decreases Progression of Polycystic Kidney Disease in the PCK Rat. J. Am. Soc. Nephrol. 2006, 17, 2220–2227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Cui, J.; Yang, C.; Rosenblum, J.S.; Zhang, Q.; Song, Q.; Pang, Y.; Fang, F.; Sun, M.; Dmitriev, P.; et al. A Transgenic Mouse Model of Pacak–Zhuang Syndrome with An Epas1 Gain-of-Function Mutation. Cancers 2019, 11, 667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, H.C.; Siroky, B.J.; Henske, E.P. Renal disease in tuberous sclerosis complex: Pathogenesis and therapy. Nat. Rev. Nephrol. 2018, 14, 704–716. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, P.; Song, L.; Bai, L.; Huen, M.S.Y.; Liu, Y.; Lu, L.Y. 53BP1 loss rescues embryonic lethality but not genomic instability of BRCA1 total knockout mice. Cell Death Differ. 2020, 27, 2552–2567. [Google Scholar] [CrossRef] [PubMed]
- Vidalin, O.; Muslmani, M.; Estienne, C.; Echchakir, H.; Abina, A.M. In vivo target validation using gene invalidation, RNA interference and protein functional knockout models: It is the time to combine. Curr. Opin. Pharmacol. 2009, 9, 669–676. [Google Scholar] [CrossRef]
- Chenouard, V.; Remy, S.; Tesson, L.; Ménoret, S.; Ouisse, L.H.; Cherifi, Y.; Anegon, I. Advances in genome editing and application to the generation of genetically modified rat models. Front. Genet. 2021, 12, 615491. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhao, J.; Zhang, Y.; Wang, L.; Zuo, L.; Niu, A.; Zhang, W.; Xue, X.; Zhao, S.; Sun, C.; et al. Generation and identification of a conditional knockout allele for the PSMD11 gene in mice. BMC Dev. Biol. 2021, 21, 4. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, Y.; Inagi, R.; Yoshihara, D.; Kugita, M.; Nagao, S.; Shimizu, A.; Takeda, N.; Wake, M.; Honda, K.; Zhou, J.; et al. Mitochondrial Abnormality Facilitates Cyst Formation in Autosomal Dominant Polycystic Kidney Disease. Mol. Cell. Biol. 2017, 37, 00337-17. [Google Scholar] [CrossRef] [Green Version]
- Cassina, L.; Chiaravalli, M.; Boletta, A. Increased mitochondrial fragmentation in polycystic kidney disease acts as a modifier of disease progression. FASEB J. 2020, 34, 6493–6507. [Google Scholar] [CrossRef]
- Yang, T.; Heng, C.; Zhou, Y.; Hu, Y.; Chen, S.; Wang, H.; Yang, H.; Jiang, Z.; Qian, S.; Wang, Y.; et al. Targeting mammalian serine/threonine-protein kinase 4 through Yes-associated protein/TEA domain transcription factor-mediated epithelial-mesenchymal transition ameliorates diabetic nephropathy orchestrated renal fibrosis. Metabolism 2020, 108, 154258. [Google Scholar] [CrossRef]
- Leuenroth, S.J.; Bencivenga, N.; Igarashi, P.; Somlo, S.; Crews, C.M. Triptolide Reduces Cystogenesis in a Model of ADPKD. J. Am. Soc. Nephrol. 2008, 19, 1659–1662. [Google Scholar] [CrossRef] [Green Version]
- Nigro, E.A.; Distefano, G.; Chiaravalli, M.; Matafora, V.; Castelli, M.; Gritti, A.P.; Bachi, A.; Boletta, A. Polycystin-1 Regulates Actomyosin Contraction and the Cellular Response to Extracellular Stiffness. Sci. Rep. 2019, 9, 16640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Formica, C.; Kunnen, S.; Dauwerse, J.G.; Mullick, A.E.; Dijkstra, K.L.; Scharpfenecker, M.; Peters, D.J.M.; the DIPAK Consortium. Reducing YAP expression in Pkd1 mutant mice does not improve the cystic phenotype. J. Cell. Mol. Med. 2020, 24, 8876–8882. [Google Scholar] [CrossRef] [PubMed]
- Casarella, A.; Nicotera, R.; Zicarelli, M.T.; Urso, A.; Presta, P.; Deodato, F.; Bolignano, D.; De Sarro, G.; Andreucci, M.; Russo, E.; et al. Autosomic dominant polycystic kidney disease and metformin: Old knowledge and new insights on retarding progression of chronic kidney disease. Med. Res. Rev. 2021, 42, 629–640. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, Y.; Dang, L.; Geng, M.; Sun, Y.; Lu, Y.; Fang, Z.; Xiong, H.; Chen, Y. Integrative Cistromic and Transcriptomic Analyses Identify CREB Target Genes in Cystic Renal Epithelial Cells. J. Am. Soc. Nephrol. 2021, 32, 2529–2541. [Google Scholar] [CrossRef]
- Schena, G.; Carmosino, M.; Chiurlia, S.; Onuchic, L.; Mastropasqua, M.; Maiorano, E.; Schena, F.P.; Caplan, M.J. β3 adrenergic receptor as potential therapeutic target in ADPKD. Physiol. Rep. 2021, 9, e15058. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Barbero, N.; Gutiérrez-Munoz, C.; Blázquez-Serra, R.; Martín-Ventura, J.L.; Blanco-Colio, L.M. Tumor necrosis factor-like weak inducer of apoptosis (TWEAK)/fibroblast growth factor-inducible 14 (Fn14) axis in cardiovascular diseases: Progress and challenges. Cells 2020, 9, 405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordido, A.; Nuñez-Gonzalez, L.; Martinez-Moreno, J.M.; Lamas-Gonzalez, O.; Rodriguez-Osorio, L.; Perez-Gomez, M.V.; Martin-Sanchez, D.; Outeda, P.; Chiaravalli, M.; Watnick, T.; et al. TWEAK Signaling Pathway Blockade Slows Cyst Growth and Disease Progression in Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2021, 32, 1913–1932. [Google Scholar] [CrossRef]
- Zimmerman, K.A.; Huang, J.; He, L.; Revell, D.Z.; Li, Z.; Hsu, J.-S.; Fitzgibbon, W.R.; Hazard, E.S.; Hardiman, G.; Mrug, M.; et al. Interferon Regulatory Factor-5 in Resident Macrophage Promotes Polycystic Kidney Disease. Kidney360 2020, 1, 179–190. [Google Scholar] [CrossRef] [Green Version]
- Gainullin, V.G.; Hopp, K.; Ward, C.J.; Hommerding, C.J.; Harris, P.C. Polycystin-1 maturation requires polycystin-2 in a dose-dependent manner. J. Clin. Investig. 2015, 125, 607–620. [Google Scholar] [CrossRef] [Green Version]
- Radadiya, P.S.; Thornton, M.M.; Puri, R.V.; Yerrathota, S.; Dinh-Phan, J.; Magenheimer, B.; Subramaniam, D.; Tran, P.V.; Zhu, H.; Bolisetty, S.; et al. Ciclopirox olamine induces ferritinophagy and reduces cyst burden in polycystic kidney disease. J. Clin. Investig. 2021, 6, e141299. [Google Scholar] [CrossRef]
- Zhou, J.; Li, X. Non-Coding RNAs in Hereditary Kidney Disorders. Int. J. Mol. Sci. 2021, 22, 3014. [Google Scholar] [CrossRef] [PubMed]
- Lakhia, R.; Yheskel, M.; Flaten, A.; Ramalingam, H.; Aboudehen, K.; Ferrè, S.; Biggers, L.; Mishra, A.; Chaney, C.; Wallace, D.P.; et al. Interstitial microRNA miR-214 attenuates inflammation and polycystic kidney disease progression. JCI Insight 2020, 5, e133785. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.C.; Valencia, T.; Allerson, C.; Schairer, A.; Flaten, A.; Yheskel, M.; Kersjes, K.; Li, J.; Gatto, S.; Takhar, M.; et al. Discovery and preclinical evaluation of anti-miR-17 oligonucleotide RGLS4326 for the treatment of polycystic kidney disease. Nat. Commun. 2019, 10, 4148. [Google Scholar] [CrossRef]
- Dwivedi, N.; Tao, S.; Jamadar, A.; Sinha, S.; Howard, C.; Wallace, D.P.; Fields, T.A.; Leask, A.; Calvet, J.P.; Rao, R. Epithelial Vasopressin Type-2 Receptors Regulate Myofibroblasts by a YAP-CCN2–Dependent Mechanism in Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2020, 31, 1697–1710. [Google Scholar] [CrossRef] [PubMed]
- Daneshgar, N.; Baguley, A.W.; Liang, P.I.; Wu, F.; Chu, Y.; Kinter, M.T.; Benavides, G.A.; Johnson, M.S.; Darley-Usmar, V.; Zhang, J.; et al. Metabolic derangement in polycystic kidney disease mouse models is ameliorated by mitochondrial-targeted antioxidants. Commun. Biol. 2021, 4, 1200. [Google Scholar] [CrossRef]
- Daneshgar, N.; Liang, P.-I.; Lan, R.S.; Horstmann, M.M.; Pack, L.; Bhardwaj, G.; Penniman, C.M.; O’Neill, B.T.; Dai, D.-F. Elamipretide treatment during pregnancy ameliorates the progression of polycystic kidney disease in maternal and neonatal mice with PKD1 mutations. Kidney Int. 2021, 101, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Pastor-Soler, N.M.; Li, H.; Pham, J.; Rivera, D.; Ho, P.-Y.; Mancino, V.; Saitta, B.; Hallows, K.R. Metformin improves relevant disease parameters in an autosomal dominant polycystic kidney disease mouse model. Am. J. Physiol. Physiol. 2022, 322, F27–F41. [Google Scholar] [CrossRef] [PubMed]
- Di Mise, A.; Wang, X.; Ye, H.; Pellegrini, L.; Torres, V.E.; Valenti, G. Pre-clinical evaluation of dual targeting of the GPCRs CaSR and V2R as therapeutic strategy for autosomal dominant polycystic kidney disease. FASEB J. 2021, 35, e21874. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, L.; Thao, K.; Sussman, C.R.; LaBranche, T.; Palmer, M.; Harris, P.C.; McKnight, G.S.; Hoeflich, K.P.; Schalm, S.; et al. Protein Kinase A Downregulation Delays the Development and Progression of Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2022, 33, 1087–1104. [Google Scholar] [CrossRef]
- Yanda, M.K.; Cebotaru, L. VX-809 mitigates disease in a mouse model of autosomal dominant polycystic kidney disease bearing the R3277C human mutation. FASEB J. 2021, 35, e21987. [Google Scholar] [CrossRef]
- Jamadar, A.; Suma, S.M.; Mathew, S.; Fields, T.A.; Wallace, D.P.; Calvet, J.P.; Rao, R. The tyrosine-kinase inhibitor Nintedanib ameliorates autosomal-dominant polycystic kidney disease. Cell Death Dis. 2021, 12, 947. [Google Scholar] [CrossRef]
- Hopp, K.; Hommerding, C.J.; Wang, X.; Ye, H.; Harris, P.C.; Torres, V.E. Tolvaptan plus Pasireotide Shows Enhanced Efficacy in a PKD1 Model. J. Am. Soc. Nephrol. 2015, 26, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Gao, J.; Yang, X.; Li, T.; Yang, B.; Aili, A. Combination of curcumin and ginkgolide B inhibits cystogenesis by regulating multiple signaling pathways. Mol. Med. Rep. 2021, 23, 195. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhou, H.; Meng, J.; Zhang, S.; Li, X.; Wang, S.; Shao, G.; Jin, W.; Geng, X.; Zhu, S.; et al. Cardamonin retards progression of autosomal dominant polycystic kidney disease via inhibiting renal cyst growth and interstitial fibrosis. Pharmacol. Res. 2020, 155, 104751. [Google Scholar] [CrossRef] [PubMed]
- Agborbesong, E.; Zhou, J.X.; Li, L.X.; Calvet, J.P.; Li, X. Antioxidant enzyme peroxiredoxin 5 regulates cyst growth and ciliogenesis via modulating Plk1 stability. FASEB J. 2022, 36, e22089. [Google Scholar] [CrossRef] [PubMed]
- Streets, A.J.; Prosseda, P.P.; Ong, A.C. Polycystin-1 regulates ARHGAP35-dependent centrosomal RhoA activation and ROCK signaling. J. Clin. Investig. 2020, 5, e135385. [Google Scholar] [CrossRef]
- Peintner, L.; Venkatraman, A.; Waeldin, A.; Hofherr, A.; Busch, T.; Voronov, A.; Viau, A.; Kuehn, E.W.; Köttgen, M.; Borner, C. Loss of PKD1/polycystin-1 impairs lysosomal activity in a CAPN (calpain)-dependent manner. Autophagy 2021, 17, 2384–2400. [Google Scholar] [CrossRef]
- Sousa, M.V.; Amaral, A.G.; Freitas, J.A.; Murata, G.M.; Watanabe, E.H.; Balbo, B.E.; Tavares, M.D.; Hortegal, R.A.; Rocon, C.; Souza, L.E.; et al. Smoking accelerates renal cystic disease and worsens cardiac phenotype in Pkd1-deficient mice. Sci. Rep. 2021, 11, 14443. [Google Scholar] [CrossRef] [PubMed]
- Cabrita, I.; Kraus, A.; Scholz, J.K.; Skoczynski, K.; Schreiber, R.; Kunzelmann, K.; Buchholz, B. Cyst growth in ADPKD is prevented by pharmacological and genetic inhibition of TMEM16A in vivo. Nat. Commun. 2020, 11, 4320. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Tian, X.; Ma, M.; Somlo, S.; Weinstein, A.M.; Wang, T. Restoration of proximal tubule flow–activated transport prevents cyst growth in polycystic kidney disease. J. Clin. Investig. 2021, 6, e146041. [Google Scholar] [CrossRef]
- Lakhia, R.; Hajarnis, S.; Williams, D.; Aboudehen, K.; Yheskel, M.; Xing, C.; Hatley, M.; Torres, V.E.; Wallace, D.P.; Vishal Patel, V. MicroRNA-21 Aggravates Cyst Growth in a Model of Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2016, 27, 2319. [Google Scholar] [CrossRef] [Green Version]
- Kashyap, S.; Hein, K.Z.; Chini, C.C.; Lika, J.; Warner, G.M.; Bale, L.K.; Torres, V.E.; Harris, P.C.; Oxvig, C.; Conover, C.A.; et al. Metalloproteinase PAPP-A regulation of IGF-1 contributes to polycystic kidney disease pathogenesis. J. Clin. Investig. 2020, 5, e135700. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Zhang, S.; Qiu, Z.; Li, X.; Huang, H.; Jin, W.; Xu, Y.; Shao, G.; Wang, L.; Meng, J.; et al. Inhibiting Focal Adhesion Kinase Ameliorates Cyst Development in Polycystin-1–Deficient Polycystic Kidney Disease in Animal Model. J. Am. Soc. Nephrol. 2021, 32, 2159–2174. [Google Scholar] [CrossRef]
- Ramalingam, H.; Kashyap, S.; Cobo-Stark, P.; Flaten, A.; Chang, C.M.; Hajarnis, S.; Hein, K.Z.; Lika, J.; Warner, G.M.; Espindola-Netto, J.M.; et al. A methionine-Mettl3-N(6)-methyladenosine axis promotes polycystic kidney disease. Cell Metab. 2021, 33, 1234–1247.e7. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Sun, Y.; Liu, Z.; Lu, Y.; Zhu, X.; Lan, B.; Mi, Z.; Dang, L.; Li, N.; Zhan, W.; et al. Activation of NRF2 ameliorates oxidative stress and cystogenesis in autosomal dominant polycystic kidney disease. Sci. Transl. Med. 2020, 12, eaba3613. [Google Scholar] [CrossRef]
- Ding, H.; Li, L.X.; Harris, P.C.; Yang, J.; Li, X. Extracellular vesicles and exosomes generated from cystic renal epithelial cells promote cyst growth in autosomal dominant polycystic kidney disease. Nat. Commun. 2021, 12, 4548. [Google Scholar] [CrossRef]
- Hopp, K.; Kleczko, E.K.; Gitomer, B.Y.; Chonchol, M.; Klawitter, J.; Christians, U.; Klawitter, J. Metabolic reprogramming in a slowly developing orthologous model of polycystic kidney disease. Am. J. Physiol. Physiol. 2022, 322, F258–F267. [Google Scholar] [CrossRef] [PubMed]
- Shan, D.; Rezonzew, G.; Mullen, S.; Roye, R.; Zhou, J.; Chumley, P.; Revell, D.Z.; Challa, A.K.; Kim, H.; Lockhart, M.E.; et al. Heterozygous Pkhd1C642* mice develop cystic liver disease and proximal tubule ectasia that mimics radiographic signs of medullary sponge kidney. Am. J. Physiol. Physiol. 2019, 316, F463–F472. [Google Scholar] [CrossRef]
- Li, D.; Hu, M.; Chen, H.; Wu, X.; Wei, X.; Lin, H.; Gao, X.; Wang, H.; Li, M.; Ong, A.C.M.; et al. An Nphp1 knockout mouse model targeting exon 2–20 demonstrates characteristic phenotypes of human nephronophthisis. Hum. Mol. Genet. 2021, 31, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Zarei, A.; Razban, V.; Hosseini, S.E.; Tabei, S.M.B. Creating cell and animal models of human disease by genome editing using CRISPR/Cas9. J. Gene Med. 2019, 21, e3082. [Google Scholar] [CrossRef]


{kind=link}
| Gene | Strain/Gene Name | Treatment/Analysis/ Administration | Results/Effects | References |
|---|---|---|---|---|
| Cy Rat | ||||
| Nphp16 | Han:SPRD-Cy/+ | 3D capsule device | Suppression of the PKD progression. | [31] |
| Nphp16 | Han:SPRD-Cy/+ | MitoQuinone, a mitochondria-specific antioxidant | Inactivation of ERK/MAPK. Reduction of intracellular superoxide. Inhibited proliferation of the epithelial cysts. | [44] |
| pcy Mouse | ||||
| Nphp3 | Pcy | RGLS4326, an anti-miR-17 oligonucleotide | Suppression of the PKD progression. | [60] |
| PCK Rat | ||||
| Pkhd1 | PCK | Lixivaptan, a novel V2R antagonist and R-568, a calcium receptor agonist | Decrease in the cAMP level. Suppression of the PKA activity. Decrease in the phosphorylated AMPK and ERK. Suppression of the PKD progression. Suppression of fibrosis. | [67] |
| KO and CKO Mice | ||||
| Pkd1 | Pkd1flox/flox; Ksp-Cre | MitoQuinone, a mitochondria-specific antioxidant | Inactivation of ERK/MAPK. Reduction of intracellular superoxide. Inhibited proliferation of epithelial cysts. | [44] |
| Pkd1 | Pkd1flox/−; Ksp-Cre | - | Disturbances in mitochondrial structure and function. Decreased expression of the fusion-promoting proteins OPA1 and MFN1. Increased expression of the mitogenic protein DRP1. | [45] |
| Mdivi-1, a DRP-1 inhibitor | Suppression of the PKD progression. Improvement of the renal function. | |||
| Pkd1 | Pkd1flox/−; Ksp-Cre | - | Increased actomyosin contraction. YAP nuclear translocation. Enhanced YAP transcriptional activity. | [48] |
| Fasudil, a protein kinase inhibitor | Inhibition of Rho kinase (ROCK)-dependent actomyosin contraction. Inhibition of YAP activity. | |||
| Pkd1 | iKspPkd1 del | Antisense oligonucleotides | Downregulation of YAP, a key transcription factor in the Hippo signaling pathway, but upregulation of downstream targets Myc, Acta2, and Vim, in the WNT and TGF-β pathways. | [49] |
| Pkd1 | Pkd1fl/fl; Cre/Esr1+ | - | Increase of phosphorylated CREB (p-CREB) and of active histone modifications (H3K4me3 and H3K27ac). | [51] |
| 666-15, a pharmacological inhibitor of CREB (cAMP response element binding protein) | Inhibition of the expansion of the cystic area. | |||
| Pkd1fl/fl; Cdh16-Cre | Genetic inhibition with a dominant-negative inhibitor of CREB (A-CREB) | Inhibition of the cystic area expansion. | ||
| Gene | Strain/Gene Name | Treatment/Analysis/ Administration | Results/Effects | References |
| KO and CKO Mice continued | ||||
| Pkd1 | Pkd1fl/fl; Pax8rtTA; TetO-Cre | SR59230A, a selective β3-adrenergic receptor antagonist | Reduction of cAMP concentration. Inhibition of the PKD progression. Partial improvement of the renal function. | [52] |
| Pkd1 | Pkd1cond/cond; Tam-Cre2, Pkd1cond/cond; Tam-Cre1 | - | Overexpression of TWEAK (Tumor necrosis factor-like weak inducer of apoptosis) and Fn14 (fibroblast growth factor-inducible 14). | [54] |
| TWEAK (Tumor necrosis factor-like weak inducer of apoptosis) anti-TWEAK | Exacerbation of PKD progression. Suppression of PKD progression. Improvement of the survival rate. Decrease in cell proliferation, NF-κB pathway activation, fibrosis, apoptosis, and macrophage infiltration. | |||
| Pkd1 | Pkd1f/f; Cre-ERTM | IRF5 (Interferon regulatory factor 5) antisense oligonucleotides | Suppression of PKD progression. Reduced number of macrophages. Reduced homeostasis. Decreased expression of the IRF5 in the macrophages. | [55] |
| Pkd1 Pkd2 | Pkd1RC/RC Pkd2+/−, Pkd1RC/RC Pkd2+/+ | - | Cystic formation. Elevated ferritin levels. | [57] |
| CPX (Ciclopirox; 6-Cyclohexyl-1-hydroxy-4-methyl-2(1H)-pyridone) or its olamine salt (CPX-O) | Suppression of PKD progression. Decreased ferritin levels. | |||
| Pkd1 Pkd2 | Ksp-Cre; Pkd1fl/fl, Pkhd1-Cre; Pkd2fl/fl, Pkd1RC/RC, Pkhd1-Cre; Pkd2fl/fl; miR-214−/− | Inhibition of miR-214 (double mutant) | Exacerbation of PKD progression. Increased Tlr4 expression (inflammatory TLR4/IFN-γ/STAT1 signaling pathway). Increased accumulation of the pericystic macrophages. | [59] |
| Pkd2 | Pkhd1-Cre; Pkd2F/F | RGLS4326, an anti-miR-17 oligonucleotide | Suppression of PKD progression. | [60] |
| Pkd1 | Pkd1f/f; Pkhd1cre, Pkd1f/f; Yapf/f; Pkhd1cre, Pkd1f/f; CCN2f/f; Pkhd1cre | Verteporfin, a YAP inhibitor Deletion of YAP gene (double mutant) Deletion of CCN2, a renal collecting duct-specific gene (double mutant) | Inhibition of renal fibrosis. | [61] |
| Pkd1 | Pkd1f/f; Pkhd1-Cre | Nintedanib, which selectively inhibits PDGFR, FGFR, and VEGFR | Suppression of PKD progression. Reduced proliferation of epithelial cysts. Decreased expression of growth factors including YAP. | [68] |
| Pkd1 | Pkd1fox/−; Ksp-Cre | Curcumin and ginkgolide B | Suppression of EGFR/ERK1/2, JNK, PI3K/mTOR, and p38 signaling pathways. | [70] |
| Pkd1 | Pkd1loxp/loxp; Ksp-Cre | Cardamomine nominated from natural product library screening | Inhibition of PKD progression. Inhibition of renal cyst development and interstitial fibrosis. | [71] |
| Pkd1 | Pkd1fl/fl; Pkhd1-Cre | Vorasertib, an inhibitor of Plk1 (Polo-like kinase 1) | Suppression of PKD progression by the antioxidant action pathway of peroxiredoxin 5 (Prdx5)-Polo-like kinase 1 (Plk1). | [72] |
| Pkd1 | Pkd1fl/fl; Pax8rtTA; TetO-Cre | Hydroxyfasudil, a ROCK (Rho-associated coiled-coil containing protein kinase) inhibitor | Suppression of PKD progression. Reduction of centrosome RhoGAP (ARHGAP). Suppression of ROCK signaling pathway. | [73] |
| Pkd1 | Pkd1fl/fl; Pax8rtTA; TetO-Cre | - | Increased activity of calcium-dependent CAPN (Calpain) protease. | [74] |
| CAPN (Calpain) inhibitor | Restoration of lysosomal function. CTSB processing/activity, autophagosome and lysosomal fusion. | |||
| Gene | Strain/Gene Name | Treatment/Analysis/ Administration | Results/Effects | References |
| KO and CKO Mice continued | ||||
| Pkd1 | Pkd1fox/fox; Nestincre, Pkd1fox/−; Nestincre | Secondhand smoke exposure | Acceleration of PKD progression. Increased tubular cell proliferation and apoptosis. Promotion of renal fibrosis. Reduction of glutathione level. Decreased contractile function and structural parameters in the heart. Noticeable reduction of body weight. | [75] |
| Pkd1 | KspCreERT2; Pkd1lox/lox | - | Increased expression of the transmembrane Protein 16A (TMEM16A) and the cystic fibrosis transmembrane conductance regulator (CFTR). Increase of the cystic area. | [76] |
| TMEM16A(transmembrane Protein 16A) antagonists niclosamide and benzbromarone TMEM16A-specific inhibitor Ani9 | Inhibition of TMEM16A. Reduced expansion of the cystic area. Suppression of the abnormal proliferation of the epithelial cysts. | |||
| Pkd2 | Pkd2−/− | DA1(dopamine receptor 1) antagonist, SCH23390 | Suppression of disease progression in PKD. Restored sensitivity of flow-activated Na+ and HCO3− transport. | [77] |
| Pkd2 | Pkhd1-Cre; Pkd2F/F; miR-21−/− | Inhibition of miR-21 (double mutant) | Suppressed expansion of cyst area by regulating apoptosis and proliferation of epithelial cells, and interstitial inflammation. | [78] |
| Pkd1 | Pkd1RC/RC | - | Increased expression of IGF-1 pathway genes. | [79] |
| Pkd1RC/RC; Pappa+/–, Pkd1RC/RC; Pappa–/– | Deletion of PAPP-A (Pregnancy Associated Plasma Protein A) gene (double mutant) | Inhibition of disease progression in PKD. | ||
| Pkd1 | Pkd1flox/flox; Ksp-Cre Pkd1flox/flox; Aqp2-Cre | - | Increased activity of the focal adhesion kinase (FAK). | [80] |
| FAK (focal adhesion kinase) inhibitors (double mutant) | Suppression of PKD progression. Inhibition of FSK/Src activity. Upregulation of ERK and mTOR signaling pathways. | |||
| Pkd1 | Pkd1F/RC | - | Increased methionine and S-adenosylmethionine (SAM). | [81] |
| Pkd1RC/RC | Dietary restriction of methionine | Dietary restriction of methionine. | ||
| Ksp-Cre; Pkd1F/RC; Mettl3F/F | Deletion of Mettl3 gene, a key component of SAM (double mutant) | Delayed expansion of cysts. | ||
| Pkd1 | KspCreERT2; Pkd1lox/lox; Tmem16alox/lox | TMEM16A gene (double mutant) | Inhibition of Ca2+ signaling pathway and cell proliferation. Increased CFTR expression. | [82] |
| Pkd1fl/fl; Cre/Esr1, | Quantitative proteomics | Promotion of the Nuclear Factor Erythroblast 2-Related Factor 2 (NRF2) degradation. | ||
| Pkd1fl/fl; Cre/Esr1; Nrf2−/− | Deletion of NRF2 gene (double mutant) | Increased ROS generation. Inhibition of the cystic area expansion. | ||
| Gene | Strain/Gene Name | Treatment/Analysis/ Administration | Results/Effects | References |
| Knock-in Mouse | ||||
| Pkd1 | Pkd1RC/null, Pkd1RC/RC | PKD1 targeted proteomic analysis | Reduction of TCA cycle, fatty acid oxidation, respiratory complexes, and endogenous antioxidants. | [62] |
| Overexpression of mitochondria-targeted catalase (mCAT) using an adeno-associated virus vector | Reduction of mitochondrial Reactive Oxygen Species (ROS) and oxidative damage. Improvement of disease progression in PKD. Partial improvement in the TCA cycle and fatty acid oxidation. | |||
| Pkd1 | Pkd1RC/RC | Eramipretide, a mit ochondrial protective tetrapeptide | Attenuated ERK1/2 phosphorylation. Improved mitochondrial supercomplex formation. Improvement of PKD progression. | [63] |
| Pkd1 | Pkd1RC/RC | Metformin | Suppression of PKD progression. Reduction of cell proliferation markers. Reduction of inflammation and injury markers. | [64] |
| Pkd1 | Pkd1RC/RC | Lixivaptan, a novel Vasopressin Receptor 2 (V2R) antagonist and R-568, a calcium receptor agonist | Reduction of the cAMP levels. Suppression of the PKA activity. Reduction of phosphorylated AMPK and ERK. Suppression of PKD progression. Suppression of fibrosis. | [65] |
| Pkd1 | Pkd1RC/RC | BLU2864, a selective PRKACA (AMP-dependent protein kinase) inhibitor | Inhibition of PKA activity. Inhibition of cyst formation, growth-promoting pathways, and cyst formation. | [66] |
| Pkd1 | Pkd1RC/RC | VX-809, a modulator of CFTR trafficking and processing | Increased basolateral membrane co-localization of CFTR. Decreased HSP27. Inhibition of PKD progression. | [67] |
| Pkd1 | Pkd1RC/RC | Nintedanib, a receptor tyrosine kinase (RTK) inhibitor | Suppression of PKD progression. Suppression of the cell proliferation. Reduction of the growth factor and fibrosis expressions. | [68] |
| Pkd1 | Pkd1RC/RC | Administration of Extracellular Vesicle (EV)/exosomes, Increased expression of EV/exosomes | Promotion of cyst formation and fibrosis. Increased phosphorylation of AKT, S6, Rb, STAT3, ERK. | [83] |
| GW4869 to inhibit exosome biogenesis/release | Suppression of cyst formation. | |||
| Pkd1 | Pkd1RC/RC | Targeted metabolomics approach | Alteration of the biosynthesis and metabolism of tryptophan and arginine. Increase of indoles, kynurenine, and polyamines. | [84] |
| Genome Editing | ||||
| Pkhd1 | Pkhd1C642* | Genome editing | Heterozygous Pkhd1C642* developed hepatic cysts. Homozygous Pkhd1C642* developed congenital hepatic fibrosis, inflammation of the portal field, fibrosis manifestations. | [85] |
| Nphp1 | Nphp1del2−20/del2−20 | Genome editing | Renal cysts. Thickening of the tubular basement membrane. Retinal degeneration. Abnormal spermatogenesis. | [86] |
| Using of AAV9 vectors | Partial rescue of both renal and retinal phenotypes. | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagao, S.; Yamaguchi, T. Review of the Use of Animal Models of Human Polycystic Kidney Disease for the Evaluation of Experimental Therapeutic Modalities. J. Clin. Med. 2023, 12, 668. https://doi.org/10.3390/jcm12020668
Nagao S, Yamaguchi T. Review of the Use of Animal Models of Human Polycystic Kidney Disease for the Evaluation of Experimental Therapeutic Modalities. Journal of Clinical Medicine. 2023; 12(2):668. https://doi.org/10.3390/jcm12020668
Chicago/Turabian StyleNagao, Shizuko, and Tamio Yamaguchi. 2023. "Review of the Use of Animal Models of Human Polycystic Kidney Disease for the Evaluation of Experimental Therapeutic Modalities" Journal of Clinical Medicine 12, no. 2: 668. https://doi.org/10.3390/jcm12020668