

Enantiodiscriminating Lipophilic Liquid Membrane-Based Assay for High-Throughput Nanomolar Enantioenrichment of Chiral Building Blocks


Abstract
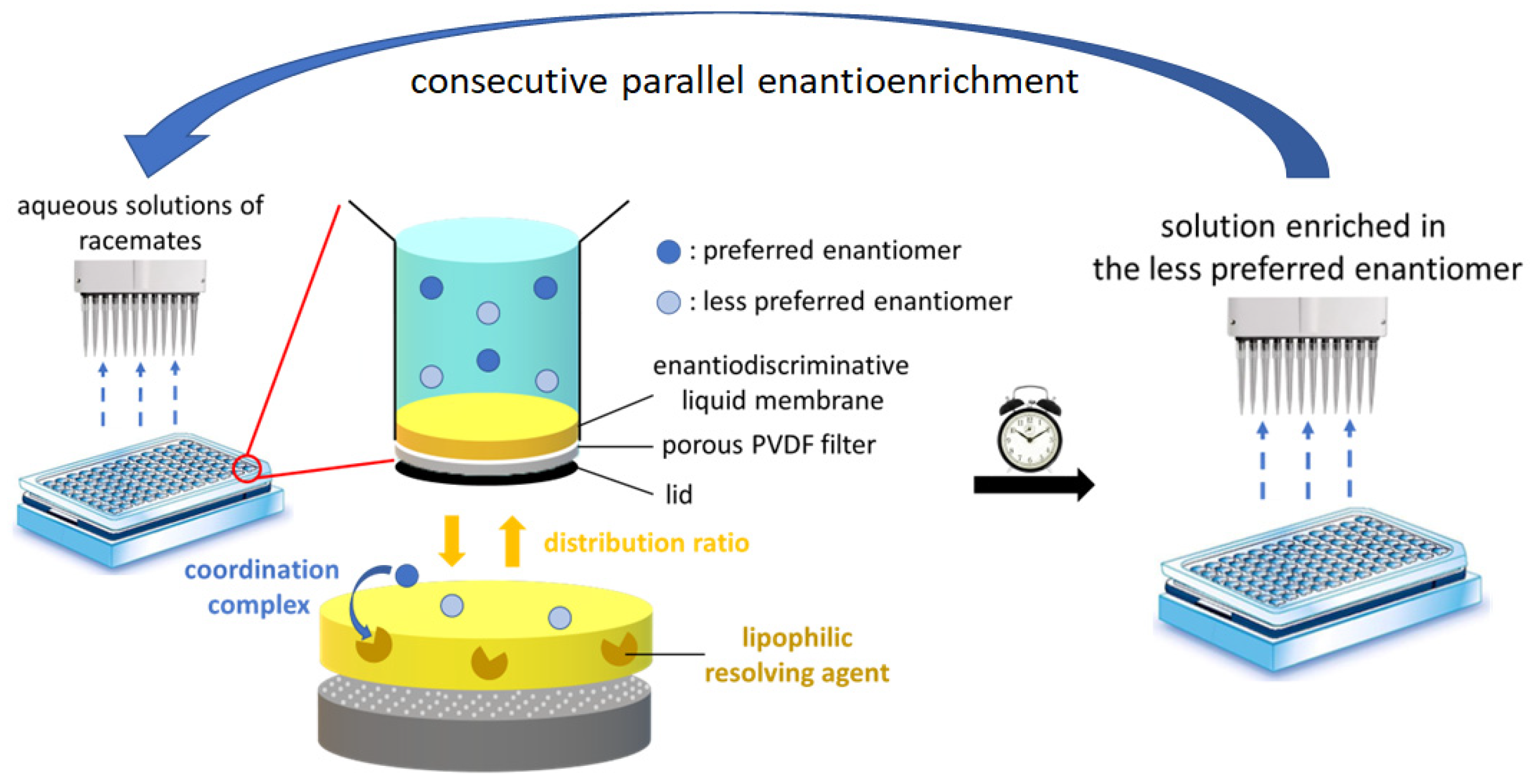
:1. Introduction
2. Experimental Section
2.1. Materials and Apparatus
2.2. Measurements
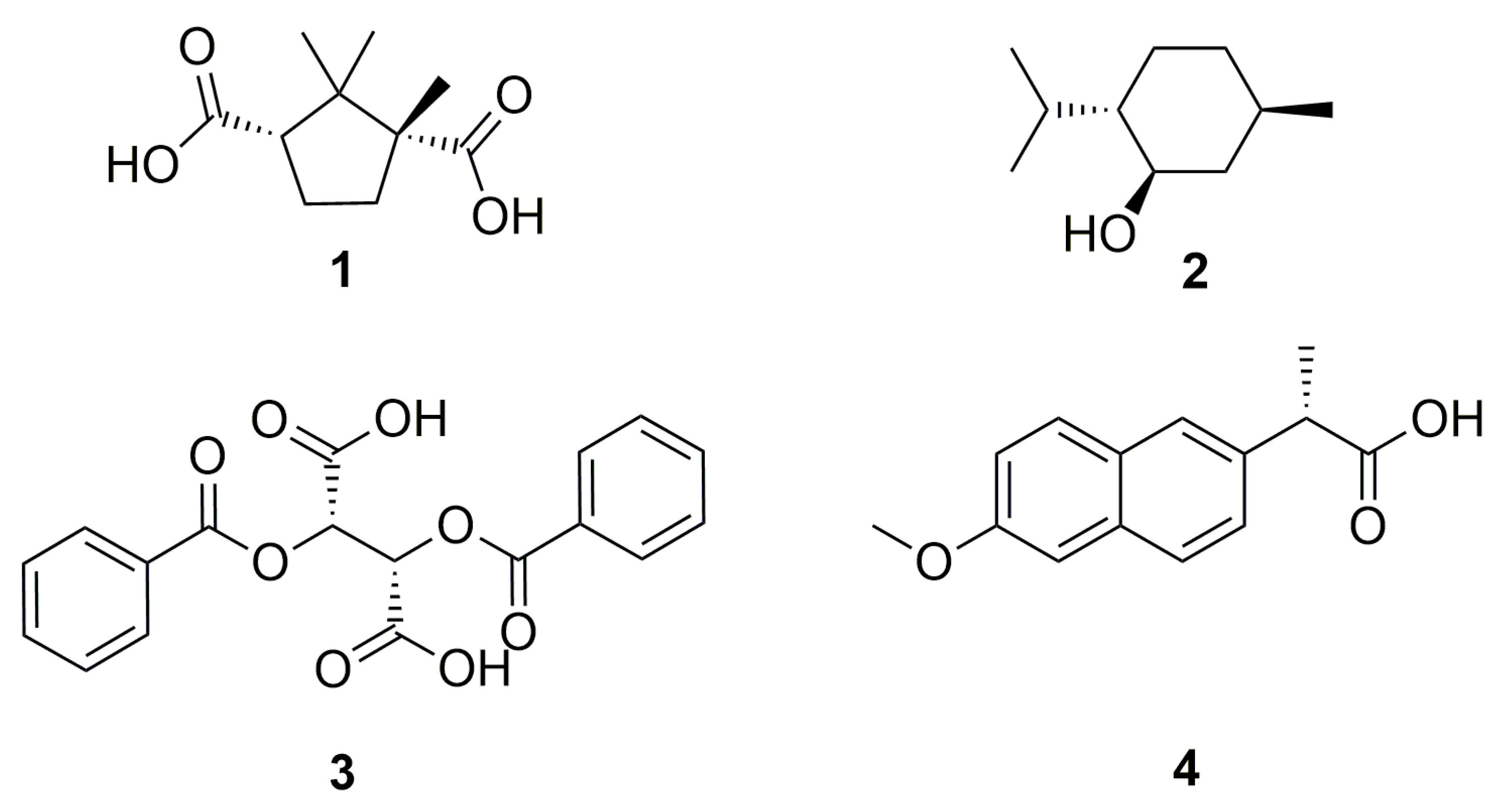
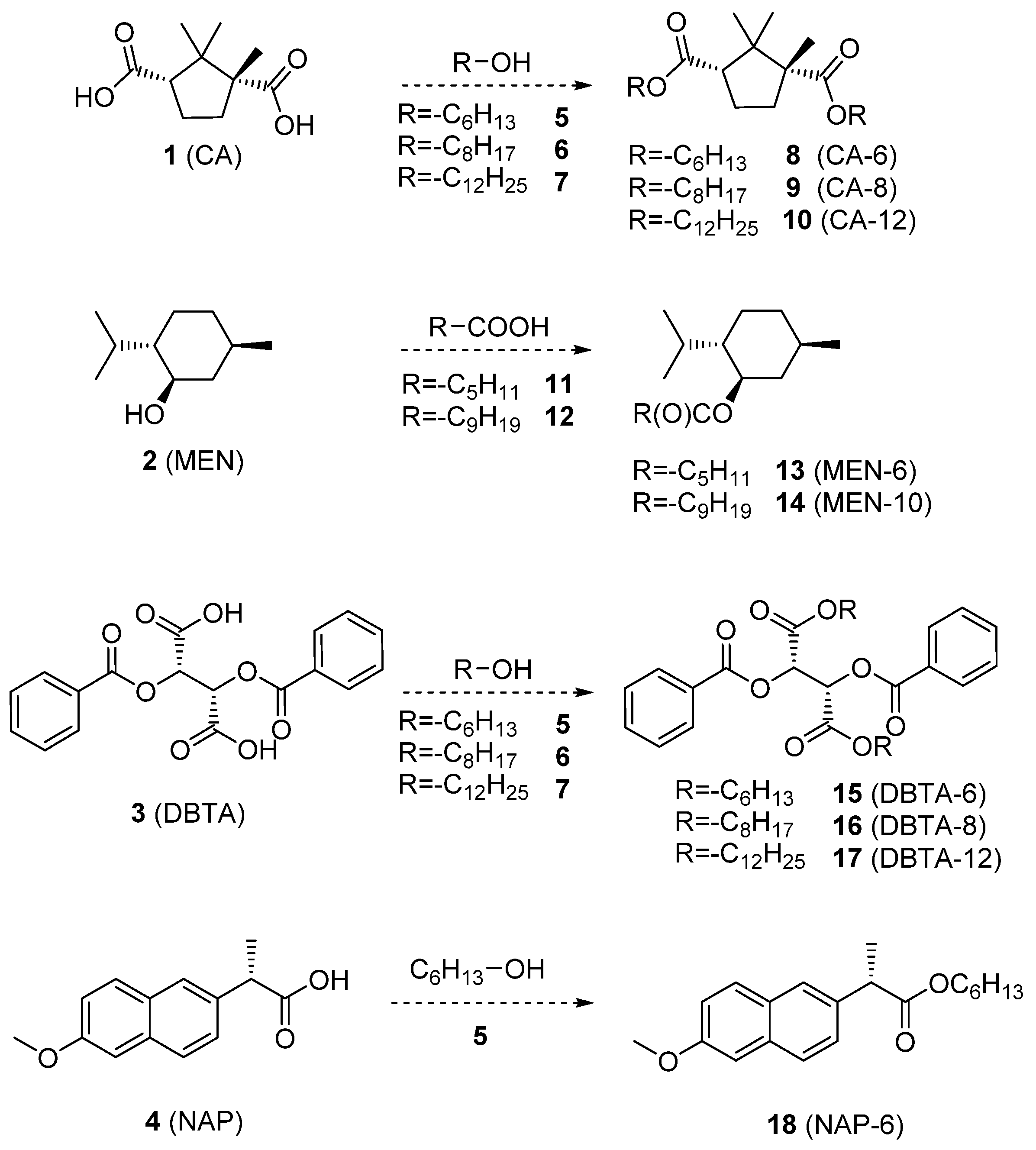
2.3. Synthesis of the Lipophilic Selector Molecules (see Scheme 1)
2.3.1. General Method for Acid-Catalyzed Direct Esterification
2.3.2. General Method for Esterification by Acid Chlorides
2.3.3. Novel Heterogeneous Catalytic Synthesis Method for Direct Esterification
2.4. Characterization of the Applied Selector Molecules
2.4.1. 1,3-Dihexyl-(1R,3S)-1,2,2-trimethylcyclopentane-1,3-dicarboxylate (CA-6, 8 in Scheme 1)
2.4.2. 1,3-Dioctyl-(1R,3S)-1,2,2-trimethylcyclopentane-1,3-dicarboxylate (CA-8, 9 in Scheme 1)
2.4.3. 1,3-Didodecyl-(1R,3S)-1,2,2-trimethylcyclopentane-1,3-dicarboxylate (CA-12, 10 in Scheme 1)
2.4.4. (1R,2S,5R)-5-Methyl-2-(propan-2-yl)cyclohexyl hexanoate (MEN-6, 13 in Scheme 1)
2.4.5. (1R,2S,5R)-5-Methyl-2-(propan-2-yl)cyclohexyl decanoate (MEN-10, 14 in Scheme 1)
2.4.6. 1,4-Dihexyl-(2S,3S)-2,3-bis(benzoyloxy)butanedioate (DBTA-6, 15 in Scheme 1)
2.4.7. 1,4-Dioctyl-(2S,3S)-2,3-bis(benzoyloxy)butanedioate (DBTA-8, 16 in Scheme 1)
2.4.8. 1,4-Didodecyl(2S,3S)-2,3-bis(benzoyloxy)butanedioate (DBTA-12, 17 in Scheme 1)
2.4.9. Hexyl-(2S)-2-(6-methoxynaphthalen-2-yl)propanoate (NAP-6, 18 in Scheme 1)
3. Results and Discussion
3.1. Synthesis of Enantiopure Lipophilic Esters Used as Chiral Selectors of Apolar Liquid Membranes

3.2. Parameter Optimization and Kinetic Properties
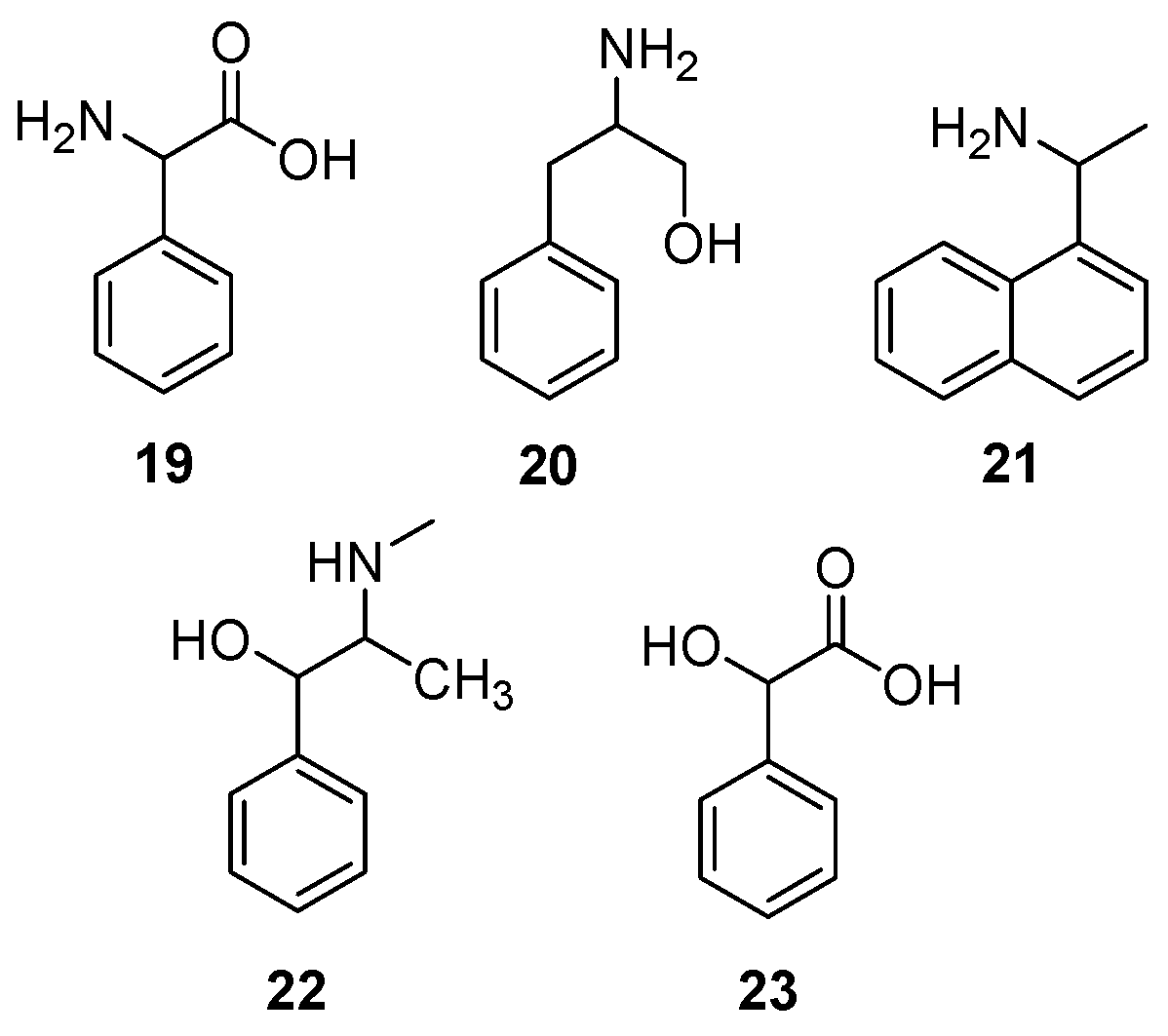
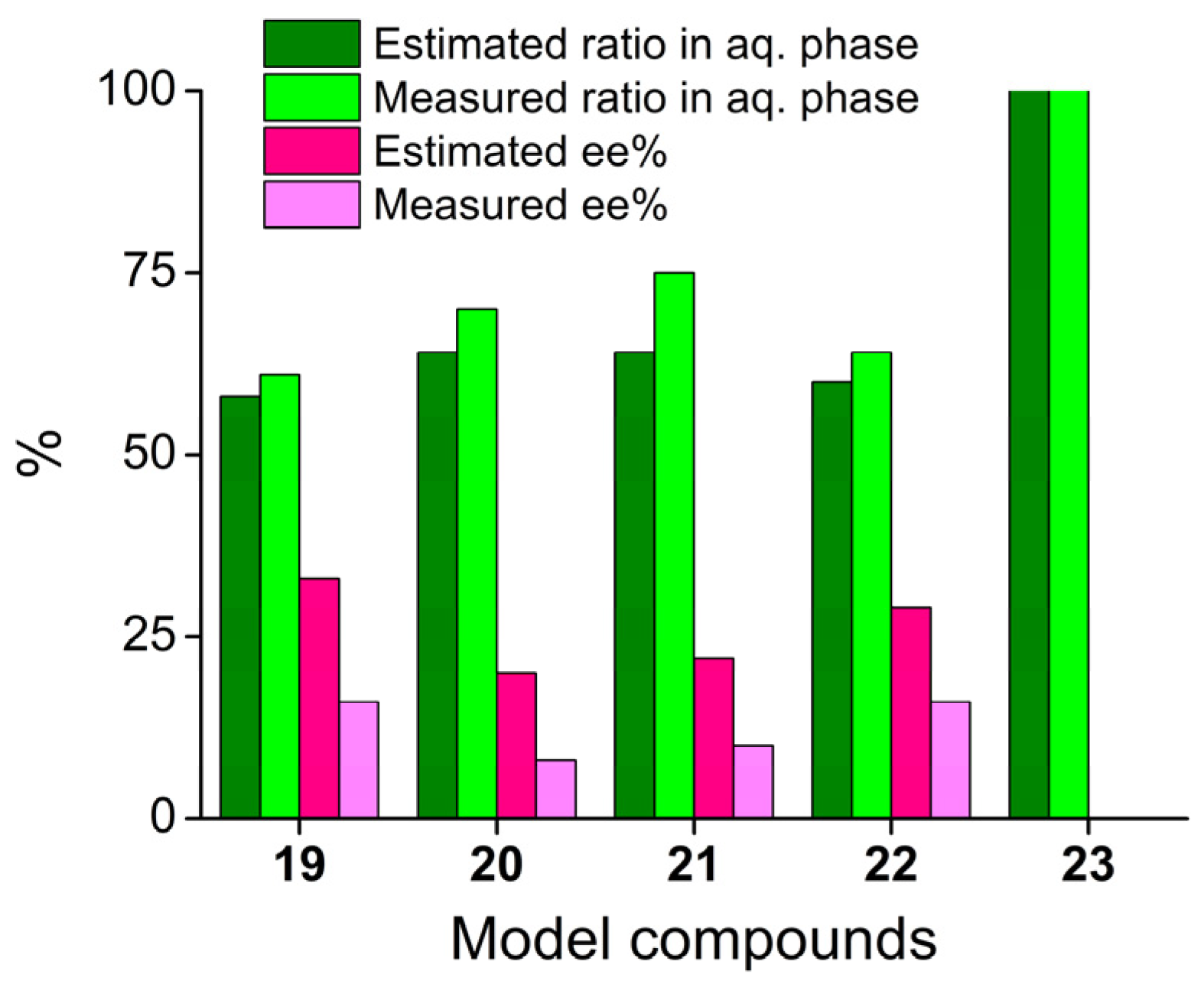
3.3. Validation of Enantioenrichment of Racemic Mixtures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Andersson, L.I. Molecular imprinting: Developments and applications in the analytical chemistry field. J. Chromatogr. B 2000, 745, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Tseng, W.B.; Hsieh, M.M.; Chiu, T.C.; Yu, P.L.; Chen, S.H. Enantioseparation of phenothiazines through capillary electrophoresis with solid phase extraction and polymer based stacking. J. Food Drug Anal. 2018, 26, 1171–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangelings, D.; Eeltink, S.; Vander Heyden, Y. Recent developments in liquid and supercritical fluid chromatographic enantioseparations. Handb. Anal. Sep. 2020, 8, 453–521. [Google Scholar] [CrossRef]
- Mazzoccanti, G.; Manetto, S.; Ricci, A.; Cabri, W.; Orlandin, A.; Catani, M.; Felletti, S.; Cavazzini, A.; Ye, M.; Ritchie, H.; et al. High-throughput enantioseparation of Nα–fluorenylmethoxycarbonyl proteinogenic amino acids through fast chiral chromatography on zwitterionic-teicoplanin stationary phases. J. Chromatogr. A 2020, 1624, 461235–461245. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Fonseca, J.R.; Rock, T.M.; Krauss-Etschmann, S.; Schmitt-Kopplin, P. Enantioseparation and selective detection of D-amino acids by ultra-high-performance liquid chromatography/mass spectrometry in analysis of complex biological samples. J. Chromatogr. A 2014, 1324, 109–114. [Google Scholar] [CrossRef] [Green Version]
- Mochizuki, T.; Taniguchi, S.; Tsutsui, H.; Min, J.Z.; Inoue, K.; Todoroki, K.; Toyo’oka, T. Relative quantification of enantiomers of chiral amines by high-throughput LC–ESI-MS/MS using isotopic variants of light and heavy l-pyroglutamic acids as the derivatization reagents. Anal. Chim. Acta 2013, 773, 76–82. [Google Scholar] [CrossRef]
- Zeng, L.; Xu, R.; Laskar, D.B.; Kassel, D.B. Parallel supercritical fluid chromatography/mass spectrometry system for high-throughput enantioselective optimization and separation. J. Chromatogr. A 2007, 1169, 193–204. [Google Scholar] [CrossRef]
- Konya, Y.; Izumi, Y.; Hamase, K.; Bamba, T. Ultrafast simultaneous chiral analysis of native amino acid enantiomers using supercritical fluid chromatography/tandem mass spectrometry. J. Chromatogr. A 2022, 1677, 463305–463312. [Google Scholar] [CrossRef]
- Nakano, Y.; Taniguchi, M.; Fukusaki, E. High-sensitive liquid chromatography-tandem mass spectrometry-based chiral metabolic profiling focusing on amino acids and related metabolites. J. Biosci. Bioeng. 2019, 127, 520–527. [Google Scholar] [CrossRef]
- Nováková, L.; Douša, M. General screening and optimization strategy for fast chiral separations in modern supercritical fluid chromatography. Anal. Chim. Acta 2017, 950, 199–210. [Google Scholar] [CrossRef]
- Konya, Y.; Taniguchi, M.; Fukusaki, E. Novel high-throughput and widely-targeted liquid chromatography-time of flight mass spectrometry method for D-amino acids in foods. J. Biosci. Bioeng. 2017, 123, 126–133. [Google Scholar] [CrossRef]
- Zawatzky, K.; Biba, M.; Regalado, E.L.; Welch, C.J. MISER chiral supercritical fluid chromatography for high throughput analysis of enantiopurity. J. Chromatogr. A 2016, 1429, 374–379. [Google Scholar] [CrossRef]
- Taniguchi, M.; Konya, Y.; Nakano, Y.; Fukusaki, E. Investigation of storage time-dependent alterations of enantioselective amino acid profiles in kimchi using liquid chromatography-time of flight mass spectrometry. J. Biosci. Bioeng. 2017, 124, 414–418. [Google Scholar] [CrossRef]
- Zhang, M.; Ye, B.C. Colorimetric chiral recognition of enantiomers using the nucleotide-capped silver nanoparticles. Anal. Chem. 2011, 83, 1504–1509. [Google Scholar] [CrossRef]
- Wei, W.; Wu, L.; Xu, C.; Ren, J.; Qu, X. A general approach using spiroborate reversible cross-linked Au nanoparticles for visual high-throughput screening of chiral vicinal diols. Chem. Sci. 2013, 4, 1156–1162. [Google Scholar] [CrossRef]
- Sancho, R.; Minguillón, C. The chromatographic separation of enantiomers through nanoscale design. Chem. Soc. Rev. 2009, 38, 797–805. [Google Scholar] [CrossRef]
- Chen, C.; Liu, W.; Hong, T. Novel approaches for biomolecule immobilization in microscale systems. Analyst 2019, 144, 3912–3924. [Google Scholar] [CrossRef]
- Lynen, F.; Saveedra, L.; Nickerson, B.; Sandra, P. Evaluation of a multiarray system for pharmaceutical analysis by microemulsion electrokinetic chromatography. Talanta 2011, 84, 724–729. [Google Scholar] [CrossRef]
- Liu, C.M.; Liang, R.P.; Wang, X.N.; Wang, J.W.; Qiu, J.D. A versatile polydopamine platform for facile preparation of protein stationary phase for chip-based open tubular capillary electrochromatography enantioseparation. J. Chromatogr. A 2013, 1294, 145–151. [Google Scholar] [CrossRef]
- Qu, P.; Lei, J.; Sheng, J.; Zhang, L.; Ju, H. Simultaneous multiple enantioseparation with a one-pot imprinted microfluidic channel by microchip capillary electrochromatography. Analyst 2011, 136, 920–926. [Google Scholar] [CrossRef]
- Wang, X.N.; Liang, R.P.; Meng, X.Y.; Qiu, J.D. One-step synthesis of mussel-inspired molecularly imprinted magnetic polymer as stationary phase for chip-based open tubular capillary electrochromatography enantioseparation. J. Chromatogr. A 2014, 1362, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Qu, P.; Zhang, L.; Sheng, J.; Lei, J.; Ju, H. Convenient enantioseparation by monolithic imprinted capillary clamped in a chip with electrochemical detection. Electrophoresis 2011, 32, 1522–1529. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, R.; Liu, J.; Ma, X.; Xiao, Y.; Wang, Y. Nacre-like ultra-robust supramolecular-functionalized graphene oxide membrane for bifunctional separation. Carbon 2021, 184, 618–626. [Google Scholar] [CrossRef]
- Meng, C.; Sheng, Y.; Chen, Q.; Tan, H.; Liu, H. Exceptional chiral separation of amino acid modified graphene oxide membranes with high-flux. J. Membr. Sci. 2017, 526, 25–31. [Google Scholar] [CrossRef]
- Meng, C.; Chen, Q.; Li, X.; Liu, H. Controlling covalent functionalization of graphene oxide membranes to improve enantioseparation performances. J. Membr. Sci. 2019, 582, 83–90. [Google Scholar] [CrossRef]
- Gunderson, S.S.; Brower, W.S.; O’Dell, J.L.; Lightfoot, E.N. Design of membrane cascades. Sep. Sci. Technol. 2007, 42, 2121–2142. [Google Scholar] [CrossRef]
- Choi, H.J.; Ahn, Y.H.; Koh, D.Y. Enantioselective mixed matrix membranes for chiral resolution. Membranes 2021, 11, 279. [Google Scholar] [CrossRef]
- Choi, H.J.; Koh, D.Y. Homochiral metal-organic framework based mixed matrix membrane for chiral resolution. Membranes 2022, 12, 357. [Google Scholar] [CrossRef]
- Li, X.; Tong, X.; Chen, Q.; Liu, H. Size effect of graphene oxide sheets on enantioseparation performances in membrane separation. Colloids Surf. A Physicochem. Eng. Asp. 2021, 618, 126464–126472. [Google Scholar] [CrossRef]
- Losacco, G.L.; Wang, H.; Haidar Ahmad, I.A.; DaSilva, J.; Makarov, A.A.; Mangion, I.; Gasparrini, F.; Lämmerhofer, M.; Armstrong, D.W.; Regalado, E.L. Enantioselective UHPLC screening combined with in silico modeling for streamlined development of ultrafast enantiopurity assays. Anal. Chem. 2021, 94, 1804–1812. [Google Scholar] [CrossRef]
- Hassan, D.S.; Kariapper, F.S.; Lynch, C.C.; Wolf, C. Accelerated asymmetric reaction screening with optical assays. Synthesis 2022, 54, 2527–2538. [Google Scholar] [CrossRef]
- Shcherbakova, E.G.; Minami, T.; Brega, V.; James, T.D.; Anzenbacher, P., Jr. Determination of enantiomeric excess in amine derivatives with molecular self-assemblies. Angew. Chem. 2015, 127, 7236–7239. [Google Scholar] [CrossRef] [Green Version]
- Nayek, A.; Ghosh, S. Enantiospecific synthesis of (+)-herbertene. Tetrahedron Lett. 2002, 43, 1313–1315. [Google Scholar] [CrossRef]
- Tan, Q.; Wang, Y.; Li, D.; Wen, J.; You, T. A novel and efficient synthesis of camphorquinone from camphoric acid. Synthesis 2003, 2003, 1869–1871. [Google Scholar] [CrossRef]
- Richers, J.; Heilmann, M.; Drees, M.; Tiefenbacher, K. Synthesis of lactones via C–H functionalization of nonactivated C(sp3)–H bonds. Org. Lett. 2016, 18, 6472–6475. [Google Scholar] [CrossRef]
- Hintermann, L.; Broggini, D.; Togni, A. Titanium lewis acids for asymmetric catalysis: Synthesis and structural characterization of dichloro[diolato(2−)-κO,κO′]bis (solvent)titanium([TiCl2(diolato)(solvent)2]) complexes. Helv. Chim. Acta 2002, 85, 1597–1612. [Google Scholar] [CrossRef]
- Bellis, E.; Kokotos, G. 4-Substituted prolines as organocatalysts for aldol reactions. Tetrahedron 2005, 61, 8669–8676. [Google Scholar] [CrossRef]
- Tian, J.; Gao, W.C.; Zhou, D.M.; Zhang, C. Recyclable hypervalent iodine(III) reagent iodosodilactone as an efficient coupling reagent for direct esterification, amidation, and peptide coupling. Org. Lett. 2012, 14, 3020–3023. [Google Scholar] [CrossRef]
- Zhao, L.; Fang, L.; Xu, Y.; Liu, S.; He, Z.; Zhao, Y. Transdermal delivery of penetrants with differing lipophilicities using O-acylmenthol derivatives as penetration enhancers. Eur. J. Pharm. Biopharm. 2008, 69, 199–213. [Google Scholar] [CrossRef]
- Zhao, L.; Li, Y.; Wang, C.; Zhuang, P.; Zheng, L.; Wang, H. D-Menthol Fatty Acid Ester Derivative Preparation Method and Application. Patent No. CN106631789, 18 October 2016. [Google Scholar]
- Chen, C.S.; Wu, S.H.; Girdaukas, G.; Sih, C.J. Quantitative analyses of biochemical kinetic resolution of enantiomers. 2. Enzyme-catalyzed esterifications in water-organic solvent biphasic systems. J. Am. Chem. Soc. 1987, 109, 2812–2817. [Google Scholar] [CrossRef]
- Kozma, D.; Böcskei, Z.; Kassai, C.; Simon, K.; Fogassy, E. Optical resolution of racemic alcohols by diastereoisomeric complex formation with O,O′-dibenzoyl-(2R,3R)-tartaric acid; The crystal structure of the (–)-(1R,2S,5R)-menthol O,O′-dibenzoyl-(2R,3R)-tartaric acid complex. Chem. Comm. 1996, 1, 753–754. [Google Scholar] [CrossRef]
- Kassai, C.; Juvancz, Z.; Bálint, J.; Fogassy, E.; Kozma, D. Optical resolution of racemic alcohols via diastereoisomeric supramolecular compound formation with O,O′-dibenzoyl-(2R,3R)-tartaric acid. Tetrahedron 2000, 56, 8355–8359. [Google Scholar] [CrossRef]
- Tan, B.; Luo, G.; Wang, J. Enantioseparation of amino acids by co-extractants with di(2-ethylhexyl)phosphoric acid and tartaric acid derivatives. Tetrahedron Asymmetry 2006, 17, 883–891. [Google Scholar] [CrossRef]
- Varga, B.; Bagi, P. Preparation of enantiomerically enriched P-stereogenic dialkyl-arylphosphine oxides via coordination mediated optical resolution. Symmetry 2020, 12, 215. [Google Scholar] [CrossRef] [Green Version]
- Parsons, A.T.; Caille, S.; Caporini, M.A.; Griffin, D.J.; Lovette, M.A.; Powazinik IV, W.; St-Pierre, G. Axial chirality in the sotorasib drug substance, Part 1: Development of a classical resolution to prepare an atropisomerically pure sotorasib intermediate. Org. Process Res. Dev. 2022, 26, 2629–2635. [Google Scholar] [CrossRef]
- Howell, B.A.; Sun, W. Biobased plasticizers from tartaric acid, an abundantly available, renewable material. Ind. Eng. Chem. Res. 2018, 57, 15234–15242. [Google Scholar] [CrossRef]
- Shili, Q.; Yangyang, S.; Xudong, H.; Hongtao, C.; Lidi, G.; Zhongyu, H.; Dongsheng, Z.; Xinyao, L.; Sibing, Z. Chiral fluorescence recognition of glutamine enantiomers by a modified Zr-based MOF based on solvent-assisted ligand incorporation. RSC Adv. 2021, 11, 37584–37594. [Google Scholar] [CrossRef]
- Ma, X.L.; Wang, Z.X.; He, X.; Shao, M.; Li, M.X. 2D double-layered dibenzoyl-tartrate chiral coordination polymer containing [Mn4L2(bpp)4] tetrahedral cage. Inorg. Chem. Commun. 2018, 92, 131–135. [Google Scholar] [CrossRef]
- Rossi, P.; Ceccarelli, J.; Milazzo, S.; Paoli, P.; Morais Missina, J.; Ciattini, S.; Ienco, A.; Tuci, G.; Valleri, M.; Giovannoni, M.P.; et al. Nonsteroidal anti-inflammatory drugs-1-phenylethylamine diastereomeric salts: A systematic solid-state investigation. Cryst. Growth Des. 2021, 21, 6947–6960. [Google Scholar] [CrossRef]
- Tumanova, N.; Seiler, V.; Tumanov, N.; Robeyns, K.; Filinchuk, Y.; Wouters, J.; Leyssens, T. Structural analysis of D-phenylglycinamide salts uncovers potential pitfalls in chiral resolution via diastereomeric salt formation. Cryst. Growth Des. 2019, 19, 3652–3659. [Google Scholar] [CrossRef]
- Assali, M.; Zaid, A.N.; Abualhasan, M.; Jaradat, N.; Tarayra, R.; Hamdan, A.; Ardah, R. Synthesis of Naproxen pro-drugs for enhanced transdermal absorption. J. Chem. Pharm. Res. 2014, 6, 1–4. [Google Scholar]
- Kalakuntala, R.; Suranani, S. Kinetics on propionic acid catalytic esterification with n-butanol over SBA-15 (Santa Barbara amorphous-15 material) catalyst. Mater. Today Proc. 2021, 47, 4814–4819. [Google Scholar] [CrossRef]
- Duan, Y.; Du, Z.; Yao, Y.; Li, R.; Wu, D. Effect of molecular sieves on lipase-catalyzed esterification of rutin with stearic acid. J. Agric. Food Chem. 2006, 54, 6219–6225. [Google Scholar] [CrossRef]
- Riddick, J.A.; Bunger, W.B.; Sakano, T.K. Organic Solvents: Physical Properties and Methods of Purification, 4th ed.; Wiley-Interscience: New York, NY, USA, 1986; pp. 1344–1400. ISBN 978-047108467-9. [Google Scholar]
- Minecka, A.; Kaminska, E.; Tarnacka, M.; Grudzka-Flak, I.; Bartoszek, M.; Wolnica, K.; Dulski, M.; Kaminski, K.; Paluch, M. Impact of intermolecular interactions, dimeric structures on the glass forming ability of naproxen, and a series of its derivatives. Mol. Pharm. 2018, 15, 4764–4776. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model System | DE,l,aq. | DE,p,aq. | β(p)1 | Significant? 2 | Estimated eeaq 3 (%) |
|---|---|---|---|---|---|
| NAP-6 & 19 | 0.56 | 0.30 | 1.89 (R) | ✓ | 22 |
| NAP-6(M) 4 & 19 | 0.35 | 0.33 | 0.95 (R) | ✗ | 0 |
| NAP-6 & 20 | 0.37 | 0.23 | 1.58 (R) | ✓ | 17 |
| NAP-6(M) 4 & 20 | 0.16 | 0.11 | 1.47 (R) | ✗ | 0 |
| NAP-6 & 21 | 2.85 | 2.00 | 1.40 (R) | ✓ | 5 |
| NAP-6(M) 4 & 21 | 0.52 | 0.52 | 1.00 (R) | ✗ | 0 |
| NAP-6 & 22 | 0.64 | 0.47 | 1.36 (1R,2S) | ✓ | 10 |
| NAP-6(M) 4 & 22 | 0.32 | 0.27 | 1.19 (1R,2S) | ✗ | 0 |
| NAP-6 & 23 | 13.3 | 10.1 | 1.31 (R) | ✗ | 0 |
| NAP-6(M) 4 & 23 | 0.82 | 0.75 | 0.92 (R) | ✗ | 0 |
| Model System | DE,l,aq. | DE,p,aq. | β(p)1 | Significant? 2 | Estimated eeaq 3 (%) |
|---|---|---|---|---|---|
| CA-12 and 19 | 1.17 | 0.89 | 1.3 (R) | ✓ | 7 |
| CA-12 and 20 | 3.35 | 3.00 | 1.12 (R) | ✗ | 0 |
| CA-12 and 21 | 99.0 | 32.3 | 3.10 (S) | ✗ | 0 |
| CA-12 and 22 | 0.45 | 0.41 | 1.10 (1R,2S) | ✗ | 0 |
| CA-12 and 23 | ~100 | ~100 | 1.00 (-) | ✗ | 0 |
| MEN-10 and 19 | 3.35 | 1.94 | 1.72 (R) | ✓ | 8 |
| MEN-10 and 20 | 5.25 | 3.17 | 1.66 (R) | ✓ | 5 |
| MEN-10 and 21 | 99.0 | 6.14 | 16.1 (R) | ✓ | 7 |
| MEN-10 and 22 | ~100 | ~100 | 1.00 (-) | ✗ | 0 |
| MEN-10 and 23 | ~100 | ~100 | 1.00 (-) | ✗ | 0 |
| DBTA-12 and 19 | 3.35 | 0.64 | 5.24 (R) | ✓ | 33 |
| DBTA-12 and 20 | 3.35 | 1.04 | 3.22 (S) | ✓ | 20 |
| DBTA-12 and 21 | 3.55 | 1.00 | 3.55 (R) | ✓ | 22 |
| DBTA-12 and 22 | 3.35 | 0.72 | 4.62 (1S,2R) | ✓ | 29 |
| DBTA-12 and 23 | ~100 | ~100 | 1.00(-) | ✗ | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jávor, B.; Vezse, P.; Golcs, Á.; Huszthy, P.; Tóth, T. Enantiodiscriminating Lipophilic Liquid Membrane-Based Assay for High-Throughput Nanomolar Enantioenrichment of Chiral Building Blocks. Membranes 2023, 13, 94. https://doi.org/10.3390/membranes13010094
Jávor B, Vezse P, Golcs Á, Huszthy P, Tóth T. Enantiodiscriminating Lipophilic Liquid Membrane-Based Assay for High-Throughput Nanomolar Enantioenrichment of Chiral Building Blocks. Membranes. 2023; 13(1):94. https://doi.org/10.3390/membranes13010094
Chicago/Turabian StyleJávor, Bálint, Panna Vezse, Ádám Golcs, Péter Huszthy, and Tünde Tóth. 2023. "Enantiodiscriminating Lipophilic Liquid Membrane-Based Assay for High-Throughput Nanomolar Enantioenrichment of Chiral Building Blocks" Membranes 13, no. 1: 94. https://doi.org/10.3390/membranes13010094