
The Role of Insulin Resistance in Fueling NAFLD Pathogenesis: From Molecular Mechanisms to Clinical Implications


 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Molecular Links between Insulin Resistance and NAFLD Pathogenesis: What Is New?
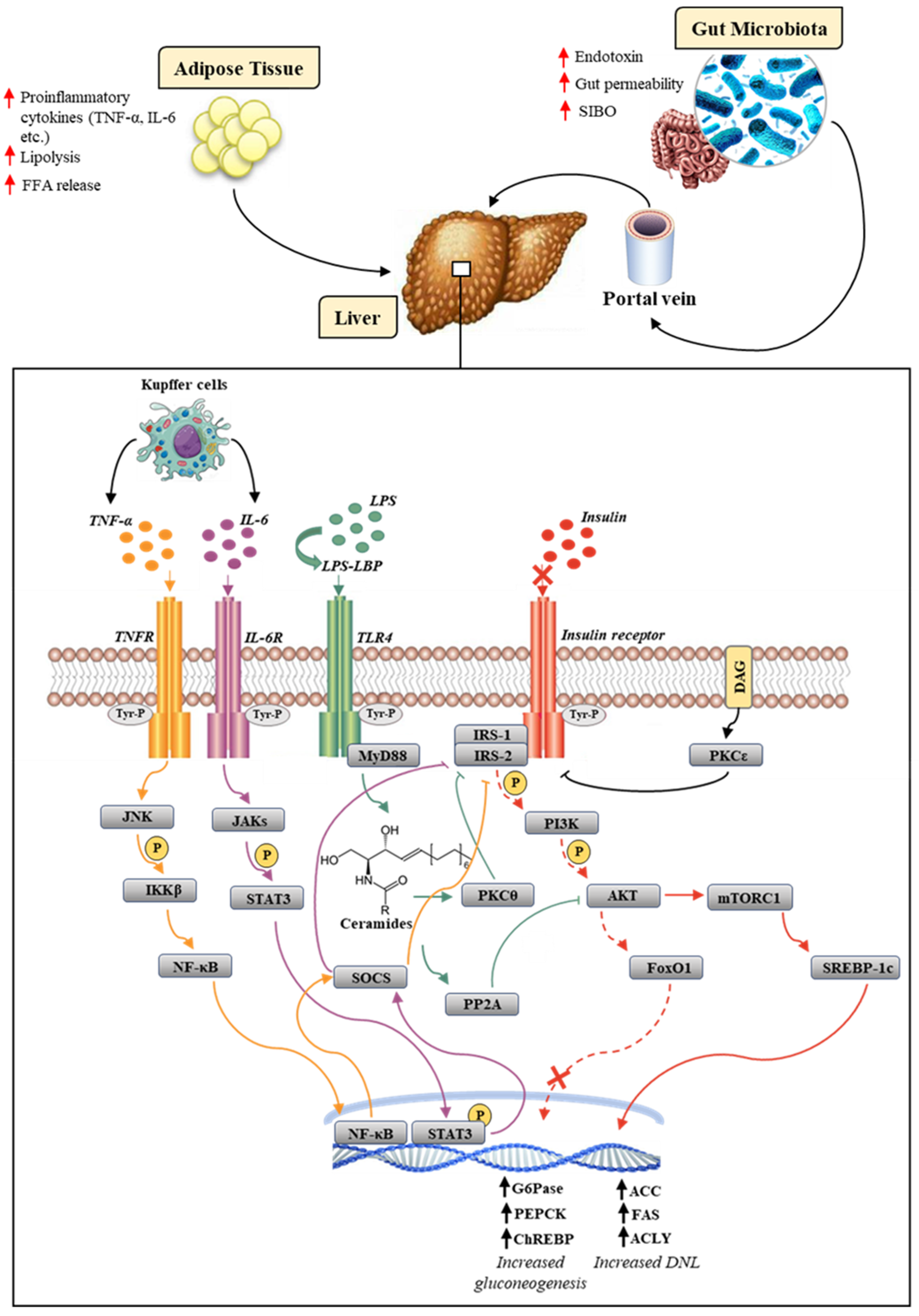
2.1. Insulin Resistance, Oxidative Stress and Inflammation: Molecular Mechanisms Linking the Three Vertices of the Same Triangle
2.2. Gut Microbiota and Kupffer Cells Influence on Insulin Resistance in NAFLD: A Novel Pathogenetic Frontier
2.3. Genetic and Epigenetic Landscape in NAFLD and IR: Molecular Aspects
2.3.1. Genetics
2.3.2. Epigenetics

{kind=link}
{kind=link}
{kind=link}
| Epigenetics | Target | Biological Significance | Reference |
|---|---|---|---|
| DNA hypomethylation | ↑ FGFR2 | Pro-fibrogenic | [96] |
| ↑ PGC1a, ↑ SREBF2, ↑ FOXA1, ↑ FOXA2 | Hepatic regulators of glucose and lipid metabolism | [97] | |
| ↓ SIRT1 | ↑ PGC-1α | Primary regulator of liver gluconeogenesis | [99] |
| ↓ mTorc2/Akt signaling | Insulin resistance | [101] | |
| ↑ miR-122 | ↑ FASN | De novo lipogenesis | [104] |
| ↑ SREBP1c | |||
| ↓ IGF-1R | |||
| ↓ miR-499 | ↓ Akt/GSK activation | Liver fat accumulation | [106] |
| ↓ PTEN | Hepatic glucose metabolism | ||
| ↑ LncRNA MALAT1 | ↑ JNK | Oxidative stress-mediated insulin resistance | [107] |
| Effect on glucose and lipid metabolism | [109] | ||
| ↑ LncRNA H19 | ↑ SREBP-1c, ↑ ACC1, ↑ SCD1, ↑ FASN, ↑ PPARγ | Development of hepatic insulin resistance | [111] |
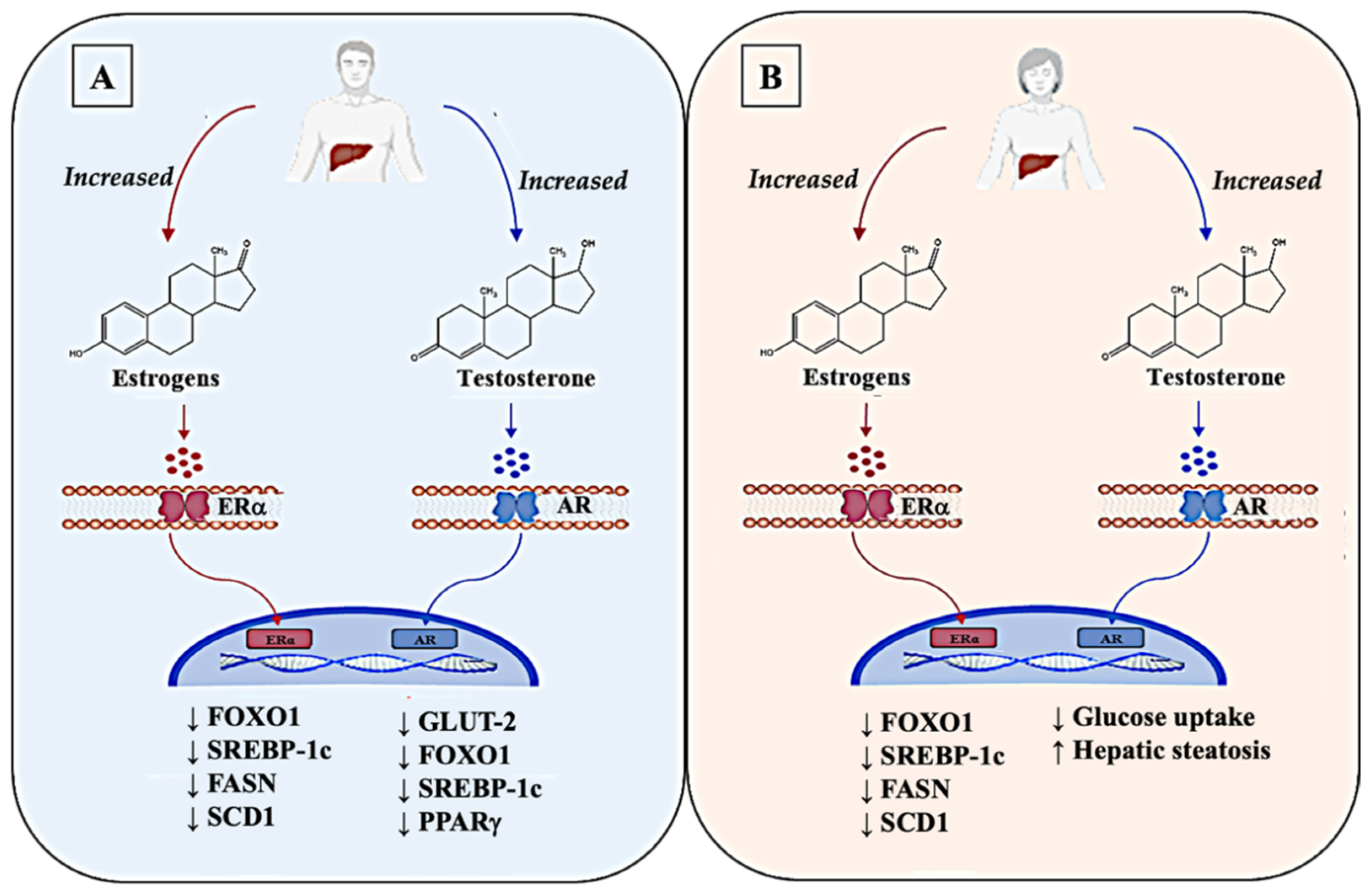
2.4. Role of Other Hormones in Fueling IR: A Matter of Sex?
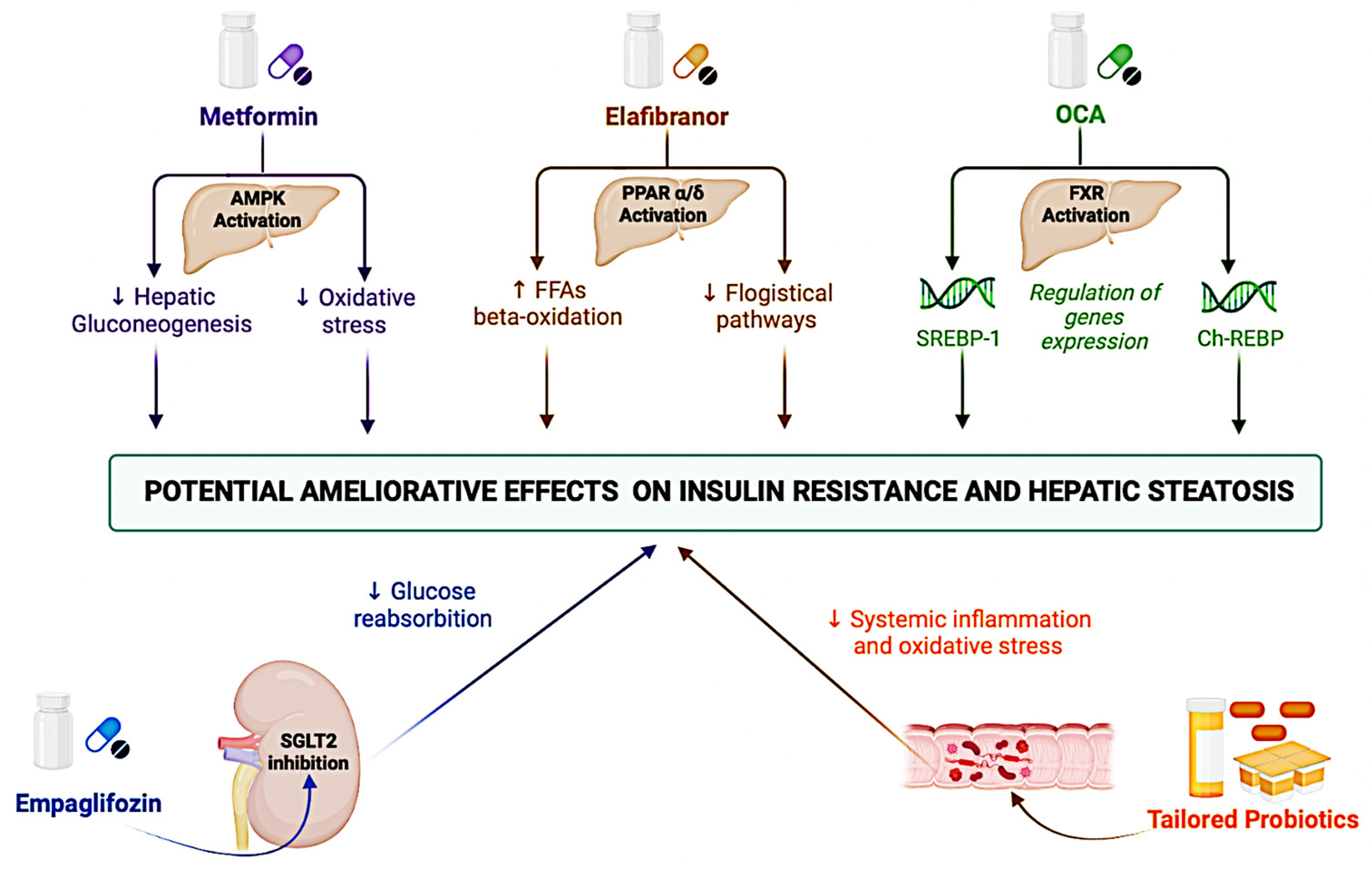
3. Insulin Resistance as the Cornerstone for the Clinical Management of NAFLD: An Overview on Diagnosis, Prognosis, and Potential Treatment Implications
4. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chalasani, N.; Younossi, Z.; LaVine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2017, 15, 11–20. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus Panel. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef]
- Utzschneider, K.M.; Kahn, S.E. The Role of Insulin Resistance in Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2006, 91, 4753–4761. [Google Scholar] [CrossRef] [Green Version]
- Rosato, V.; Masarone, M.; Dallio, M.; Federico, A.; Aglitti, A.; Persico, M. NAFLD and Extra-Hepatic Comorbidities: Current Evidence on a Multi-Organ Metabolic Syndrome. Int. J. Environ. Res. Public Health 2019, 16, 3415. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Park, S.Y.; Choi, C.S. Insulin Resistance: From Mechanisms to Therapeutic Strategies. Diabetes Metab. J. 2022, 46, 15–37. [Google Scholar] [CrossRef]
- Yki-Järvinen, H.; Luukkonen, P.K.; Hodson, L.; Moore, J.B. Dietary carbohydrates and fats in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 770–786. [Google Scholar] [CrossRef]
- Geng, Y.; Faber, K.N.; de Meijer, V.E.; Blokzijl, H.; Moshage, H. How does hepatic lipid accumulation lead to lipotoxicity in non-alcoholic fatty liver disease? Hepatol. Int. 2021, 15, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Sekizkardes, H.; Chung, S.T.; Chacko, S.; Haymond, M.W.; Startzell, M.; Walter, M.; Walter, P.J.; Lightbourne, M.; Brown, R.J. Free fatty acid processing diverges in human pathologic insulin resistance conditions. J. Clin. Investig. 2020, 130, 3592–3602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakurai, Y.; Kubota, N.; Yamauchi, T.; Kadowaki, T. Role of Insulin Resistance in MAFLD. Int. J. Mol. Sci. 2021, 22, 4156. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Kubota, N.; Kadowaki, T. Imbalanced Insulin Actions in Obesity and Type 2 Diabetes: Key Mouse Models of Insulin Signaling Pathway. Cell Metab. 2017, 25, 797–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.-Y.; Cai, C.-Z.; Yu, M.-L.; Feng, Z.-M.; Zhang, Y.-W.; Liu, P.-H.; Zeng, H.; Yu, C.-H. LB100 ameliorates nonalcoholic fatty liver disease via the AMPK/Sirt1 pathway. World J. Gastroenterol. 2019, 25, 6607–6618. [Google Scholar] [CrossRef]
- Dewidar, B.; Kahl, S.; Pafili, K.; Roden, M. Metabolic liver disease in diabetes—From mechanisms to clinical trials. Metabolism 2020, 111, 154299. [Google Scholar] [CrossRef]
- Otero, Y.F.; Stafford, J.M.; McGuinness, O.P. Pathway-selective insulin resistance and metabolic disease: The importance of nutrient flux. J. Biol. Chem. 2014, 289, 20462–20469. [Google Scholar] [CrossRef] [Green Version]
- Mu, W.; Cheng, X.-F.; Liu, Y.; Lv, Q.-Z.; Liu, G.-L.; Zhang, J.-G.; Li, X.-Y. Potential Nexus of Non-alcoholic Fatty Liver Disease and Type 2 Diabetes Mellitus: Insulin Resistance Between Hepatic and Peripheral Tissues. Front. Pharmacol. 2019, 9, 1566. [Google Scholar] [CrossRef] [Green Version]
- Vatner, D.F.; Petersen, M.C.; Li, X.; Rogers, J.C.; Cline, G.; Samuel, V.; Shulman, G.I. Evidence against Pathway-Selective Hepatic Insulin Resistance in Mice. Diabetes 2018, 67, 1766-P. [Google Scholar] [CrossRef]
- Roden, M.; Shulman, G.I. The integrative biology of type 2 diabetes. Nature 2019, 576, 51–60. [Google Scholar] [CrossRef] [Green Version]
- ter Horst, K.W.; Vatner, D.F.; Zhang, D.; Cline, G.W.; Ackermans, M.T.; Nederveen, A.J.; Verheij, J.; Demirkiran, A.; van Wagensveld, B.A.; Dallinga-Thie, G.M.; et al. Hepatic Insulin Resistance Is Not Pathway Selective in Humans with Nonalcoholic Fatty Liver Disease. Diabetes Care 2020, 44, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Linden, A.G.; Li, S.; Choi, H.Y.; Fang, F.; Fukasawa, M.; Uyeda, K.; Hammer, R.E.; Horton, J.D.; Engelking, L.J.; Liang, G. Interplay between ChREBP and SREBP-1c coordinates postprandial glycolysis and lipogenesis in livers of mice. J. Lipid Res. 2018, 59, 475–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, M.; Hibi, M.; Nakagawa, T.; Hayakawa, T.; Field, C.J. High-fructose diet-induced hepatic expression of the Scd1 gene is associated with increased acetylation of histones H3 and H4 and the binding of ChREBP at the Scd1 promoter in rats. Biomed. Res. 2021, 42, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Bugianesi, E.; Zannoni, C.; Vanni, E.; Marzocchi, R.; Marchesini, G. Non-alcoholic fatty liver and insulin resistance: A cause–effect relationship? Dig. Liver Dis. 2004, 36, 165–173. [Google Scholar] [CrossRef]
- Yki-Järvinen, H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014, 2, 901–910. [Google Scholar] [CrossRef]
- Tanase, D.M.; Gosav, E.M.; Costea, C.F.; Ciocoiu, M.; Lacatusu, C.M.; Maranduca, M.A.; Ouatu, A.; Floria, M. The Intricate Relationship between Type 2 Diabetes Mellitus (T2DM), Insulin Resistance (IR), and Nonalcoholic Fatty Liver Disease (NAFLD). J. Diabetes Res. 2020, 2020, 3920196. [Google Scholar] [CrossRef]
- Fujii, H.; Kawada, N. Japan Study Group of Nafld Jsg-Nafld. The Role of Insulin Resistance and Diabetes in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 3863. [Google Scholar] [CrossRef]
- Barazzoni, R.; Cappellari, G.G.; Ragni, M.; Nisoli, E. Insulin resistance in obesity: An overview of fundamental alterations. Eat. Weight Disord. 2018, 23, 149–157. [Google Scholar] [CrossRef]
- Rehman, K.; Akash, M.S.H.; Liaqat, A.; Kamal, S.; Qadir, M.I.; Rasul, A. Role of Interleukin-6 in Development of Insulin Resistance and Type 2 Diabetes Mellitus. Crit. Rev. Eukaryot. Gene Expr. 2017, 27, 229–236. [Google Scholar] [CrossRef]
- Grimble, R.F. Inflammatory status and insulin resistance. Curr. Opin. Clin. Nutr. Metab. Care 2002, 5, 551–559. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Tabas, I.; Pajvani, U.B. Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis. Gastroenterology 2020, 158, 1913–1928. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Meroni, M.; Baselli, G.A.; Bassani, G.A.; Rametta, R.; Pietrelli, A.; Maggioni, M.; Facciotti, F.; Trunzo, V.; Badiali, S.; et al. Insulin resistance promotes Lysyl Oxidase Like 2 induction and fibrosis accumulation in non-alcoholic fatty liver disease. Clin. Sci. 2017, 131, 1301–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranjbar, G.; Mikhailidis, D.P.; Sahebkar, A. Effects of newer antidiabetic drugs on nonalcoholic fatty liver and steatohepatitis: Think out of the box! Metabolism 2019, 101, 154001. [Google Scholar] [CrossRef]
- Svegliati-Baroni, G.; Ridolfi, F.; Di Sario, A.; Casini, A.; Marucci, L.; Gaggiotti, G.; Orlandoni, P.; Macarri, G.; Perego, L.; Benedetti, A.; et al. Insulin and insulin-like growth factor-1 stimulate proliferation and type I collagen accumulation by human hepatic stellate cells: Differential effects on signal transduction pathways. Hepatology 1999, 29, 1743–1751. [Google Scholar] [CrossRef] [PubMed]
- Villar-Lorenzo, A.; Rada, P.; Rey, E.; Marañón, P.; Arroba, A.I.; Santamaría, B.; Sáiz, J.; Rupérez, F.J.; Barbas, C.; García-Monzón, C.; et al. Insulin receptor substrate 2 (IRS2)-deficiency delays liver fibrosis associated to cholestatic injury. Dis. Model. Mech. 2019, 12, dmm038810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar] [CrossRef]
- Song, Q.; Zhang, X. The Role of Gut-Liver Axis in Gut Microbiome Dysbiosis Associated NAFLD and NAFLD-HCC. Biomedicines 2022, 10, 524. [Google Scholar] [CrossRef]
- Wijarnpreecha, K.; Lou, S.; Watthanasuntorn, K.; Kroner, P.T.; Cheungpasitporn, W.; Lukens, F.J.; Pungpapong, S.; Keaveny, A.P.; Ungprasert, P. Small intestinal bacterial overgrowth and nonalcoholic fatty liver disease: A systematic review and meta-analysis. Eur. J. Gastroenterol. Hepatol. 2020, 32, 601–608. [Google Scholar] [CrossRef]
- Augustyn, M.; Grys, I.; Kukla, M. Small intestinal bacterial overgrowth and nonalcoholic fatty liver disease. Clin. Exp. Hepatol. 2019, 5, 1–10. [Google Scholar] [CrossRef]
- Kuang, L.; Zhou, W.; Jiang, Y. Association of small intestinal bacterial overgrowth with nonalcoholic fatty liver disease in children: A meta-analysis. PLoS ONE 2021, 16, e0260479. [Google Scholar] [CrossRef]
- Mikolasevic, I.; Delija, B.; Mijic, A.; Stevanovic, T.; Skenderevic, N.; Sosa, I.; Krznaric-Zrnic, I.; Abram, M.; Krznaric, Z.; Domislovic, V.; et al. Small intestinal bacterial overgrowth and non-alcoholic fatty liver disease diagnosed by transient elastography and liver biopsy. Int. J. Clin. Pract. 2021, 75, e13947. [Google Scholar] [CrossRef] [PubMed]
- Kapil, S.; Duseja, A.; Sharma, B.K.; Singla, B.; Chakraborti, A.; Das, A.; Ray, P.; Dhiman, R.K.; Chawla, Y. Small intestinal bacterial overgrowth and toll-like receptor signaling in patients with non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2016, 31, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Sharpton, S.; Schnabl, B.; Knight, R.; Loomba, R. Current Concepts, Opportunities, and Challenges of Gut Microbiome-Based Personalized Medicine in Nonalcoholic Fatty Liver Disease. Cell Metab. 2020, 33, 21–32. [Google Scholar] [CrossRef]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fianchi, F.; Liguori, A.; Gasbarrini, A.; Grieco, A.; Miele, L. Nonalcoholic Fatty Liver Disease (NAFLD) as Model of Gut-Liver Axis Interaction: From Pathophysiology to Potential Target of Treatment for Personalized Therapy. Int. J. Mol. Sci. 2021, 22, 6485. [Google Scholar] [CrossRef]
- Martín-Mateos, R.; Albillos, A. The Role of the Gut-Liver Axis in Metabolic Dysfunction-Associated Fatty Liver Disease. Front. Immunol. 2021, 12, 660179. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Dallio, M.; Godos, J.; Loguercio, C.; Salomone, F. Targeting gut-liver axis for the treatment of nonalcoholic steatohepatitis: Translational and clinical evidence. Transl. Res. 2015, 167, 116–124. [Google Scholar] [CrossRef]
- Dabke, K.; Hendrick, G.; Devkota, S. The gut microbiome and metabolic syndrome. J. Clin. Investig. 2019, 129, 4050–4057. [Google Scholar] [CrossRef]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.-M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Cho, Y.; Kim, D.; Seo, W.; Gao, B.; Yoo, S.; Song, B. Fructose Promotes Leaky Gut, Endotoxemia, and Liver Fibrosis Through Ethanol-Inducible Cytochrome P450-2E1–Mediated Oxidative and Nitrative Stress. Hepatology 2019, 73, 2180–2195. [Google Scholar] [CrossRef]
- Shi, A.; Li, T.; Zheng, Y.; Song, Y.; Wang, H.; Wang, N.; Dong, L.; Shi, H. Chlorogenic Acid Improves NAFLD by Regulating gut Microbiota and GLP-1. Front. Pharmacol. 2021, 12, 693048. [Google Scholar] [CrossRef] [PubMed]
- Sakai, Y.; Arie, H.; Ni, Y.; Zhuge, F.; Xu, L.; Chen, G.; Nagata, N.; Suzuki, T.; Kaneko, S.; Ota, T.; et al. Lactobacillus pentosus strain S-PT84 improves steatohepatitis by maintaining gut permeability. J. Endocrinol. 2020, 247, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Mascianà, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Kessoku, T.; Kobayashi, T.; Imajo, K.; Tanaka, K.; Yamamoto, A.; Takahashi, K.; Kasai, Y.; Ozaki, A.; Iwaki, M.; Nogami, A.; et al. Endotoxins and Non-Alcoholic Fatty Liver Disease. Front. Endocrinol. 2021, 12, 770986. [Google Scholar] [CrossRef]
- Dallio, M.; Sangineto, M.; Romeo, M.; Villani, R.; Romano, A.D.; Loguercio, C.; Serviddio, G.; Federico, A. Immunity as Cornerstone of Non-Alcoholic Fatty Liver Disease: The Contribution of Oxidative Stress in the Disease Progression. Int. J. Mol. Sci. 2021, 22, 436. [Google Scholar] [CrossRef]
- El-Bendary, M.; Naemattalah, M.; Yassen, A.; Mousa, N.; Elhammady, D.; Sultan, A.M.; Abdel-Wahab, M. Interrelationship between Toll-like receptors and infection after orthotopic liver transplantation. World J. Transplant. 2020, 10, 162–172. [Google Scholar] [CrossRef]
- Gabarin, R.S.; Li, M.; Zimmel, P.A.; Marshall, J.C.; Li, Y.; Zhang, H. Intracellular and Extracellular Lipopolysaccharide Signaling in Sepsis: Avenues for Novel Therapeutic Strategies. J. Innate Immun. 2021, 13, 323–332. [Google Scholar] [CrossRef]
- Johnston, A.M.; Pirola, L.; Van Obberghen, E. Molecular mechanisms of insulin receptor substrate protein-mediated modulation of insulin signalling. FEBS Lett. 2003, 546, 32–36. [Google Scholar] [CrossRef] [Green Version]
- Carpino, G.; Del Ben, M.; Pastori, D.; Carnevale, R.; Baratta, F.; Overi, D.; Francis, H.; Cardinale, V.; Onori, P.; Safarikia, S.; et al. Increased Liver Localization of Lipopolysaccharides in Human and Experimental NAFLD. Hepatology 2019, 72, 470–485. [Google Scholar] [CrossRef]
- Galbo, T.; Perry, R.J.; Jurczak, M.J.; Camporez, J.-P.G.; Alves, T.C.; Kahn, M.; Guigni, B.A.; Serr, J.; Zhang, D.; Bhanot, S.; et al. Saturated and unsaturated fat induce hepatic insulin resistance independently of TLR-4 signaling and ceramide synthesis in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 12780–12785. [Google Scholar] [CrossRef] [Green Version]
- Holland, W.L.; Bikman, B.T.; Wang, L.-P.; Yuguang, G.; Sargent, K.M.; Bulchand, S.; Knotts, T.A.; Shui, G.; Clegg, D.J.; Wenk, M.R.; et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J. Clin. Investig. 2011, 121, 1858–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, G.C. Signalling links in the liver: Knitting SOCS with fat and inflammation. J. Hepatol. 2005, 43, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Rivera, C.A.; Adegboyega, P.; van Rooijen, N.; Tagalicud, A.; Allman, M.; Wallace, M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J. Hepatol. 2007, 47, 571–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spruss, A.; Kanuri, G.; Wagnerberger, S.; Haub, S.; Bischoff, S.C.; Bergheim, I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology 2009, 50, 1094–1104. [Google Scholar] [CrossRef]
- Larter, C.Z.; Farrell, G.C. Insulin resistance, adiponectin, cytokines in NASH: Which is the best target to treat? J. Hepatol. 2006, 44, 253–261. [Google Scholar] [CrossRef]
- Sugita, H.; Kaneki, M.; Tokunaga, E.; Sugita, M.; Koike, C.; Yasuhara, S.; Tompkins, R.G.; Martyn, J.A.J. Inducible nitric oxide synthase plays a role in LPS-induced hyperglycemia and insulin resistance. Am. J. Physiol. Metab. 2002, 282, E386–E394. [Google Scholar] [CrossRef] [Green Version]
- Carvalho-Filho, M.A.; Ueno, M.; Hirabara, S.M.; Seabra, A.B.; Carvalheira, J.B.; de Oliveira, M.G.; Velloso, L.A.; Curi, R.; Saad, M.J. S-nitrosation of the insulin receptor, insulin receptor substrate 1, and protein kinase B/Akt: A novel mechanism of insulin resistance. Diabetes 2005, 54, 959–967. [Google Scholar] [CrossRef] [Green Version]
- Shinozaki, S.; Choi, C.S.; Shimizu, N.; Yamada, M.; Kim, M.; Zhang, T.; Dong, H.H.; Kim, Y.-B.; Kaneki, M. Liver-specific Inducible Nitric-oxide Synthase Expression Is Sufficient to Cause Hepatic Insulin Resistance and Mild Hyperglycemia in Mice. J. Biol. Chem. 2011, 286, 34959–34975. [Google Scholar] [CrossRef] [Green Version]
- Zanotto, T.M.; Quaresma, P.G.; Guadagnini, D.; Weissmann, L.; Santos, A.C.; Vecina, J.F.; Calisto, K.; dos Santos, A.; Prada, P.D.O.; Saad, M.J. Blocking iNOS and endoplasmic reticulum stress synergistically improves insulin resistance in mice. Mol. Metab. 2016, 6, 206–218. [Google Scholar] [CrossRef]
- Umano, G.R.; Martino, M.; Santoro, N. The association between pediatric NAFLD and common genetic variants. Children 2017, 4, 49. [Google Scholar] [CrossRef]
- Roshandel, G.; Khoshnia, M.; Poustchi, H.; Hemming, K.; Kamangar, F.; Gharavi, A.; Ostovaneh, M.R.; Nateghi, A.; Majed, M.; Navabakhsh, B.; et al. Effectiveness of polypill for primary and secondary prevention of cardiovascular diseases (PolyIran): A pragmatic, cluster-randomised trial. Lancet 2019, 394, 672–683. [Google Scholar] [CrossRef]
- Pingitore, P.; Dongiovanni, P.; Motta, B.M.; Meroni, M.; Lepore, S.M.; Mancina, R.M.; Pelusi, S.; Russo, C.; Caddeo, A.; Rossi, G.; et al. PNPLA3 overexpression results in reduction of proteins predisposing to fibrosis. Hum. Mol. Genet. 2016, 25, 5212–5222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; He, S.; Li, J.Z.; Seo, Y.-K.; Osborne, T.F.; Cohen, J.C.; Hobbs, H.H. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc. Natl. Acad. Sci. USA 2010, 107, 7892–7897. [Google Scholar] [CrossRef] [Green Version]
- Meroni, M.; Longo, M.; Tria, G.; Dongiovanni, P. Genetics Is of the Essence to Face NAFLD. Biomedicines 2021, 9, 1359. [Google Scholar] [CrossRef]
- Buch, S.; Stickel, F.; Trepo, E. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat. Genet. 2015, 47, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Que, S.; Zhou, L.; Zheng, S.; Romeo, S.; Mardinoglu, A.; Valenti, L. The effect of the TM6SF2 E167K variant on liver steatosis and fibrosis in patients with chronic hepatitis C: A meta-analysis. Sci. Rep. 2017, 7, 9273. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Kory, N.; Basuray, S.; Cohen, J.C.; Hobbs, H.H. PNPLA3, CGI-58, and Inhibition of Hepatic Triglyceride Hydrolysis in Mice. Hepatology 2019, 69, 2427–2441. [Google Scholar] [CrossRef] [Green Version]
- Basuray, S.; Smagris, E.; Cohen, J.C.; Hobbs, H.H. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology 2017, 66, 1111–1124. [Google Scholar] [CrossRef] [Green Version]
- Donati, B.; Motta, B.M.; Pingitore, P.; Meroni, M.; Pietrelli, A.; Alisi, A.; Petta, S.; Xing, C.; Dongiovanni, P.; del Menico, B.; et al. The rs2294918 E434K variant modulates patatin-like phospholipase domain-containing 3 expression and liver damage. Hepatology 2016, 63, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Manco, M. Insulin Resistance and NAFLD: A Dangerous Liaison beyond the Genetics. Children 2017, 4, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meroni, M.; Longo, M.; Fracanzani, A.L.; Dongiovanni, P. MBOAT7 down-regulation by genetic and environmental factors predisposes to MAFLD. eBioMedicine 2020, 57, 102866. [Google Scholar] [CrossRef] [PubMed]
- Zarini, S.; Hankin, J.A.; Murphy, R.C.; Gijón, M.A. Lysophospholipid acyltransferases and eicosanoid biosynthesis in zebrafish myeloid cells. Prostaglandins Other Lipid Mediat. 2014, 113–115, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Stickel, F.; Moreno, C.; Hampe, J.; Morgan, M.Y. The genetics of alcohol dependence and alcohol-related liver disease. J. Hepatol. 2017, 66, 195–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luukkonen, P.K.; Zhou, Y.; Hyotylainen, T.; Leivonen, M.; Arola, J.; Orho-Melander, M.; Oresic, M.; Yki-Jarvinen, H. The MBOAT7 variant rs641738 alters hepatic phosphatidyl inositols and increases severity of non-alcoholic fatty liver disease in humans. J. Hepatol. 2016, 65, 1263–1265. [Google Scholar] [CrossRef] [Green Version]
- Donati, B.; Dongiovanni, P.; Romeo, S.; Meroni, M.; McCain, M.; Miele, L.; Petta, S.; Maier, S.; Rosso, C.; de Luca, L.; et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Sci. Rep. 2017, 7, 4492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sookoian, S.; Flichman, D.; Garaycoechea, M.E.; Gazzi, C.; Martino, J.S.; Castaño, G.O.; Pirola, C.J. Lack of evidence supporting a role of TMC4-rs641738 missense variant-MBOAT7- intergenic downstream variant-in the Susceptibility to Nonalcoholic Fatty Liver Disease. Sci. Rep. 2018, 8, 5097. [Google Scholar] [CrossRef] [Green Version]
- Helsley, R.N.; Varadharajan, V.; Brown, A.L.; Gromovsky, A.D.; Schugar, R.C.; Ramachandiran, I.; Fung, K.; Kabbany, M.N.; Banerjee, R.; Neumann, C.K.; et al. Obesity-linked suppression of membrane-bound O-acyltransferase 7 (MBOAT7) drives non-alcoholic fatty liver disease. eLife 2019, 8, 8. [Google Scholar] [CrossRef]
- Umano, G.R.; Caprio, S.; Di Sessa, A.; Chalasani, N.; Dykas, D.J.; Pierpont, B.; Bale, A.E.; Santoro, N. The rs626283 Variant in the MBOAT7 Gene is Associated with Insulin Resistance and Fatty Liver in Caucasian Obese Youth. Am. J. Gastroenterol. 2018, 113, 376–383. [Google Scholar] [CrossRef]
- Ehrhardt, N.; Doche, M.E.; Chen, S.; Mao, H.Z.; Walsh, M.T.; Bedoya, C.; Guindi, M.; Xiong, W.; Irudayam, J.I.; Iqbal, J.; et al. Hepatic Tm6sf2 overexpression affects cellular ApoB-trafficking, plasma lipid levels, hepatic steatosis and atherosclerosis. Hum. Mol. Genet. 2017, 26, 2719–2731. [Google Scholar] [CrossRef]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musso, G.; Cipolla, U.; Cassader, M.; Pinach, S.; Saba, F.; De Michieli, F.; Paschetta, E.; Bongiovanni, D.; Framarin, L.; Leone, N.; et al. TM6SF2 rs58542926 variant affects postprandial lipoprotein metabolism and glucose homeostasis in NAFLD. J. Lipid Res. 2017, 58, 1221–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, A.P.; Voight, B.F.; Teslovich, T.M.; Ferreira, T.; Segrè, A.V.; Steinthorsdottir, V.; Strawbridge, R.J.; Khan, H.; Grallert, H.; Mahajan, A.; et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat. Genet. 2012, 44, 981–990. [Google Scholar] [PubMed]
- Musso, G.; Paschetta, E.; Gambino, R.; Cassader, M.; Molinaro, F. Interactions among bone, liver, and adipose tissue predisposing to diabesity and fatty liver. Trends Mol. Med. 2013, 19, 522–535. [Google Scholar] [CrossRef] [PubMed]
- Maude, H.; Sanchez-Cabanillas, C.; Cebola, I. Epigenetics of Hepatic Insulin Resistance. Front. Endocrinol. 2021, 12, 681356. [Google Scholar] [CrossRef]
- Murphy, S.; Yang, H.; Moylan, C.A.; Pang, H.; Dellinger, A.; Abdelmalek, M.; Garrett, M.E.; Ashley-Koch, A.; Suzuki, A.; Tillmann, H.L.; et al. Relationship between Methylome and Transcriptome in Patients with Nonalcoholic Fatty Liver Disease. Gastroenterology 2013, 145, 1076–1087. [Google Scholar] [CrossRef] [Green Version]
- Ahrens, M.; Ammerpohl, O.; von Schönfels, W.; Kolarova, J.; Bens, S.; Itzel, T.; Teufel, A.; Herrmann, A.; Brosch, M.; Hinrichsen, H.; et al. DNA Methylation Analysis in Nonalcoholic Fatty Liver Disease Suggests Distinct Disease-Specific and Remodeling Signatures After Bariatric Surgery. Cell Metab. 2013, 18, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef]
- Rodgers, J.T.; Puigserver, P. Fasting-Dependent Glucose and Lipid Metabolic Response Through Hepatic Sirtuin 1. Proc. Natl. Acad. Sci. USA 2007, 104, 12861–12866. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.-H.; Kim, H.-S.; Xiao, C.; Xu, X.; Gavrilova, O.; Deng, C.-X. Hepatic Sirt1 deficiency in mice impairs mTorc2/Akt signaling and results in hyperglycemia, oxidative damage, and insulin resistance. J. Clin. Investig. 2011, 121, 4477–4490. [Google Scholar] [CrossRef] [Green Version]
- Bricambert, J.; Alves-Guerra, M.-C.; Esteves, P.; Prip-Buus, C.; Bertrand-Michel, J.; Guillou, H.; Chang, C.J.; Wal, M.N.V.; Canonne-Hergaux, F.; Mathurin, P.; et al. The histone demethylase Phf2 acts as a molecular checkpoint to prevent NAFLD progression during obesity. Nat. Commun. 2018, 9, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandiera, S.; Pfeffer, S.; Baumert, T.F.; Zeisel, M.B. miR-122—A key factor and therapeutic target in liver disease. J. Hepatol. 2015, 62, 448–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowey, B.; Hertz, L.; Chiu, S.; Valdez, K.; Li, Q.; Liang, T.J. Hepatitis C virus infection induces hepatic expression of NF-κB-inducing kinase and lipogenesis by downregulating miR-122. MBio 2019, 10, e01617-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, J.-K.; Dai, W.; Zheng, Y.-W.; Zhao, S.-P. miR-122 promotes hepatic lipogenesis via inhibiting the LKB1/AMPK pathway by targeting Sirt1 in non-alcoholic fatty liver disease. Mol. Med. 2019, 25, 26. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Wang, T.; Chen, X.; Jiang, J.; Song, N.; Li, R.; Xin, Y.; Xuan, S. Inhibition of miR-499-5p expression improves nonalcoholic fatty liver disease. Ann. Hum. Genet. 2020, ahg.12374. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, N.; Pan, H.-P.; Wang, Z.; Cao, Z.-Y. MiR-499-5p Contributes to Hepatic Insulin Resistance by Suppressing PTEN. Cell. Physiol. Biochem. 2015, 36, 2357–2365. [Google Scholar] [CrossRef] [Green Version]
- Puthanveetil, P.; Chen, S.; Feng, B.; Gautam, A.; Chakrabarti, S. Long non-coding RNA MALAT1 regulates hyperglycaemia induced inflammatory process in the endothelial cells. J. Cell. Mol. Med. 2015, 19, 1418–1425. [Google Scholar] [CrossRef]
- Goyal, N.; Sivadas, A.; Shamsudheen, K.V.; Jayarajan, R.; Verma, A.; Sivasubbu, S.; Scaria, V.; Datta, M. RNA sequencing of db/db mice liver identifies lncRNA H19 as a key regulator of gluconeogenesis and hepatic glucose output. Sci. Rep. 2017, 7, 9312. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wu, H.; Wang, F.; Ye, M.; Zhu, H.; Bu, S. Long non-coding RNA MALAT1 expression in patients with gestational diabetes mellitus. Int. J. Gynecol. Obstet. 2018, 140, 164–169. [Google Scholar] [CrossRef]
- Nilsson, E.; Matte, A.; Perfilyev, A.; de Mello, V.; Käkelä, P.; Pihlajamäki, J.; Ling, C. Epigenetic Alterations in Human Liver From Subjects With Type 2 Diabetes in Parallel With Reduced Folate Levels. J. Clin. Endocrinol. Metab. 2015, 100, E1491–E1501. [Google Scholar] [CrossRef]
- Liu, J.; Tang, T.; Wang, G.D.; Liu, B. LncRNA-H19 promotes hepatic lipogenesis by directly regulating miR-130a/PPARγ axis in non-alcoholic fatty liver disease. Biosci. Rep. 2019, 39, BSR20181722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meroni, M.; Longo, M.; Dongiovanni, P. The Role of Probiotics in Nonalcoholic Fatty Liver Disease: A New Insight into Therapeutic Strategies. Nutrients 2019, 11, 2642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallio, M.; Romeo, M.; Gravina, A.; Masarone, M.; Larussa, T.; Abenavoli, L.; Persico, M.; Loguercio, C.; Federico, A. Nutrigenomics and Nutrigenetics in Metabolic- (Dysfunction) Associated Fatty Liver Disease: Novel Insights and Future Perspectives. Nutrients 2021, 13, 1679. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.-M.; Noworolski, S.M.; Wen, M.J.; Dyachenko, A.; Prior, J.L.; Weinberg, M.E.; Herraiz, L.A.; Tai, V.W.; Bergeron, N.; Bersot, T.P.; et al. Effect of a High-Fructose Weight-Maintaining Diet on Lipogenesis and Liver Fat. J. Clin. Endocrinol. Metab. 2015, 100, 2434–2442. [Google Scholar] [CrossRef] [PubMed]
- Jornayvaz, F.R.; Shulman, G.I. Diacylglycerol Activation of Protein Kinase Cε and Hepatic Insulin Resistance. Cell Metab. 2012, 15, 574–584. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Fox, C.S.; Jacques, P.F.; Speliotes, E.K.; Hoffmann, U.; Smith, C.E.; Saltzman, E.; McKeown, N.M. Sugar-sweetened beverage, diet soda, and fatty liver disease in the Framingham Heart Study cohorts. J. Hepatol. 2015, 63, 462–469. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Tang, T.; Wang, G.D.; Liu, B. Omega-3 polyunsaturated fatty acids as a treatment strategy for nonalcoholic fatty liver disease. Pharmacol. Ther. 2018, 181, 108–125. [Google Scholar]
- Han, H.-S.; Kang, G.; Kim, J.S.; Choi, B.H.; Koo, S.-H. Regulation of glucose metabolism from a liver-centric perspective. Exp. Mol. Med. 2016, 48, e218. [Google Scholar] [CrossRef] [Green Version]
- Magkos, F.; Wang, X.; Mittendorfer, B. Metabolic Actions of Insulin in Men and Women. Nutrition 2010, 26, 686–693. [Google Scholar] [CrossRef] [Green Version]
- Mauvais-Jarvis, F. Gender differences in glucose homeostasis and diabetes. Physiol. Behav. 2018, 187, 20–23. [Google Scholar] [CrossRef]
- Bazhan, N.; Jakovleva, T.; Feofanova, N.; Denisova, E.; Dubinina, A.; Sitnikova, N.; Makarova, E. Sex Differences in Liver, Adipose Tissue, and Muscle Transcriptional Response to Fasting and Refeeding in Mice. Cells 2019, 8, 1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meda, C.; Barone, M.; Mitro, N.; Lolli, F.; Pedretti, S.; Caruso, D.; Maggi, A.; Della Torre, S. Hepatic ERα accounts for sex differences in the ability to cope with an excess of dietary lipids. Mol. Metab. 2019, 32, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Della Torre, S.; Mitro, N.; Meda, C.; Lolli, F.; Pedretti, S.; Barcella, M.; Ottobrini, L.; Metzger, D.; Caruso, D.; Maggi, A. Short-Term Fasting Reveals Amino Acid Metabolism as a Major Sex-Discriminating Factor in the Liver. Cell Metab. 2018, 28, 256–267.e5. [Google Scholar] [CrossRef] [PubMed]
- Della Torre, S.; Benedusi, V.; Pepe, G.; Meda, C.; Rizzi, N.; Uhlenhaut, N.H.; Maggi, A. Dietary essential amino acids restore liver metabolism in ovariectomized mice via hepatic estrogen receptor α. Nat. Commun. 2021; in press. [Google Scholar]
- Dahlman-Wright, K.; Cavailles, V.; Fuqua, S.A.; Jordan, V.C.; Katzenellenbogen, J.A.; Korach, K.; Maggi, A.; Muramatsu, M.; Parker, M.G.; Gustafsson, J. International Union of Pharmacology. LXIV. Estrogen Receptors. Pharmacol. Rev. 2006, 58, 773–781. [Google Scholar] [CrossRef] [Green Version]
- Tramunt, B.; Smati, S.; Grandgeorge, N.; Lenfant, F.; Arnal, J.-F.; Montagner, A.; Gourdy, P. Sex differences in metabolic regulation and diabetes susceptibility. Diabetologia 2020, 63, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Yang, W.; Zhou, F.; Li, X.; Pan, Q.; Shen, Z.; Han, G.; Newell-Fugate, A.; Tian, Y.; Majeti, R.; et al. Estrogen Improves Insulin Sensitivity and Suppresses Gluconeogenesis via the Transcription Factor Foxo1. Diabetes 2019, 68, 291–304. [Google Scholar] [CrossRef] [Green Version]
- Della Torre, S.; Mitro, N.; Fontana, R.; Gomaraschi, M.; Favari, E.; Recordati, C.; Lolli, F.; Quagliarini, F.; Meda, C.; Ohlsson, C.; et al. An Essential Role for Liver ERα in Coupling Hepatic Metabolism to the Reproductive Cycle. Cell Rep. 2016, 15, 360–371. [Google Scholar] [CrossRef] [Green Version]
- Paquette, A.; Chapados, N.; Bergeron, R.; Lavoie, J.-M. Fatty Acid Oxidation is Decreased in the Liver of Ovariectomized Rats. Horm. Metab. Res. 2009, 41, 511–515. [Google Scholar] [CrossRef]
- Bryzgalova, G.; Gao, H.; Ahren, B.; Zierath, J.R.; Galuska, D.; Steiler, T.L.; Dahlman-Wright, K.; Nilssosn, S.; Gustafsson, J.A.; Efendic, S.; et al. Evidence that oestrogen receptor-α plays an important role in the regulation of glucose homeostasis in mice: Insulin sensitivity in the liver. Diabetologia 2006, 49, 588–597. [Google Scholar] [CrossRef] [Green Version]
- Macut, D.; Tziomalos, K.; Božić-Antić, I.; Bjekić-Macut, J.; Katsikis, I.; Papadakis, E.; Andrić, Z.; Panidis, D. Non-Alcoholic Fatty Liver Disease Is Associated with Insulin Resistance and Lipid Accumulation Product in Women with Polycystic Ovary Syndrome. Hum. Reprod. 2016, 31, 1347–1353. [Google Scholar] [CrossRef]
- Qiu, S.; Vazquez, J.T.; Boulger, E.; Liu, H.; Xue, P.; Hussain, M.A.; Wolfe, A. Hepatic estrogen receptor α is critical for regulation of gluconeogenesis and lipid metabolism in males. Sci. Rep. 2017, 7, 1661. [Google Scholar] [CrossRef] [Green Version]
- Courant, F.; Aksglaede, L.; Antignac, J.-P.; Monteau, F.; Sorensen, K.; Andersson, A.-M.; Skakkebaek, N.E.; Juul, A.; Le Bizec, B. Assessment of Circulating Sex Steroid Levels in Prepubertal and Pubertal Boys and Girls by a Novel Ultrasensitive Gas Chromatography-Tandem Mass Spectrometry Method. J. Clin. Endocrinol. Metab. 2010, 95, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Grossmann, M.; Wierman, M.E.; Angus, P.; Handelsman, D.J. Reproductive Endocrinology of Nonalcoholic Fatty Liver Disease. Endocr. Rev. 2018, 40, 417–446. [Google Scholar] [CrossRef]
- Navarro, G.; Allard, C.; Xu, W.; Mauvais-Jarvis, F. The Role of Androgens in Metabolism, Obesity, and Diabetes in Males and Females: Androgens and Diabetes. Obesity 2015, 23, 713–719. [Google Scholar] [CrossRef] [Green Version]
- Pal, M.; Gupta, S. Testosterone Supplementation Improves Glucose Homeostasis despite Increasing Hepatic Insulin Resistance in Male Mouse Model of Type 2 Diabetes Mellitus. Nutr. Diabetes 2016, 6, e236. [Google Scholar] [CrossRef] [Green Version]
- Azziz, R.; Carmina, E.; Chen, Z.; Dunaif, A.; Laven, J.S.; Legro, R.S.; Lizneva, D.; Natterson-Horowtiz, B.; Teede, H.J.; Yildiz, B.O. Polycystic Ovary Syndrome. Nat. Rev. Dis. Primers 2016, 2, 16057. [Google Scholar] [CrossRef]
- Kelly, D.M.; Nettleship, J.E.; Akhtar, S.; Muraleedharan, V.; Sellers, D.J.; Brooke, J.C.; McLaren, D.S.; Channer, K.S.; Jones, T.H. Testosterone suppresses the expression of regulatory enzymes of fatty acid synthesis and protects against hepatic steatosis in cholesterol-fed androgen deficient mice. Life Sci. 2014, 109, 95–103. [Google Scholar] [CrossRef]
- Seidu, T.; McWhorter, P.; Myer, J.; Alamgir, R.; Eregha, N.; Bogle, D.; Lofton, T.; Ecelbarger, C.; Andrisse, S. DHT causes liver steatosis via transcriptional regulation of SCAP in normal weight female mice. J. Endocrinol. 2021, 250, 49–65. [Google Scholar] [CrossRef]
- Lonardo, A.; Nascimbeni, F.; Ballestri, S.; Fairweather, D.; Win, S.; Than, T.A.; Abdelmalek, M.F.; Suzuki, A. Sex Differences in Nonalcoholic Fatty Liver Disease: State of the Art and Identification of Research Gaps. Hepatology 2019, 70, 1457–1469. [Google Scholar] [CrossRef]
- Matsuo, K.; Gualtieri, M.R.; Cahoon, S.S.; Jung, C.E.; Paulson, R.J.; Shoupe, D.; Muderspach, L.I.; Wakatsuki, A.; Wright, J.D.; Roman, L.D. Surgical menopause and increased risk of nonalcoholic fatty liver disease in endometrial cancer. Menopause 2016, 23, 189–196. [Google Scholar] [CrossRef]
- Kamada, Y.; Kiso, S.; Yoshida, Y.; Chatani, N.; Kizu, T.; Hamano, M.; Tsubakio, M.; Takemura, T.; Ezaki, H.; Hayashi, N.; et al. Estrogen Deficiency Worsens Steatohepatitis in Mice Fed High-Fat and High-Cholesterol Diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G1031–G1043. [Google Scholar] [CrossRef] [Green Version]
- Goossens, G.H.; Jocken, J.W.E.; Blaak, E.E. Sexual Dimorphism in Cardiometabolic Health: The Role of Adipose Tissue, Muscle and Liver. Nat. Rev. Endocrinol. 2020, 17, 47–66. [Google Scholar] [CrossRef]
- Jastrzębska, S.; Walczak-Jędrzejowska, R.; Kramek, E.; Marchlewska, K.; Oszukowska, E.; Filipiak, E.; Kula, K.; Słowikowska-Hilczer, J. Relationship between sexual function, body mass index and levels of sex steroid hormones in young men (Abstract). Endokrynol. Pol. 2014, 65, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Damiano, F.; Rochira, A.; Gnoni, A.; Siculella, L. Action of Thyroid Hormones, T3 and T2, on Hepatic Fatty Acids: Differences in Metabolic Effects and Molecular Mechanisms. Int. J. Mol. Sci. 2017, 18, 744. [Google Scholar] [CrossRef]
- Huang, Y.Y.; Gusdon, A.M.; Qu, S. Cross-talk between the thyroid and liver: A new target for nonalcoholic fatty liver disease treatment. World J. Gastroenterol. 2013, 19, 8238–8246. [Google Scholar] [CrossRef]
- Jornayvaz, F.R.; Lee, H.Y.; Jurczak, M.J.; Alves, T.C.; Guebre-Egziabher, F.; Guigni, B.A.; Zhang, D.; Samuel, V.T.; Silva, J.E.; Shulman, G.I. Thyroid hormone receptor-alpha gene knockout mice are protected from diet-induced hepatic insulin resistance. Endocrinology 2012, 153, 583–591. [Google Scholar] [CrossRef] [Green Version]
- D’Ambrosio, R.; Campi, I.; Maggioni, M.; Perbellini, R.; Giammona, E.; Stucchi, R.; Borghi, M.; Degasperi, E.; De Silvestri, A.; Persani, L.; et al. The relationship between liver histology and thyroid function tests in patients with non-alcoholic fatty liver disease (NAFLD). PLoS ONE 2021, 16, e0249614. [Google Scholar] [CrossRef]
- Gariani, K.; Jornayvaz, F.R. Pathophysiology of NASH in endocrine diseases. Endocr. Connect. 2021, 10, R52–R65. [Google Scholar] [CrossRef]
- Mansouri, A.; Gattolliat, C.H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef] [Green Version]
- Sinha, R.A.; Singh, B.K.; Yen, P.M. Direct effects of thyroid hormones on hepatic lipid metabolism. Nat. Rev. Endocrinol. 2018, 14, 259–269. [Google Scholar] [CrossRef]
- Eslam, M.; Sarin, S.K.; Wong, V.W.-S.; Fan, J.-G.; Kawaguchi, T.; Ahn, S.H.; Zheng, M.-H.; Shiha, G.; Yilmaz, Y.; Gani, R.; et al. The Asian Pacific Association for the Study of the Liver clinical practice guidelines for the diagnosis and management of metabolic associated fatty liver disease. Hepatol. Int. 2020, 14, 889–919. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K. Hyperinsulinemic-euglycemic clamp to assess insulin sensitivity in vivo. Methods Mol. Biol. 2009, 560, 221–238. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Kim, M.K.; Kang, S.; Park, K.; Kim, J.H.; Baik, S.J.; Nam, J.S.; Ahn, C.W.; Park, J.S. Triglyceride Glucose Index Is Superior to the Homeostasis Model Assessment of Insulin Resistance for Predicting Nonalcoholic Fatty Liver Disease in Korean Adults. Endocrinol. Metab. 2019, 34, 179–186. [Google Scholar] [CrossRef]
- Aller, R.; Sigüenza, R.; Pina, M.; Laserna, C.; Antolín, B.; Burgueño, B.; Durà, M.; Izaola, O.; Primo, D.; de Luis, D.A. Insulin resistance is related with liver fibrosis in type 2 diabetic patients with non-alcoholic fatty liver disease proven biopsy and Mediterranean diet pattern as a protective factor. Endocrine 2020, 68, 557–563. [Google Scholar] [CrossRef]
- Zambrano-Huailla, R.; Guedes, L.; Stefano, J.T.; de Souza, A.A.A.; Marciano, S.; Yvamoto, E.; Michalczuk, M.T.; Vanni, D.S.; Rodriguez, H.; Carrilho, F.J.; et al. Diagnostic performance of three non-invasive fibrosis scores (Hepamet, FIB-4, NAFLD fibrosis score) in NAFLD patients from a mixed Latin American population. Ann. Hepatol. 2020, 19, 622–626. [Google Scholar] [CrossRef]
- Abenavoli, L.; Boccuto, L.; Federico, A.; Dallio, M.; Loguercio, C.; Di Renzo, L.; De Lorenzo, A. Diet and Non-Alcoholic Fatty Liver Disease: The Mediterranean Way. Int. J. Environ. Res. Public Health 2019, 16, 3011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podhorecka, M.; Ibanez, B.; Dmoszyńska, A. Metformin-its potential anti-cancer and anti-aging effects. Postepy Hig. Med. Dosw. 2017, 71, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Cassader, M.; Rosina, F.; Gambino, R. Impact of current treatments on liver disease, glucose metabolism and cardiovascular risk in non-alcoholic fatty liver disease (NAFLD): A systematic review and meta-analysis of randomised trials. Diabetologia 2012, 55, 885–904. [Google Scholar] [CrossRef]
- Li, Y.; Liu, L.; Wang, B.; Wang, J.; Chen, D. Metformin in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Biomed. Rep. 2012, 1, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Botta, M.; Audano, M.; Sahebkar, A.; Sirtori, C.R.; Mitro, N.; Ruscica, M. PPAR Agonists and Metabolic Syndrome: An Established Role? Int. J. Mol. Sci. 2018, 19, 1197. [Google Scholar] [CrossRef] [Green Version]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-alpha and -delta, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 2016, 150, 1147–1159.e1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porez, G.; Prawitt, J.; Gross, B.; Staels, B. Bile acid receptors as targets for the treatment of dyslipidemia and cardiovascular disease. J. Lipid Res. 2012, 53, 1723–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.-U.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E.; et al. Efficacy and Safety of the Farnesoid X Receptor Agonist Obeticholic Acid in Patients with Type 2 Diabetes and Nonalcoholic Fatty Liver Disease. Gastroenterology 2013, 145, 574–582.e1. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef] [Green Version]
- Mudaliar, S.; Polidori, D.; Zambrowicz, B.; Henry, R.R. Sodium-Glucose Cotransporter Inhibitors: Effects on Renal and Intestinal Glucose Transport: From Bench to Bedside. Diabetes Care 2015, 38, 2344–2353. [Google Scholar] [CrossRef] [Green Version]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Kuchay, M.S.; Krishan, S.; Mishra, S.K.; Farooqui, K.J.; Singh, M.K.; Wasir, J.S.; Bansal, B.; Kaur, P.; Jevalikar, G.; Gill, H.K.; et al. Effect of Empagliflozin on Liver Fat in Patients with Type 2 Diabetes and Nonalcoholic Fatty Liver Disease: A Randomized Controlled Trial (E-LIFT Trial). Diabetes Care 2018, 41, 1801–1808. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.Y.; Li, L.; Yu, C.H.; Shen, Z.; Chen, L.H.; Li, Y.M. Effects of probiotics on nonalcoholic fatty liver disease: A meta-analysis. World J. Gastroenterol. 2013, 19, 6911–6918. [Google Scholar] [CrossRef]
- Dallio, M.; Masarone, M.; Romeo, M.; Tuccillo, C.; Morisco, F.; Persico, M.; Loguercio, C.; Federico, A. PNPLA3, TM6SF2, and MBOAT7 Influence on Nutraceutical Therapy Response for Non-alcoholic Fatty Liver Disease: A Randomized Controlled Trial. Front. Med. 2021, 8, 734847. [Google Scholar] [CrossRef]



| Gene | Predisposing Variant | Biological Significance | Reference |
|---|---|---|---|
| PNPLA3 | rs738409 C > G | Hepatic fat accumulation | [74] |
| MBOAT7 | rs641738 C > T | Susceptibility for hepatic damage | [75] |
| TM6SF2 | rs58542926 C > T | Hepatic steatosis, steatohepatitis, and fibrosis | [76] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palma, R.; Pronio, A.; Romeo, M.; Scognamiglio, F.; Ventriglia, L.; Ormando, V.M.; Lamazza, A.; Pontone, S.; Federico, A.; Dallio, M. The Role of Insulin Resistance in Fueling NAFLD Pathogenesis: From Molecular Mechanisms to Clinical Implications. J. Clin. Med. 2022, 11, 3649. https://doi.org/10.3390/jcm11133649
Palma R, Pronio A, Romeo M, Scognamiglio F, Ventriglia L, Ormando VM, Lamazza A, Pontone S, Federico A, Dallio M. The Role of Insulin Resistance in Fueling NAFLD Pathogenesis: From Molecular Mechanisms to Clinical Implications. Journal of Clinical Medicine. 2022; 11(13):3649. https://doi.org/10.3390/jcm11133649
Chicago/Turabian StylePalma, Rossella, Annamaria Pronio, Mario Romeo, Flavia Scognamiglio, Lorenzo Ventriglia, Vittorio Maria Ormando, Antonietta Lamazza, Stefano Pontone, Alessandro Federico, and Marcello Dallio. 2022. "The Role of Insulin Resistance in Fueling NAFLD Pathogenesis: From Molecular Mechanisms to Clinical Implications" Journal of Clinical Medicine 11, no. 13: 3649. https://doi.org/10.3390/jcm11133649