
Ovocystatin Induced Changes in Expression of Alzheimer’s Disease Relevant Proteins in APP/PS1 Transgenic Mice

 , , , , , and
, , , , , and 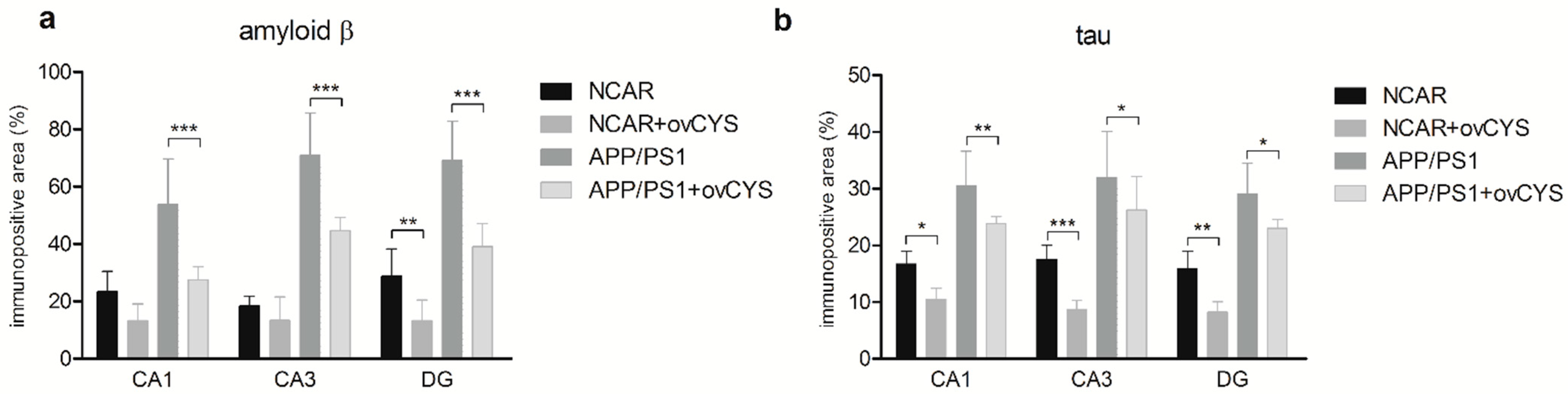{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents—Isolation and Characterization of Ovocystatin
2.2. Animals—APP/PS1 Mice
2.3. Animals Grouping, Intervention, and Sample Preparation
2.4. Immunohistochemistry
2.5. Image Analysis
2.6. Statistical Analysis
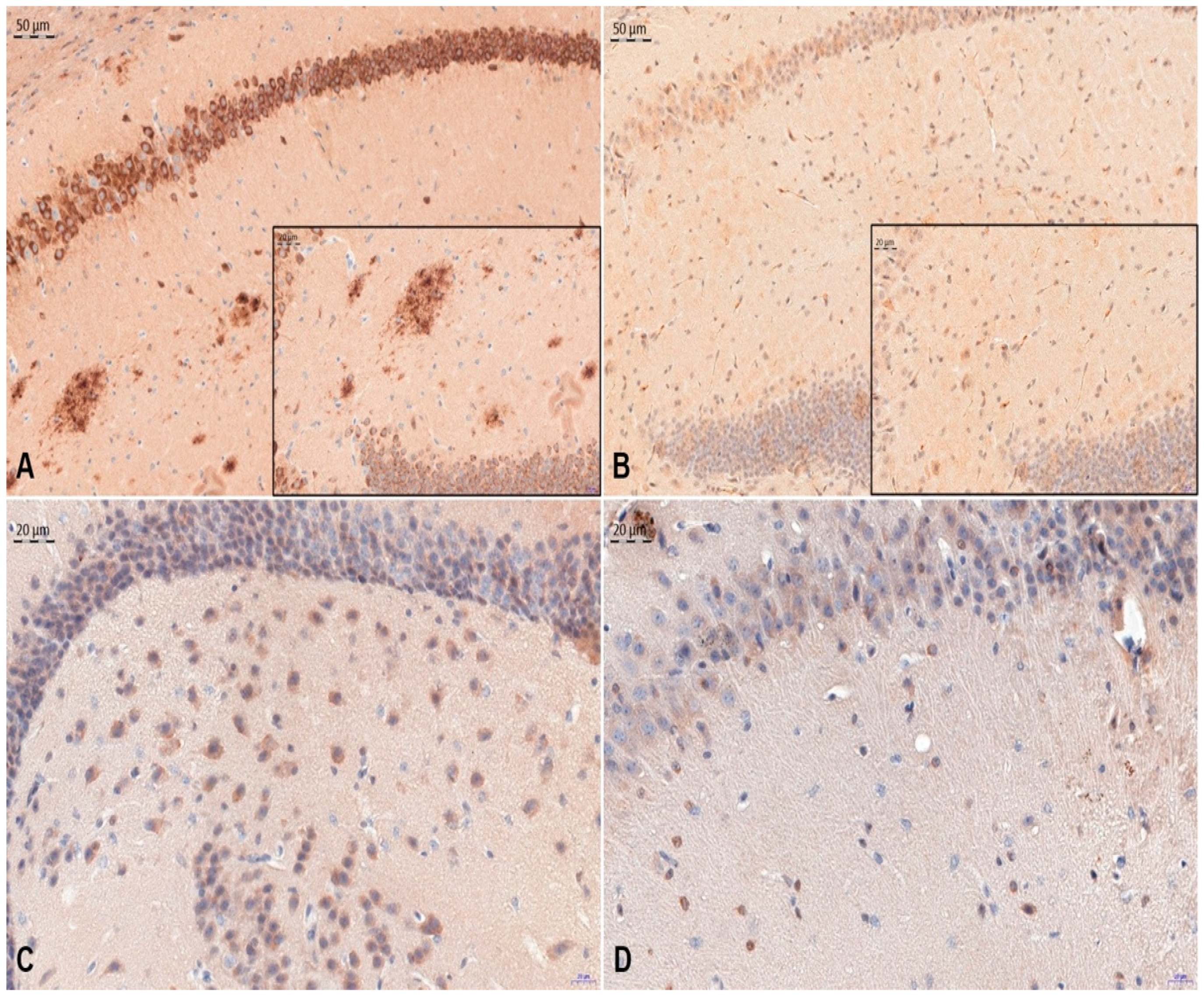
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eleti, S. Drugs in Alzheimer’s disease Dementia: An overview of current pharmacological management and future directions. Psychiatr. Danub. 2016, 28, 136–140. [Google Scholar] [PubMed]
- Ihl, R.; Bunevicius, R.; Frölich, L.; Winblad, B.; Schneider, L.S.; Dubois, B.; Burns, A.; Thibaut, F.; Kasper, S.; Möller, H.-J.; et al. World Federation of Societies of Biological Psychiatry guidelines for the pharmacological treatment of dementias in primary care. Int. J. Psychiatry Clin. Pract. 2014, 19, 2–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Prim. 2015, 1, 15056. [Google Scholar] [CrossRef]
- Trevisan, K.; Cristina-Pereira, R.; Silva-Amaral, D.; Aversi-Ferreira, T.A. Theories of Aging and the Prevalence of Alzheimer’s Disease. BioMed Res. Int. 2019, 2019, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Sochocka, M.; Zwolińska, K.; Leszek, J. The Infectious Etiology of Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 996–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selkoe, D.J. Alzheimer’s disease: Genotypes, phenotype, and treatments. Science 1997, 275, 630–631. [Google Scholar] [CrossRef]
- Finckh, U.; Kuschel, C.; Anagnostouli, M.; Patsouris, E.; Pantes, G.V.; Gatzonis, S.; Kapaki, E.; Davaki, P.; Lamszus, K.; Stavrou, D.; et al. Novel mutations and repeated findings of mutations in familial Alzheimer disease. Neurogenetics 2005, 6, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimer’s Dement. 2016, 12, 292–323. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020, 16, 327–406.
- Prince, M.; Wimo, A.; Guerchet, M.; Gemma-Claire, A.; Wu, Y.-T.; Prina, M. World Alzheimer Report 2015: The Global Impact of Dementia—An Analysis of Prevalence, Incidence, Cost and Trends; Alzheimer’s Disease International: London, UK, 2015. [Google Scholar]
- Yu, T.W.; Lane, H.Y.; Lin, C.H. Novel therapeutic approaches for Alzheimer’s disease: An updated review. Int. J. Mol. Sci. 2021, 22, 8208. [Google Scholar] [CrossRef]
- Adefegha, A. Functional Foods and Nutraceuticals as Dietary Intervention in Chronic Diseases; Novel Perspectives for Health Promotion and Disease Prevention. J. Diet. Suppl. 2017, 15, 977–1009. [Google Scholar] [CrossRef] [PubMed]
- Hasanbasic, S.; Jahic, A.; Karahmet, E.; Sejranic, A.; Prnjavorac, A. The Role of Cysteine Protease in Alzheimer Disease. Mater. Socio Medica 2016, 28, 235–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, E. Cystatin C: A potential target for Alzheimer’s treatment. Expert Rev. Neurother. 2008, 8, 687–689. [Google Scholar] [CrossRef] [PubMed]
- Mathews, P.; Levy, E. Cystatin C in aging and in Alzheimer’s disease. Ageing Res. Rev. 2016, 32, 38–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobek, L.A.; Levine, M.J. Cystatins—Inhibitors of Cysteine Proteinases. Crit. Rev. Oral Biol. Med. 1992, 3, 307–332. [Google Scholar] [CrossRef] [PubMed]
- Abrahamson, M.; Alvarez-Fernandez, M.; Nathanson, C.M. Cystatins. Biochem Symp. 2003, 70, 179–199. [Google Scholar]
- Kaur, G.; Levy, E. Cystatin C in Alzheimer’s disease. Front. Mol. Neurosci. 2012, 5, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, E.; Gauthier, S.; Kaur, G.; Mi, W.; Tizon, B. Protective mechanisms by cystatin C in neurodegenerative diseases. Front. Biosci. 2011, S3, 541–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, E.; Sastre, M.; Kumar, A.; Gallo, G.; Piccardo, P.; Ghetti, B.; Tagliavini, F. Codeposition of Cystatin C with Amyloid-β Protein in the Brain of Alzheimer Disease Patients. J. Neuropathol. Exp. Neurol. 2001, 60, 94–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sastre, M.; Calero, M.; Pawlik, M.; Mathews, P.M.; Kumar, A.; Danilov, V.; Schmidt, S.D.; Nixon, R.A.; Frangione, B.; Levy, E. Binding of cystatin C to Alzheimer’s amyloid beta inhibits in vitro amyloid fibril formation. Neurobiol. Aging 2004, 25, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Selenica, M.L.; Wang, X.; Ostergaard-Pedersen, L.; Westlind-Danielsson, A.; Grubb, A. Cystatin C reduces the in vitro formation of soluble A-beta1-42 oligomers and protofibrils. Scand. J. Clin. Lab. Invest. 2007, 67, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Hook, G.; Hook, V.; Kindy, M. The cysteine protease inhibitor, E64d, reduces brain amyloid-β and improves memory deficits in alzheimer’s disease animal models by inhibiting cathepsin B, but not BACE1, β-secretase activity. J. Alzheimer’s Dis. 2011, 26, 387–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoka, V.; Turk, V.; Turk, B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res. Rev. 2016, 32, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Shamsi, A.; Bano, B. Journey of cystatins from being mere thiol protease inhibitors to at heart of many pathological conditions. Int. J. Biol. Macromol. 2017, 102, 674–693. [Google Scholar] [CrossRef]
- Engh, R.A.; Dieckmann, T.; Bode, W.; Auerswald, E.A.; Turk, V.; Huber, R.; Oschkinat, H. Conformational Variability of Chicken Cystatin: Comparison of Structures Determined by X-ray Diffraction and NMR Spectroscopy. J. Mol. Biol. 1993, 234, 1060–1069. [Google Scholar] [CrossRef] [PubMed]
- Barrett, A.J.; Salvesen, G. Proteinase Inhibitors; Elsevier: Amsterdam, The Netherlands, 1986; pp. 515–569. [Google Scholar]
- Gołab, K.; Gburek, J.; Juszczyńska, K.; Trziszka, T.; Polanowski, A. Stabilization of monomeric chicicen egg white cystatin. Przem. Chem. 2012, 91, 741–744. [Google Scholar]
- Stańczykiewicz, B.; Jakubik-Witkowska, M.; Polanowski, A.; Trziszka, T.; Rymaszewska, J. An animal model of the procognitive properties of cysteine protease inhibitor and immunomodulatory peptides based on colostrum. Adv. Clin. Exp. Med. 2017, 26, 563–569. [Google Scholar] [CrossRef] [Green Version]
- Stańczykiewicz, B.; Rutkowska, M.; Lemieszewska, M.; Jakubik-Witkowska, M.; Gburek, J.; Gołąb, K.; Juszczyńska, K.; Trziszka, T.; Rymaszewska, J. Potential protective effect of ovocystatin on aging- related cognitive impairment in rats Wpływ ovocystatyny na funkcje poznawcze szczurów. Postep. Hig. Med. Dosw. 2017, 71, 1202–1208. [Google Scholar]
- Stańczykiewicz, B.; Jakubik-Witkowska, M.; Rutkowska, M.; Polanowski, A.; Gburek, J.; Gołąb, K.; Juszczyńska, K.; Trziszka, T.; Rymaszewska, J. Beneficial effect of ovocystatin on the cognitive decline in APP/PS1 transgenic mice. Adv. Med. Sci. 2018, 64, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Barrett, A. A new assay for cathepsin B1 and other thiol proteinases. Anal. Biochem. 1972, 47, 280–293. [Google Scholar] [CrossRef]
- Schägger, H.; von Jagow, G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 1987, 166, 368–379. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Slunt, H.H.; Ratovitski, T.; A Jenkins, N.; Copeland, N.G.; Borchelt, D.R. Co-expression of multiple transgenes in mouse CNS: A comparison of strategies. Biomol. Eng. 2001, 17, 157–165. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Fadale, D.J.; Anderson, J.; Xu, G.M.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Lee, M.K.; Younkin, L.H.; Wagner, S.L.; et al. Mutant presenilins specifically elevate the levels of the 42 residue β-amyloid peptide in vivo: Evidence for augmentation of a 42-specific γ secretase. Hum. Mol. Genet. 2004, 13, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Bavisotto, C.C.; Scalia, F.; Gammazza, A.M.; Carlisi, D.; Bucchieri, F.; De Macario, E.C.; Macario, A.J.L.; Cappello, F.; Campanella, C. Extracellular Vesicle-Mediated Cell–Cell Communication in the Nervous System: Focus on Neurological Diseases. Int. J. Mol. Sci. 2019, 20, 434. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, S.H.; Andrews, A.M.; Paul, D.; Pachter, J.S. Extracellular vesicles: Mediators and biomarkers of pathology along CNS barriers. Fluids Barriers CNS 2018, 15, 19. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, J.; Stewart, T.; Banks, W.A.; Zhang, J. The Transport Mechanism of Extracellular Vesicles at the Blood-Brain Barrier. Curr. Pharm. Des. 2018, 23, 6206–6214. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhang, X.; Kang, A.; Ran, C.; Wang, G.; Hao, H. Thinking outside the brain for cognitive improvement: Is peripheral immunomodulation on the way? Neuropharmacology 2015, 96, 94–104. [Google Scholar] [CrossRef]
- Dixit, R.; Ross, J.L.; Goldman, Y.E.; Holzbaur, E.L.F. Differential Regulation of Dynein and Kinesin Motor Proteins by Tau. Science 2008, 319, 1086–1089. [Google Scholar] [CrossRef] [Green Version]
- Oddo, S.; Caccamo, A.; Green, K.N.; Liang, K.; Tran, L.; Chen, Y.; Leslie, F.M.; LaFerla, F.M. Chronic nicotine administration exacerbates tau pathology in a transgenic model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 3046–3051. [Google Scholar] [CrossRef] [Green Version]
- Tizon, B.; Ribe, E.M.; Mi, W.; Troy, C.M.; Levy, E. Cystatin C protects neuronal cells from amyloid-beta-induced toxicity. J. Alzheimers. Dis. 2010, 19, 885–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaeser, S.A.; Herzig, M.C.; Coomaraswamy, J.; Kilger, E.; Selenica, M.-L.; Winkler, D.T.; Staufenbiel, M.; Levy, E.; Grubb, A.; Jucker, M. Cystatin C modulates cerebral beta-amyloidosis. Nat. Genet. 2007, 39, 1437–1439. [Google Scholar] [CrossRef]
- Mi, W.; Pawlik, M.; Sastre, M.; Jung, S.S.; Radvinsky, D.S.; Klein, A.M.; Sommer, J.; Schmidt, S.D.; Nixon, R.A.; Mathews, P.M.; et al. Cystatin C inhibits amyloid-beta deposition in Alzheimer’s disease mouse models. Nat. Genet. 2007, 39, 1440–1442. [Google Scholar] [CrossRef] [PubMed]
- Deng, A.; Irizarry, M.C.; Nitsch, R.M.; Growdon, J.H.; Rebeck, G.W. Elevation of Cystatin C in Susceptible Neurons in Alzheimer’s Disease. Am. J. Pathol. 2001, 159, 1061–1068. [Google Scholar] [CrossRef]
- Sun, B.; Zhou, Y.; Halabisky, B.; Lo, I.; Cho, S.-H.; Mueller-Steiner, S.; Devidze, N.; Wang, X.; Grubb, A.; Gan, L. Cystatin C-Cathepsin B Axis Regulates Amyloid Beta Levels and Associated Neuronal Deficits in an Animal Model of Alzheimer’s Disease. Neuron 2008, 60, 247–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heneka, M.T.; Carson, M.J.; Khoury, J.E.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Nixon, R.A. Autophagy, amyloidogenesis and Alzheimer disease. J. Cell Sci. 2007, 120, 4081–4091. [Google Scholar] [CrossRef] [Green Version]
- Tizon, B.; Sahoo, S.; Yu, W.H.; Gauthier, S.; Kumar, A.R.; Mohan, P.; Figliola, M.; Pawlik, M.; Grubb, A.; Uchiyama, Y.; et al. Induction of Autophagy by Cystatin C: A Mechanism That Protects Murine Primary Cortical Neurons and Neuronal Cell Lines. PLoS ONE 2010, 5, e9819. [Google Scholar] [CrossRef]
- Duan, J.; Marcellus, K.A.; Qin, X.; Wang, Y.; Paudel, H.K. Cystatin C promotes tau protein phosphorylation and causes microtubule instability by inhibiting intracellular turnover of GSK3β in neurons. Mol. Cell. Neurosci. 2018, 89, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Congdon, E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Hung, A.; Cui, H.; Dawkins, E.; Bolos, M.; Foa, L.; Young, K.; Small, D.H. Role of Cystatin C in Amyloid Precursor Protein-induced Proliferation of Neural Stem/Progenitor Cells. J. Biol. Chem. 2013, 288, 18853–18862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blankenvoorde, M.F.; Hof, W.V.; Walgreen-Weterings, E.; Van Steenbergen, T.J.; Brand, H.S.; Veerman, E.C.; Amerongen, A.V.N. Cystatin and cystatin-derived peptides have antibacterial activity against the pathogen Porphyromonas gingivalis. Biol. Chem. 1998, 379, 1371–1376. [Google Scholar] [PubMed]
- Blankenvoorde, M.F.; Henskens, Y.M.; Hof, W.V.; Veerman, E.C.; Amerongen, A.V.N. Inhibition of the growth and cysteine proteinase activity of Porphyromonas gingivalis by human salivary cystatin S and chicken cystatin. Biol. Chem. 1996, 377, 847–850. [Google Scholar]
- Cohen, J.B.; Ryseck, L.P. Cystatins: Protease Inhibitors, Biomarkers and Immunomodulators; Nova Science Pub Inc.: Hauppauge, NY, USA, 2011; pp. 1–269. [Google Scholar]
- Hook, G.; Jacobsen, J.S.; Grabstein, K.; Kindy, M.S.; Hook, V.Y.H. Cathepsin B is a New Drug Target for Traumatic Brain Injury Therapeutics: Evidence for E64d as a Promising Lead Drug Candidate. Front. Neurol. 2015, 6, 178. [Google Scholar] [CrossRef] [Green Version]
- Hook, V.Y.; Kindy, M.; Hook, G. Inhibitors of cathepsin B improve memory and reduce Abeta in transgenic Alzheimer’s Disease mice expressing the wild-type, but not the Swedish mutant, beta -secretase APP site. J. Biol. Chem. 2008, 283, 7745–7753. [Google Scholar] [CrossRef] [Green Version]
- Nagai, A.; Ryu, J.K.; Terashima, M.; Tanigawa, Y.; Wakabayashi, K.; McLarnon, J.G.; Kobayashi, S.; Masuda, J.; Kim, S.U. Neuronal cell death induced by cystatin C in vivo and in cultured human CNS neurons is inhibited with cathepsin B. Brain Res. 2005, 1066, 120–128. [Google Scholar] [CrossRef]
- Watanabe, S.; Hayakawa, T.; Wakasugi, K.; Yamanaka, K. Cystatin C protects neuronal cells against mutant copper-zinc superoxide dismutase-mediated toxicity. Cell Death Dis. 2014, 5, e1497. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stanczykiewicz, B.; Gburek, J.; Rutkowska, M.; Lemieszewska, M.; Gołąb, K.; Juszczyńska, K.; Piotrowska, A.; Trziszka, T.; Dzięgiel, P.; Podhorska-Okołów, M.; et al. Ovocystatin Induced Changes in Expression of Alzheimer’s Disease Relevant Proteins in APP/PS1 Transgenic Mice. J. Clin. Med. 2022, 11, 2372. https://doi.org/10.3390/jcm11092372
Stanczykiewicz B, Gburek J, Rutkowska M, Lemieszewska M, Gołąb K, Juszczyńska K, Piotrowska A, Trziszka T, Dzięgiel P, Podhorska-Okołów M, et al. Ovocystatin Induced Changes in Expression of Alzheimer’s Disease Relevant Proteins in APP/PS1 Transgenic Mice. Journal of Clinical Medicine. 2022; 11(9):2372. https://doi.org/10.3390/jcm11092372
Chicago/Turabian StyleStanczykiewicz, Bartlomiej, Jakub Gburek, Maria Rutkowska, Marta Lemieszewska, Krzysztof Gołąb, Katarzyna Juszczyńska, Aleksandra Piotrowska, Tadeusz Trziszka, Piotr Dzięgiel, Marzenna Podhorska-Okołów, and et al. 2022. "Ovocystatin Induced Changes in Expression of Alzheimer’s Disease Relevant Proteins in APP/PS1 Transgenic Mice" Journal of Clinical Medicine 11, no. 9: 2372. https://doi.org/10.3390/jcm11092372