
Cardioplegia between Evolution and Revolution: From Depolarized to Polarized Cardiac Arrest in Adult Cardiac Surgery

 , , , ,
, , , , {kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Dawn of Cardiac Surgery: Making the Impossible a Reality
“No surgeon who wished to preserve the respect of his colleagues would ever attempt to suture a wound of the heart”Theodor Billroth (1881) [1]
1.2. Two Revolutions: Hypothermia and the Heart-Lung Machine
1.3. Cardioplegia: The Third Revolution
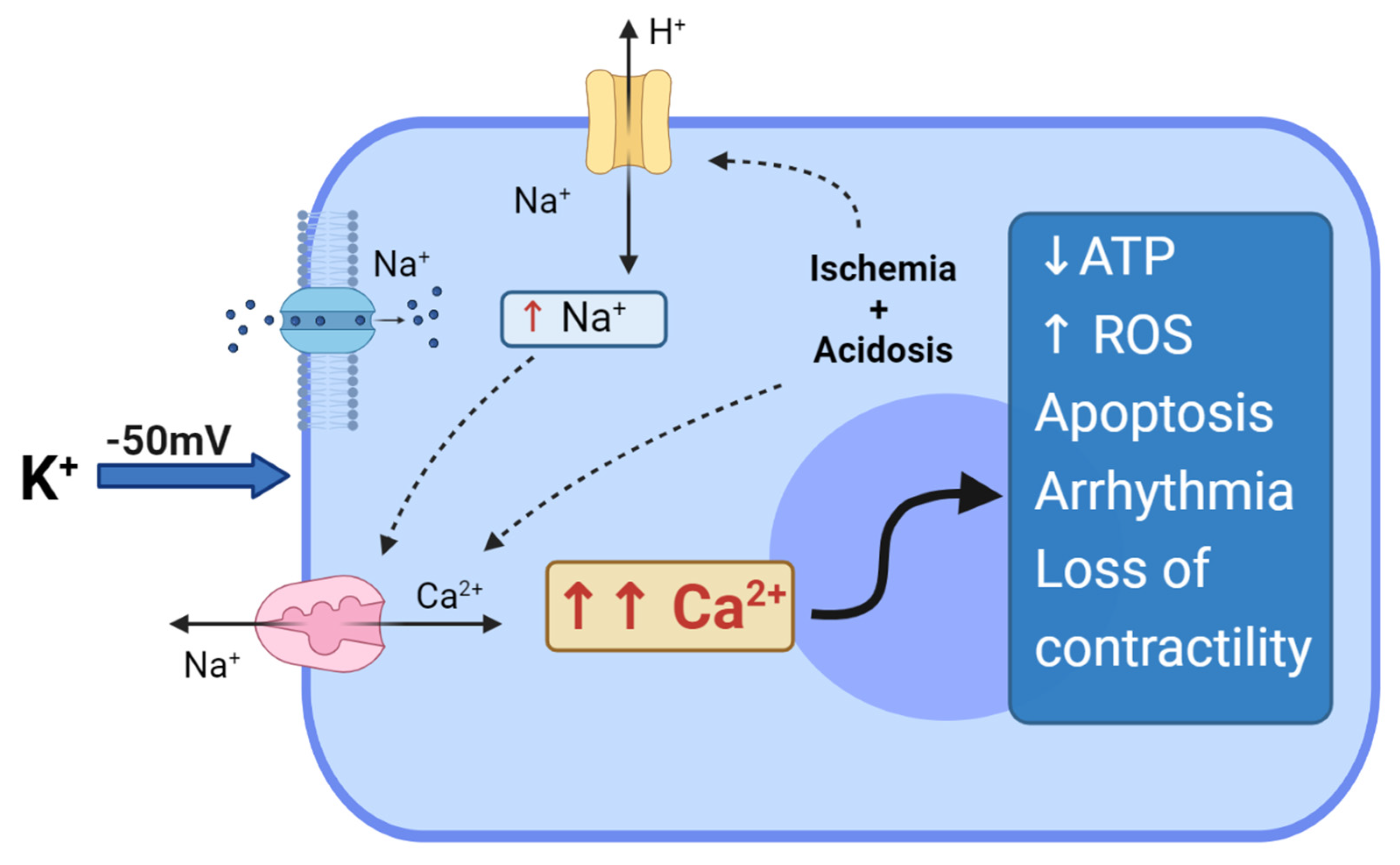
2. Hyperkalemic Arrest: What Are the Limits?
3. Polarized Arrest as a Clinical Alternative
3.1. From the Isolated Heart to Animal Studies:
3.2. Humans Trials: Are We Getting Closer to Change?
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Alexi-Meskishvili, V.; Böttcher, W. Suturing of penetrating wounds to the heart in the nineteenth century: The beginnings of heart surgery. Ann. Thorac. Surg. 2011, 92, 1926–1931. [Google Scholar] [CrossRef]
- Dalton, H.C., III. Report of a Case of Stab-Wound of the Pericardium, Terminating in Recovery after Resection of a Rib and Suture of the Pericardium. Ann. Surg. 1895, 21, 147–152. [Google Scholar] [CrossRef]
- Shumacker, H.B., Jr. The Evolution of Cardiac Surgery; Indiana University Press: Bloomington, Indiana, 1992. [Google Scholar]
- Westaby, S.; Bosher, C. Landmarks in Cardiac Surgery; Isis Medical Media Ltd.: Oxford, UK, 1997. [Google Scholar]
- Ellis, H. The early days of surgery for cardiac injuries. J. Perioper. Pract. 2017, 27, 268. [Google Scholar] [CrossRef]
- Cohn, L.H. The first successful surgical treatment of mitral stenosis: The 70th anniversary of Elliot Cutler’s mitral commissurotomy. Ann. Thorac. Surg. 1993, 56, 1187–1190. [Google Scholar] [CrossRef]
- Cooley, D.A.; Frazier, O.H. The past 50 years of cardiovascular surgery. Circulation 2000, 102 (Suppl. 4), IV87–IV93. [Google Scholar] [CrossRef]
- Sealy, W.C. Hypothermia: Its possible role in cardiac surgery. Ann. Thorac. Surg. 1989, 47, 788–791. [Google Scholar] [CrossRef]
- Lewis, F.J.; Taufic, M. Closure of atrial septal defects with the aid of hypothermia; experimental accomplishments and the report of one successful case. Surgery 1953, 33, 52–59. [Google Scholar]
- Gibbon, J.H., Jr. The development of the heart-lung apparatus. Am. J. Surg. 1978, 135, 608–619. [Google Scholar] [CrossRef]
- Kirklin, J.W.; Dushane, J.W.; Patrick, R.T.; Donald, D.E.; Hetzel, P.S.; Harshbarger, H.G.; Wood, E.H. Intracardiac surgery with the aid of a mechanical pump-oxygenator system (gibbon type): Report of eight cases. Proc. Staff Meet. Mayo Clin. 1955, 30, 201–206. [Google Scholar]
- Brewer, D.L.; Bilbro, R.H.; Bartel, A.G. Myocardial infarction as a complication of coronary bypass surgery. Circulation 1973, 47, 58. [Google Scholar] [CrossRef] [Green Version]
- Roberts, W.C.; Bulkley, B.H.; Morrow, A.G. Pathologic anatomy of cardiac valve replacement: A study of 224 necropsy patients. Prog. Cardiovasc. Dis. 1973, 15, 539. [Google Scholar] [CrossRef]
- Hultgren, H.N.; Miyagawa, M.; Buch, W.; Angell, W.W. Ischemic myocardial injury during cardiopulmonary bypass surgery. Am. Heart J. 1973, 85, 167. [Google Scholar] [CrossRef]
- Lam, C.R.; Gahagan, T.; Sergant, C.; Green, E. Clinical experiences with Induced cardiac arrest during intracardiac surgical procedures. Ann. Surg. 1957, 146, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Ringer, S. A further contribution regarding the influence of the different csonstituent of the blood on the contraction of the heart. J. Physiol. 1883, 4, 29–42. [Google Scholar] [CrossRef]
- Lam, C.R.; Geoghegan, T.; Lepore, A. Induced cardiac arrest for intracardiac surgical procedures; an experimental study. J. Thorac. Surg. 1955, 30, 620–625. [Google Scholar] [CrossRef]
- Melrose, D.G.; Dreyer, B.; Bentall, H.H.; Baker, J.B.E. Elective cardiac arrest. Lancet 1955, 2, 21–22. [Google Scholar] [CrossRef]
- Gerbode, F.; Melrose, D.G. The use of K+ arrest in open cardiac surgery. Am. J. Surg. 1958, 96, 221–227. [Google Scholar] [CrossRef]
- Effler, D.B.; Groves, L.K.; Sones, F.M.J.; Kolff, W.J. Elective cardiac arrest in open-heart surgery; report of three cases. Cleve. Clin. Q. 1956, 23, 105–114. [Google Scholar] [CrossRef]
- Schaff, H.V.; Dombroff, R.; Flaherty, J.T.; Bulkley, B.H.; Hutchins, G.M.; Goldman, R.A.; Gott, V.L. Effect of K+ cardioplegia on myocardial ischemia and post arrest ventricular function. Circulation 1978, 58, 240–249. [Google Scholar] [CrossRef] [Green Version]
- Allen, P.; Lillehei, C.W. Use of induced cardiac arrest in open heart surgery; results in seventy patients. Minn. Med. 1957, 40, 672–676. [Google Scholar]
- Nunn, D.D.; Belisle, C.A.; Lee, W.H.; Parker, E.F., Jr. A comparative study of aortic occlusion alone and of K+ citrate arrest during cardio pulmonary bypass. Surgery 1959, 45, 848–851. [Google Scholar] [PubMed]
- Wasserman, F.; Wolcott, M.W.; Wherrry, C.G.; Brodsky, L. Comparative effect of 5 per cent K+ chloride and 30 percent K+ citrate in resuscitation from ventricular fibrillation following acute myocardial infarction; an experimental study. J. Thorac. Cardiovasc. Surg. 1959, 38, 30–39. [Google Scholar] [CrossRef]
- Willman, V.L.; Cooper, T.; Zafiracopoulos, P.; Hanlon, C.R. Depression of ventricular function following elective cardiac arrest with K+ citrate. Surgery 1959, 46, 792–796. [Google Scholar] [PubMed]
- Bjork, V.O.; Fors, B. Induced cardiac arrest. J. Thorac. Cardiovasc. Surg. 1961, 41, 387–394. [Google Scholar] [CrossRef]
- Hufnagel, C.A.; Conrad, P.W.; Schanno, J.; Pifarre, R. Profound cardiac hypothermia. Ann. Surg. 1961, 153, 790. [Google Scholar] [CrossRef] [PubMed]
- Griepp, R.B.; Stinson, E.B.; Shumway, N.E. Profound local hypothermia for myocardial protection during open-heart surgery. J. Thorac Cardiovasc. Surg. 1973, 66, 731. [Google Scholar] [CrossRef]
- Shumway, N.E.; Lower, R.R. Topical cardiac hypothermia for extended periods of anoxic arrest. Surg. Forum. 1960, 10, 563. [Google Scholar]
- Shumway, N.E.; Lower, R.R.; Stofer, R.C. Selective hypothermia of the heart in anoxic cardiac arrest. Surg. Gynec. Obstet. 1959, 109, 750. [Google Scholar]
- Cooley, D.A.; Reul, G.J.J.; Wukasch, D.C. Ischemic myocardial contracture (“stone heart”). A complication of cardiac surgery. Isr. J. Med. Sci. 1975, 11, 203–210. [Google Scholar]
- Bretschneider, H.J. Survival time and recuperative time of the heart in normothermia and hypothermia. Verh. Dtsch. Ges. Kreislaufforsch. 1964, 30, 11–34. [Google Scholar]
- Bretschneider, H.J.; Hubner, G.; Knoll, D.; Lohr, B.; Nordbeck, H.; Spieckermann, P.G. Myocardial resistance and tolerance to ischemia: Physiological and biochemical basis. J. Cardiovasc. Surg. 1975, 16, 241–260. [Google Scholar]
- Preusse, C.J. Cardioplegia with an intracellular formulation. In Ischemia-Reperfusion in Cardiac Surgery; Piper, H.M., Preusse, C.J., Eds.; Springer: Dordrecht, Netherlands, 1993. [Google Scholar]
- Mokbel, M.; Zamani, H.; Lei, I.; Chen, Y.E.; Romano, M.A.; Aaronson, K.D.; Haft, J.W.; Pagani, F.D.; Tang, P.C. Histidine-Tryptophan-Ketoglutarate Solution for Donor Heart Preservation Is Safe for Transplantation. Ann. Thorac. Surg. 2020, 109, 763–770. [Google Scholar] [CrossRef]
- Hearse, D.J.; Braimbridge, M.V.; Jynge, P. Protection of the Ischemic Myocardium: Cardioplegia; Raven Press: New York, NY, USA, 2012. [Google Scholar]
- Jynge, P.; Hearse, D.J.; Feuvray, D.; Mahalu, W.; Canković-Darracott, S.; O’Brien, K.; Braimbridge, M.V. The St. Thomas’ hospital cardioplegic solution: A characterization in two species. Scand. J. Thorac. Cardiovasc. Surg. Suppl. 1981, 30, 1–28. [Google Scholar] [PubMed]
- Braimbridge, M.V.; Chayen, J.; Bitensky, L.; Hearse, D.J.; Jynge, P.; Cankovic-Darracott, S. Cold cardioplegia or continuous coronary perfusion? Report on preliminary clinical experience as assessed cytochemically. J. Thorac. Cardiovasc. Surg. 1977, 74, 900–906. [Google Scholar] [CrossRef]
- Buckberg, G.D. A proposed “solution” to the cardioplegic controversy. J. Thorac. Cardiovasc. Surg. 1979, 77, 803. [Google Scholar] [CrossRef]
- Buckberg, G.D. Antegrade/retrograde blood cardioplegia to ensure cardioplegic distribution: Operative techniques and objectives. J. Card. Surg. 1989, 4, 216. [Google Scholar] [CrossRef] [PubMed]
- Karthik, S.; Grayson, A.D.; Oo, A.Y.; Fabri, B.M. A survey of current myocardial protection practices during coronary artery bypass grafting. Ann. R Coll. Surg. Engl. 2004, 86, 413–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, J.M.; Miles, L.F.; Abu-Omar, Y.; Galhardo, C.; Falter, F. Global Cardioplegia Practices: Results from the Global Cardiopulmonary Bypass Survey. J. Extra. Corpor. Technol. 2018, 50, 83–93. [Google Scholar] [PubMed]
- Tyers, G.F.; Todd, G.J.; Niebauer, I.M.; Manley, N.J.; Waldhausen, J.A. The mechanism of myocardial damage following K+ citrate (Melrose) cardioplegia. Surgery. 1975, 78, 45–53. [Google Scholar]
- Dobson, G.P.; Faggian, G.; Onorati, F.; Vinten-Johansen, J. Hyperkalemic cardioplegia for adult and pediatric surgery: End of an era? Front. Physiol. 2013, 4, 228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bers, D.M.; Despa, S. Cardiac myocytes Ca2+ and Na+ regulation in normal and failing hearts. J. Pharmacol. Sci. 2006, 100, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Bers, D.M.; Despa, S. Na+ transport in cardiac myocytes; Implications for excitation-contraction coupling. IUBMB Life 2009, 61, 215–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, R.J.; Mavroudis, C.; Gardner, C.; Turley, K.; Ullyot, D.; Ebert, P.A. Relationship between atrioventricular arrhythmias and the concentration of K+ ion in cardioplegic solution. J. Thorac. Cardiovasc. Surg. 1980, 80, 517–526. [Google Scholar] [CrossRef]
- Heyndrickx, G.R.; Millard, R.W.; McRitchie, R.J.; Maroko, P.R.; Vatner, S.F. Regional myocardial functional and electrophysiological alterations after brief coronary artery occlusion in conscious dogs. J. Clin. Invest. 1975, 56, 978–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsutsumi, T.; Wyatt, R.F.; Abildskov, J.A. Effects of hyperkalemia on local changes of repolarization duration in canine left ventricle. J. Electrocardiol. 1983, 16, 1–6. [Google Scholar] [CrossRef]
- Braunwald, E.; Kloner, R.A. The stunned myocardium: Prolonged, postischemic ventricular dysfunction. Circulation 1982, 66, 1146–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloner, R.A.; Przyklenk, K.; Kay, G.L. Clinical evidence for stunned myocardium after coronary artery bypass surgery. J. Card Surg. 1994, 9, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Damiano, R.J., Jr.; Cohen, N.M. Hyperpolarized arrest attenuates myocardial stunning following global surgical ischemia: An alternative to traditional hyperkalemic cardioplegia? J. Card Surg. 1994, 9, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Bolli, R.; Patel, B.S.; Jeroudi, M.O.; Lai, E.K.; McCay, P.B. Demonstration of free radical generation in "stunned" myocardium of intact dogs with the use of the spin trap alpha-phenyl N-tert-butyl nitrone. J. Clin. Invest. 1988, 82, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Parolari, A.; Rubini, P.; Cannata, A.; Bonati, L.; Alamanni, F.; Tremoli, E.; Biglioli, P. Endothelial damage during myocardial preservation and storage. Ann. Thorac. Surg. 2002, 73, 682–690. [Google Scholar] [CrossRef]
- Verma, S.; Anderson, T.J. Fundamentals of endothelial function for the clinical cardiologist. Circulation. 2002, 105, 546–549. [Google Scholar] [CrossRef] [Green Version]
- Higashi, Y.; Noma, K.; Yoshizumi, M.; Kihara, Y. Endothelial function and oxidative stress in cardiovascular diseases. Circ. J. 2009, 73, 411–418. [Google Scholar] [CrossRef] [Green Version]
- Siess, W. Molecular mechanisms of platelet activation. Physiol. Rev. 1989, 69, 58–178. [Google Scholar] [CrossRef]
- Algarni, K.D.; Maganti, M.; Yau, T.M. Predictors of low cardiac output syndrome after isolated coronary artery bypass surgery:trends over 20 years. Ann. Thorac. Surg. 2011, 92, 1678–1684. [Google Scholar] [CrossRef]
- Sellke, F.W.; Boyle, E.M., Jr.; Verrier, E.D. Endothelial cell injury in cardiovascular surgery: The pathophysiology of vasomotor dysfunction. Ann. Thorac. Surg. 1996, 62, 1222–1228. [Google Scholar] [CrossRef]
- He, G.W. Endothelial function related to vascular tone in cardiac surgery. Heart Lung Circ. 2005, 14, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Conti, V.R.; Ware, D.L. Cardiac arrhythmias in cardiothoracic surgery. Chest Surg. Clin. N Am. 2002, 12, 439–460. [Google Scholar] [CrossRef]
- Gundry, S.R.; Sequeira, A.; Coughlin, T.R.; McLaughlin, J.S. Postoperative conduction disturbances: A comparison of blood and crystalloid cardioplegia. Ann. Thorac. Surg. 1989, 47, 384–390. [Google Scholar] [CrossRef]
- Rousou, J.A.; Meeran, M.K.; Engelman, R.M.; Breyer, R.H.; Lemeshow, S. Does the type of venous drainage or cardioplegia affect postoperative conduction and atrial arrhythmias? Circulation 1985, 72, II259–II263. [Google Scholar] [PubMed]
- Chambers, D.J.; Hearse, D.J. Cardioplegia and surgical ischemia. In Heart Physiology and Pathophysiology; Sperelakis, N., Kurachi, Y., Terzic, A., Cohen, M.V., Eds.; Academic Press: San Diego, CA, USA, 2001; pp. 887–925. [Google Scholar]
- Lubbe, W.F.; Podzuweit, T.; Opie, L.H. Potential arrhythmogenic role of cyclic adenosine monophosphate (AMP) and cytosolic calcium overload: Implications for prophylactic effects of beta-blockers in myocardial infarction and proarrhythmic effects of phosphodiesterase inhibitors. J. Am. Coll. Cardiol. 1992, 19, 1622–1633. [Google Scholar] [CrossRef] [Green Version]
- Paterson, D.J.; Rogers, J.; Powell, T.; Brown, H.F. Effect of catecholamines on the ventricular myocyte action potential in raised extracellular K+. Acta. Physiol. Scand. 1993, 148, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Vittone, L.; Said, M.; Mattiazzi, A. beta 2-Adrenergic stimulation is involved in the contractile dysfunction of the stunned heart. Naunyn. Schmiedebergs. Arch. Pharmacol. 2006, 373, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Lameris, T.W.; de Zeeuw, S.; Alberts, G.; Boomsma, F.; Duncker, D.J.; Verdouw, P.D.; Veld, A.J.; van Den Meiracker, A.H. Time course and mechanism of myocardial catecholamine release during transient ischemia in vivo. Circulation 2000, 101, 2645–2650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; He, G.W. Effect of cardioplegic and organ preservation solutions and their components on coronary endothelium-derived relaxing factors. Ann. Thorac. Surg. 2005, 80, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Chambers, D.J. Mechanisms and alternative methods of achieving cardiac arrest. Ann. Thorac. Surg. 2003, 75, S661–S666. [Google Scholar] [CrossRef]
- Dobson, G.P. Membrane polarity: A target for myocardial protection and reduced inflammation in adult and pediatric cardiothoracic surgery. J. Thorac. Cardiovasc. Surg. 2010, 140, 1213–1217. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Jiang, R.; Dobson, G.P.; Vinten-Johansen, J. The novel non-depolarizing, normokalemic cardioplegia formulation adenosine–lidocaine (Adenocaine) exerts superior anti-neutrophil effects by synergistic actions of its components. J. Thorac. Cardiovasc. Surg. 2012, 143, 1167–1175. [Google Scholar] [CrossRef] [Green Version]
- Granfeldt, A.; Letson, H.L.; Dobson, G.P.; Shi, W.; Vinten-Johansen, J.; Tønnesen, E. Adenosine, lidocaine and Mg2+ improves cardiac and pulmonary function, induces reversible hypotension and exerts anti-inflammatory effects in an endotoxemic porcine model. Crit. Care. 2014, 18, 682. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Xing, J.; Miao, N.; Li, W.; Liu, W.; Lai, Y.-Q.; Luo, Y.; Ji, B. The myocardial protective effect of adenosine as an adjunct to intermittent blood cardioplegia during open heart surgery. Eur. J. Cardio-Thorac. Surg. 2009, 36, 1018–1023. [Google Scholar] [CrossRef]
- Snabaitis, A.K.; Shattock, M.J.; Chambers, D.J. Comparison of polarized and depolarized arrest in the isolated rat heart for long-term preservation. Circulation 1997, 96, 3148–3156. [Google Scholar] [CrossRef]
- Sternbergh, W.C.; Brunsting, L.A.; Abd-Elfattah, A.S.; Wechsler, A.S. Basal metabolic energy requirements of polarized and depolarized arrest in rat heart. Am. J. Physiol. 1989, 256, H846–H851. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.D. Local anesthetics. In Basic and Clinical Pharmacology; Katzung, B.G., Ed.; Appleton & Lange: Stamford, CT, USA, 1998; pp. 425–433. [Google Scholar]
- Brown, D.L.; Ransom, D.M.; Hall, J.A.; Leicht, C.H.; Schroeder, D.R.; Offord, K.P. Regional anesthesia and local anesthetic-induced systemic toxicity: Seizure frequency and accompanying cardiovascular changes. Anesth. Analg. 1995, 81, 321–328. [Google Scholar]
- Attwell, D.; Cohen, I.; Eisner, D.; Ohba, M.; Ojeda, C. The steady state TTX-sensitive (‘window’) Na+ current in cardiac Purkinje fibres. Pflugers Arch. 1979, 379, 137–142. [Google Scholar] [CrossRef]
- Tyers, G.F.; Todd, G.J.; Niebauer, I.M.; Manley, N.J.; Waldhausen, J.A. Effect of intracoronary tetrodotoxin on recovery of the isolated working rat heart from sixty minutes of ischemia. Circulation 1974, 50, II175–II180. [Google Scholar]
- Tyers, G.F.; Todd, G.J.; Neely, J.R.; Waldhausen, J.A. The mechanism of myocardial protection from ischemic arrest by intracoronary tetrodotoxin administration. J. Thorac. Cardiovasc. Surg. 1975, 69, 190–195. [Google Scholar] [CrossRef]
- Narahashi, T. Tetrodotoxin: A brief history. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2008, 84, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Noma, A. ATP-regulated K+ channels in cardiac muscle. Nature 1983, 305, 147–148. [Google Scholar] [CrossRef] [PubMed]
- Hearse, D.J. Activation of ATP-sensitive K+ channels: A novel pharmacological approach to myocardial protection? Cardiovasc. Res. 1995, 30, 1–17. [Google Scholar] [CrossRef]
- Nichols, C.G. Adenosine Triphosphate-Sensitive Potassium Currents in Heart Disease and Cardioprotection. Card Electrophysiol. Clin. 2016, 8, 323–335. [Google Scholar] [CrossRef] [Green Version]
- McPherson, C.D.; Pierce, G.N.; Cole, W.C. Ischemic cardioprotection by ATP-sensitive K1 channels involves high-energy phosphate preservation. Am. J. Physiol. 1993, 265, H1809–H1818. [Google Scholar]
- Cohen, N.M.; Wise, R.M.; Wechsler, A.S.; Damiano, R.J. Elective cardiac arrest with a hyperpolarizing adenosine triphosphate-sensitive K+ channel opener. A novel form of myocardial protection? J. Thorac. Cardiovasc. Surg. 1993, 106, 317–328. [Google Scholar] [CrossRef]
- Chi, L.; Uprichard, A.C.; Lucchesi, B.R. Profibrillatory actions of pinacidil in a conscious canine model of sudden coronary death. J. Cardiovasc. Pharmacol. 1990, 15, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Bessho, R.; Chambers, D.J. Myocardial protection: The efficacy of an ultrashort-acting beta-blocker, esmolol, as a cardioplegic agent. J. Thorac. Cardiovasc. Surg. 2001, 122, 993–1003. [Google Scholar] [CrossRef] [Green Version]
- Bessho, R.; Chambers, D.J. Myocardial protection with oxygenated esmolol cardioplegia during prolonged normothermic ischemia in the rat. J. Thorac. Cardiovasc. Surg. 2002, 124, 340–351. [Google Scholar] [CrossRef] [Green Version]
- Arlock, P.; Wohlfart, B.; Sjoberg, T.; Steen, S. The negative inotropic effect of esmolol on isolated cardiac muscle. Scand. Cardiovasc. J. 2005, 39, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Fallouh, H.B.; McLatchie, L.M.; Shattock, M.J.; Chambers, D.J.; Kentish, J.C. Esmolol as a cardioplegic agent: An effect beyond (beta)-blockade. Circulation 2007, 116, II-323–II-324. [Google Scholar]
- Fallouh, H.B.; McLatchie, L.M.; Bardswell, S.C.; Shattock, M.J.; Chambers, D.J.; Kentish, J.C. Myocardial arrest by esmolol: Negative inotropy induced by calcium and sodium channel blockade. J. Mol. Cell. Cardiol. 2008, 44, S49–S50. [Google Scholar]
- Deng, C.Y.; Lin, S.G.; Zhang, W.C.; Kuang, S.J.; Qian, W.M.; Wu, S.L.; Shan, Z.X.; Yang, M.; Yu, X.Y. Esmolol inhibits Na+ current in rat ventricular myocytes. Methods Find. Exp. Clin. Pharmacol. 2006, 28, 697–702. [Google Scholar] [CrossRef]
- Pirk, J.; Kolár, F.; Ost’ádal, B.; Sedivý, J.; Stambergová, A.; Kellovský, P. The effect of the ultrashort beta-blocker esmolol on cardiac function recovery: An experimental study. Eur. J. Cardio-Thorac. Surg. 1999, 15, 199–203. [Google Scholar] [CrossRef] [Green Version]
- Fujii, M.; Chambers, D.J. Cardioprotection with esmolol cardioplegia: Efficacy as a blood-based solution. Eur. J. Cardio-Thorac. Surg. 2013, 43, 619–627. [Google Scholar] [CrossRef]
- Nishina, D.; Chambers, D.J. Efficacy of esmolol cardioplegia during hypothermic ischaemia. Eur. J. Cardio-Thorac. Surg. 2018, 53, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Belardinelli, L.; Giles, W.R.; West, A. Ionic mechanisms o adenosine actions in pacemaker cells from rabbit heart. J. Physiol. 1988, 405, 615–633. [Google Scholar] [CrossRef] [Green Version]
- Schubert, T.; Vetter, H.; Owen, P.; Reichart, B.; Opie, L.H. Adenosine cardioplegia. Adenosine versus K+ cardioplegia:effects on cardiac arrest and postischemic recovery in the isolated rat heart. J. Thorac. Cardiovasc. Surg. 1989, 98, 1057–1065. [Google Scholar] [CrossRef]
- De Jong, J.W.; van der Meer, P.; van Loon, H.; Owen, P.; Opie, L.H. Adenosine as adjunct to K+ cardioplegia: Effect on function, energy metabolism, and electrophysiology. J. Thorac. Cardiovasc. Surg. 1990, 100, 445–454. [Google Scholar] [CrossRef]
- Dobson, G.P. Organ arrest, protection and preservation: Natural hibernation to cardiac surgery. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2004, 139, 469–485. [Google Scholar] [CrossRef] [PubMed]
- Rudd, D.M.; Dobson, G.P. Eight hours of cold static storage with adenosine and lidocaine (Adenocaine) heart preservation solutions: Toward therapeutic suspended animation. J. Thorac. Cardiovasc. Surg. 2011, 142, 1552–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, M.J.; Letson, H.L.; Dobson, G.P. Adenosine, lidocaine and Mg2+ (ALM) induces a reversible hypotensive state, reduces lung edema and prevents coagulopathy in the rat model of polymicrobial sepsis. J. Trauma Acute Care Surg. 2014, 77, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Vinten-Johansen, J.; Thourani, V.H.; Ronson, R.S.; Jordan, J.E.; Zhao, Z.Q.; Nakamura, M.; Velez, D.; Guyton, R.A. Broad-spectrum cardioprotection with adenosine. Ann. Thorac. Surg. 1999, 68, 1942–1948. [Google Scholar] [CrossRef]
- Boehm, D.H.; Human, P.A.; von Oppell, U.; Owen, P.; Reichenspurner, H.; Opie, L.H.; Rose, A.G.; Reichart, B. Adenosine cardioplegia: Reducing reperfusion injury of the ischaemic myocardium? Eur. J. Cardio-Thorac. Surg. 1991, 5, 542–545. [Google Scholar] [CrossRef]
- Dobson, G.P.; Jones, M.W. Adenosine and lidocaine: A new concept in non depolarizing surgical myocardial arrest, protection, and preservation. J. Thorac. Cardiovasc. Surg. 2004, 127, 794–805. [Google Scholar] [CrossRef] [Green Version]
- Djabir, Y.; Letson, H.L.; Dobson, G.P. Adenosine, lidocaine, and Mg2+ (ALM™) increases survival and corrects coagulopathy after eight-minute asphyxial cardiac arrest in the rat. Shock 2013, 40, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Sloots, K.L.; Dobson, G.P. Normokalemic adenosine-lidocaine cardioplegia: Importance of maintaining a polarized myocardium for optimal arrest and reanimation. J. Thorac. Cardiovasc. Surg. 2010, 139, 1576–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mentzer, R.M., Jr.; Rahko, P.S.; Molina-Viamonte, V.; Canver, C.C.; Chopra, P.S.; Love, R.B.; Cook, T.D.; Hegge, J.O.; Lasley, R.D. Safety, tolerance, and efficacy of adenosine as an additive to blood cardioplegia in humans during coronary artery bypass surgery. Am. J. Cardiol. 1997, 79, 38–43. [Google Scholar] [CrossRef]
- Abdelwahab, A.A.; Sabry, M.; Elshora, H.A.; Arafat, A.A. Effect of Fast Cardioplegic Arrest Induced by Adenosine on Cardiac Troponin Levels After Heart Valve Surgery. Heart Lung Circ. 2019, 28, 1714–1719. [Google Scholar] [CrossRef] [PubMed]
- Onorati, F.; Santini, F.; Dandale, R.; Ucci, G.; Pechlivanidis, K.; Menon, T.; Chiominto, B.; Mazzucco, A.; Faggian, G. “Polarizing” microplegia improves cardiac cycle efficiency after CABG for unstable angina. Int. J. Cardiol. 2013, 167, 2739–2746. [Google Scholar] [CrossRef] [PubMed]
- Baraka, A.; Hirt, N.; Dabbous, A.; Taha, S.; Rouhana, C.; el-Khoury, N.; Ghabash, M.; Jamhoury, M.; Sibaii, A. Lidocaine cardioplegia for prevention of reperfusion ventricular fibrillation. Ann. Thorac. Surg. 1993, 55, 1529–1533. [Google Scholar] [CrossRef]
- Ramani, J.; Malhotra, A.; Wadhwa, V.; Sharma, P.; Garg, P.; Tarsaria, M.; Pandya, H. Single-Dose Lignocaine-Based Blood Cardioplegia in Single Valve Replacement Patients. Braz. J. Cardiovasc. Surg. 2017, 32, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.R.; Baker, A.B. Incidence of ventricular fibrillation after aortic cross-clamp release using lignocaine cardioplegia. Anaesth. Intensive Care 1994, 22, 442–446. [Google Scholar] [CrossRef]
- Mustonen, P.K.; Hippeläinen, M.J.; Pöyhönen, M.J.; Rehnberg, L.S. Procaine and timing of aortic declamping affect ventricular reperfusion fibrillation. Ann. Chir. Gynaecol. 1996, 85, 52. [Google Scholar]
- Sellevold, O.F.; Berg, E.M.; Levang, O.W. Procaine is effective for minimizing postischemic ventricular fibrillation in cardiac surgery. Anesth. Analg. 1995, 81, 932–938. [Google Scholar] [PubMed]
- Hayashi, Y.; Sawa, Y.; Ohtake, S.; Nishimura, M.; Ichikawa, H.; Matsuda, H. Controlled nicorandil administration for myocardial protection during coronary artery bypass grafting under cardiopulmonary bypass. J. Cardiovasc. Pharmacol. 2001, 38, 21–28. [Google Scholar] [CrossRef]
- Chinnan, N.K.; Puri, G.D.; Thingnam, S.K. Myocardial protection by nicorandil during open-heart surgery under cardiopulmonary bypass. Eur. J. Anaesthesiol. 2007, 24, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Iguchi, A.; Tsuru, Y.; Nakame, T.; Satou, K.; Tabayashi, K. Nicorandil pretreatment and improved myocardial protection during cold blood cardioplegia. Jpn. J. Thorac. Cardiovasc. Surg. 2000, 48, 24–29. [Google Scholar] [CrossRef]
- Scorsin, M.; Mebazaa, A.; Al Attar, N.; Medini, B.; Callebert, J.; Raffoul, R.; Ramadan, R.; Maillet, J.M.; Ruffenach, A.; Simoneau, F.; et al. Efficacy of esmolol as a myocardial protective agent during continuous retrograde blood cardioplegia. J. Thorac. Cardiovasc. Surg. 2003, 125, 1022–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zangrillo, A.; Bignami, E.; Noè, B.; Nardelli, P.; Licheri, M.; Gerli, C.; Crivellari, M.; Oriani, A.; Di Prima, A.L.; Fominskiy, E.; et al. Esmolol in Cardiac Surgery: A Randomized Controlled Trial. J. Cardio-Thorac. Vasc. Anesth. 2021, 35, 1106–1114. [Google Scholar] [CrossRef] [PubMed]
- Bignami, E.; Guarnieri, M.; Franco, A.; Gerli, C.; De Luca, M.; Monaco, F.; Landoni, G.; Zangrillo, A. Esmolol before cardioplegia and as cardioplegia adjuvant reduces cardiac troponin release after cardiac surgery. A Randomized Trial. Perfusion. 2017, 32, 313–320. [Google Scholar] [CrossRef]
- Rinne, T.; Harmoinen, A.; Kaukinen, S. Esmolol cardioplegia in unstable coronary revascularisation patients. A randomised clinical trial. Acta Anaesthesiol. Scand. 2000, 44, 727–732. [Google Scholar] [CrossRef]
- Jin, Z.X.; Zhang, S.L.; Wang, X.M.; Bi, S.H.; Xin, M.; Zhou, J.J.; Cui, Q.; Duan, W.X.; Wang, H.B.; Yi, D.H. The myocardial protective effects of a moderate-K+ adenosine-lidocaine cardioplegia in pediatric cardiac surgery. J. Thorac. Cardiovasc. Surg. 2008, 136, 1450–1455. [Google Scholar] [CrossRef] [Green Version]
- Jakobsen, Ø.; Næsheim, T.; Aas, K.N.; Sørlie, D.; Steensrud, T. Adenosine instead of supranormal K+ in cardioplegia: It is safe, efficient, and reduces the incidence of postoperative atrial fibrillation. A randomized clinical trial. J. Thorac. Cardiovasc. Surg. 2013, 145, 812–818. [Google Scholar] [CrossRef] [Green Version]
- Jakobsen, Ø.; Muller, S.; Aarsaether, E.; Steensrud, T.; Sørlie, D.G. Adenosine instead of supranormal K+ in cardioplegic solution improves cardioprotection. Eur. J. Cardio-Thorac. Surg. 2007, 32, 493–500. [Google Scholar] [CrossRef]
- Vinten-Johansen, J.; Dobson, G.P. Adenosine-procaine cardioplegia and adenosine-lidocaine cardioplegia: Two sides of the same coin? J. Thorac. Cardiovasc. Surg. 2013, 145, 1684–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onorati, F.; Dobson, G.P.; San Biagio, L.; Abbasciano, R.; Fanti, D.; Covajes, C.; Menon, T.; Gottin, L.; Biancari, F.; Mazzucco, A.; et al. Superior Myocardial Protection Using "Polarizing" Adenosine, Lidocaine, and Mg2+ Cardioplegia in Humans. J. Am. Coll. Cardiol. 2016, 67, 1751–1753. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Francica, A.; Tonelli, F.; Rossetti, C.; Tropea, I.; Luciani, G.B.; Faggian, G.; Dobson, G.P.; Onorati, F. Cardioplegia between Evolution and Revolution: From Depolarized to Polarized Cardiac Arrest in Adult Cardiac Surgery. J. Clin. Med. 2021, 10, 4485. https://doi.org/10.3390/jcm10194485
Francica A, Tonelli F, Rossetti C, Tropea I, Luciani GB, Faggian G, Dobson GP, Onorati F. Cardioplegia between Evolution and Revolution: From Depolarized to Polarized Cardiac Arrest in Adult Cardiac Surgery. Journal of Clinical Medicine. 2021; 10(19):4485. https://doi.org/10.3390/jcm10194485
Chicago/Turabian StyleFrancica, Alessandra, Filippo Tonelli, Cecilia Rossetti, Ilaria Tropea, Giovanni Battista Luciani, Giuseppe Faggian, Geoffrey Phillip Dobson, and Francesco Onorati. 2021. "Cardioplegia between Evolution and Revolution: From Depolarized to Polarized Cardiac Arrest in Adult Cardiac Surgery" Journal of Clinical Medicine 10, no. 19: 4485. https://doi.org/10.3390/jcm10194485