
Aptamer-Modified Erythrocyte Membrane-Coated pH-Sensitive Nanoparticles for c-Met-Targeted Therapy of Glioblastoma Multiforme

 , and
, and 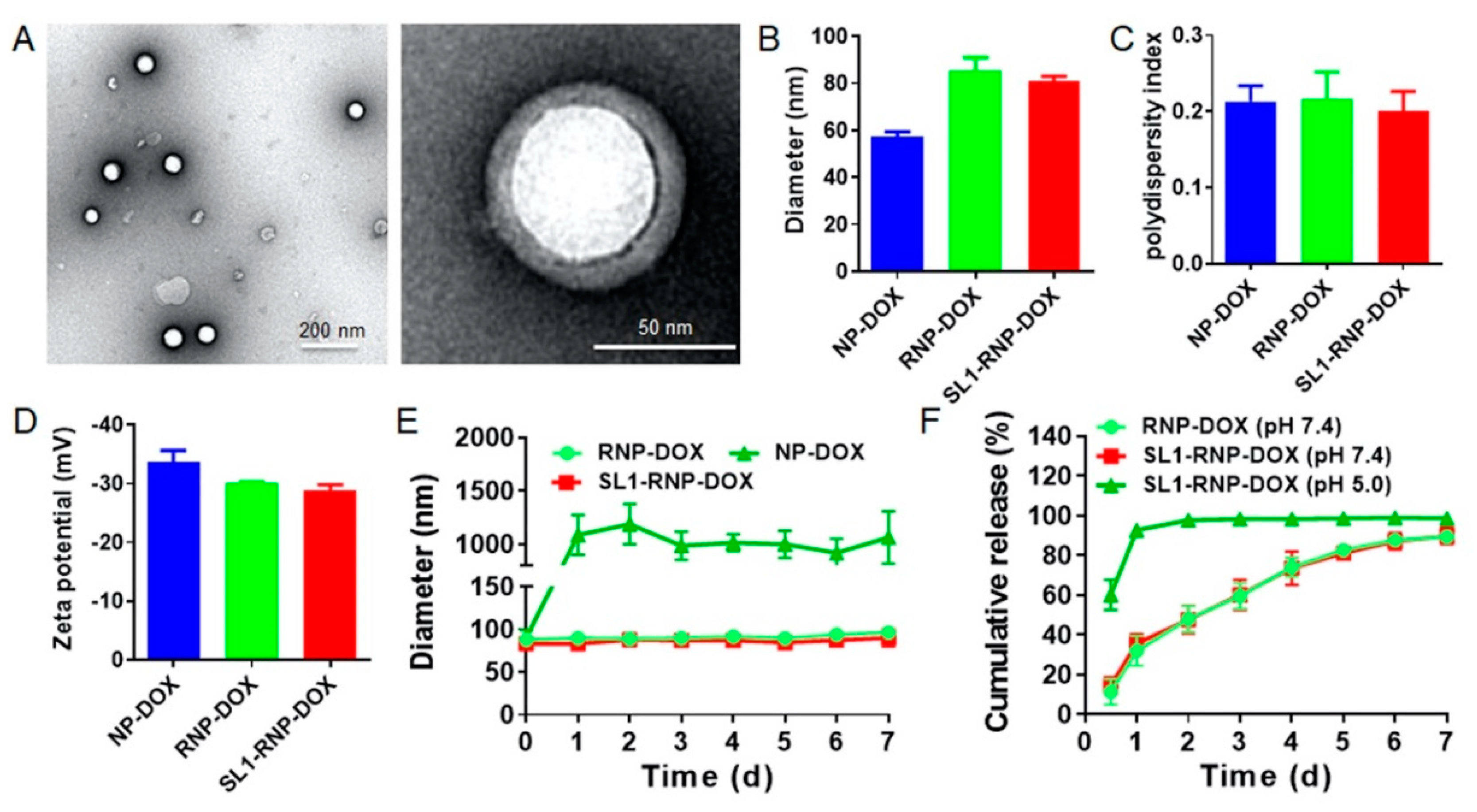{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cells and Animals
2.3. Preparation of Nanoparticle Cores
2.4. Preparation of SL1-Modified RBC Membranes
2.5. Preparation and Characterization of SL1-RNP-DOX
2.6. Drug Loading and Drug Release
2.7. In Vitro Cytotoxicity Assay
2.8. Cell Apoptosis Assay
2.9. In Vitro Cellular Uptake
2.10. In Vivo Fluorescence Imaging and Biodistribution
2.11. Anti-GBM Efficacy in Vivo
2.12. Statistical Analysis
3. Results
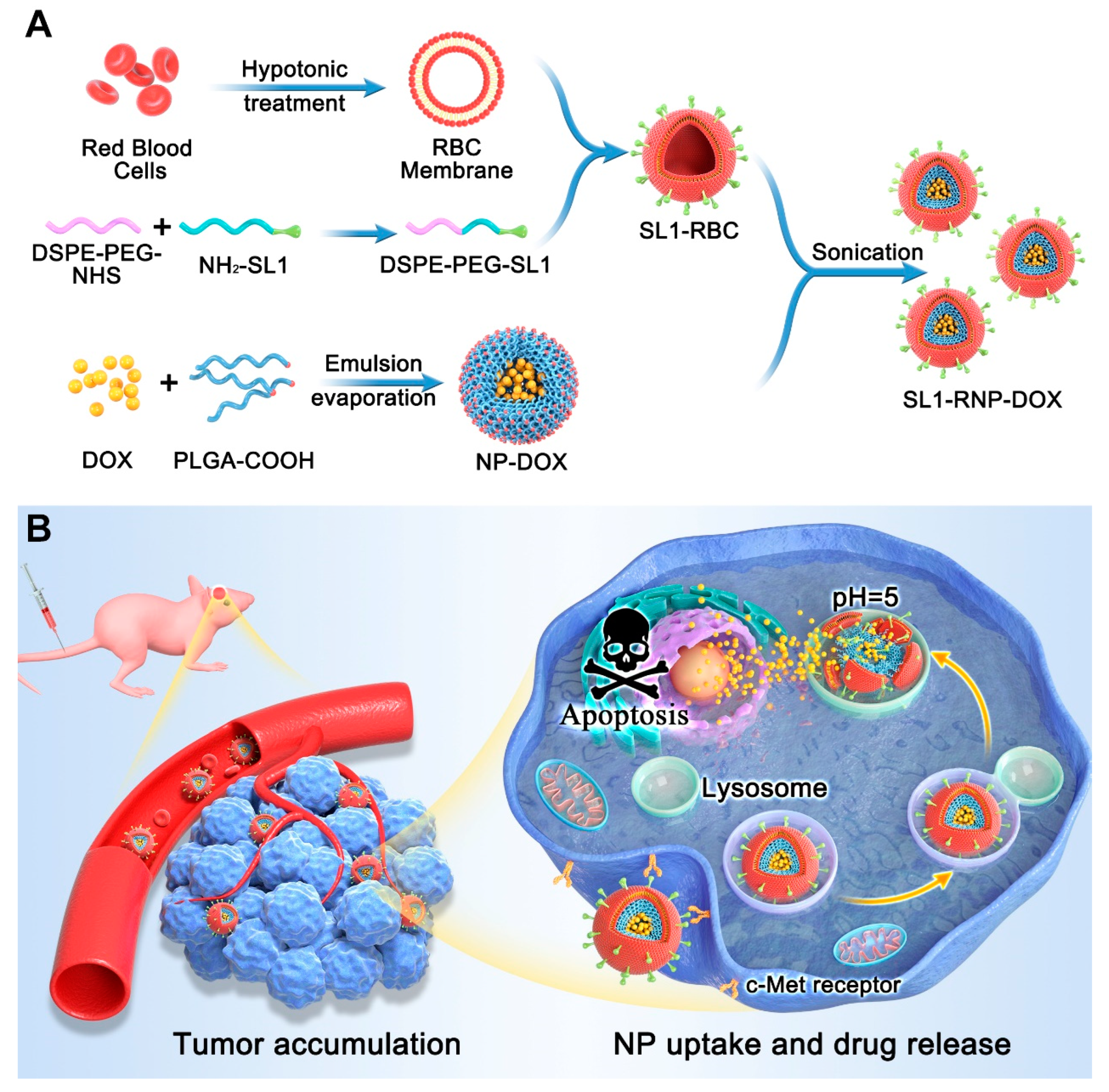
3.1. Characterization of SL1-RNP-DOX
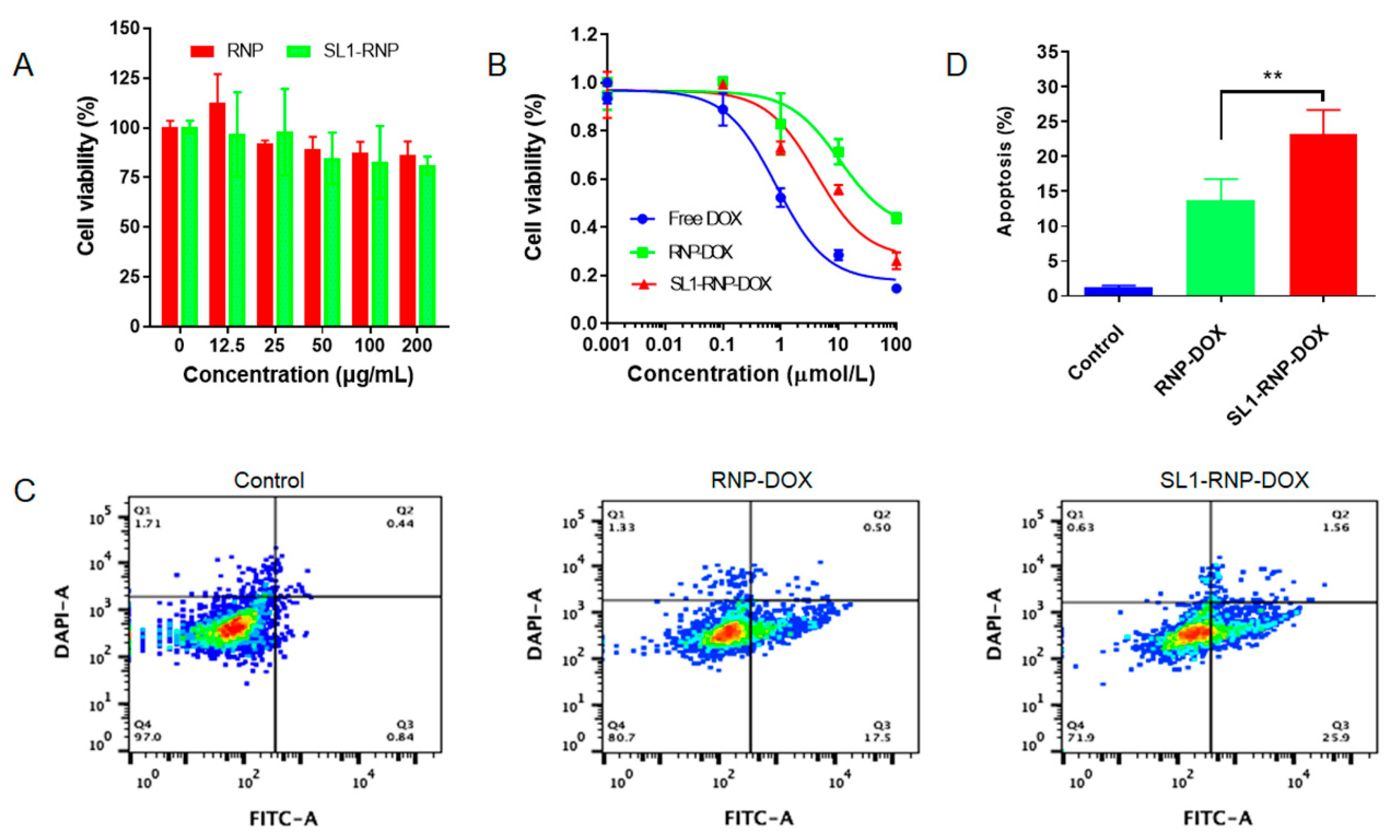
3.2. Cytotoxicity Assay and Cell Apoptosis Assay
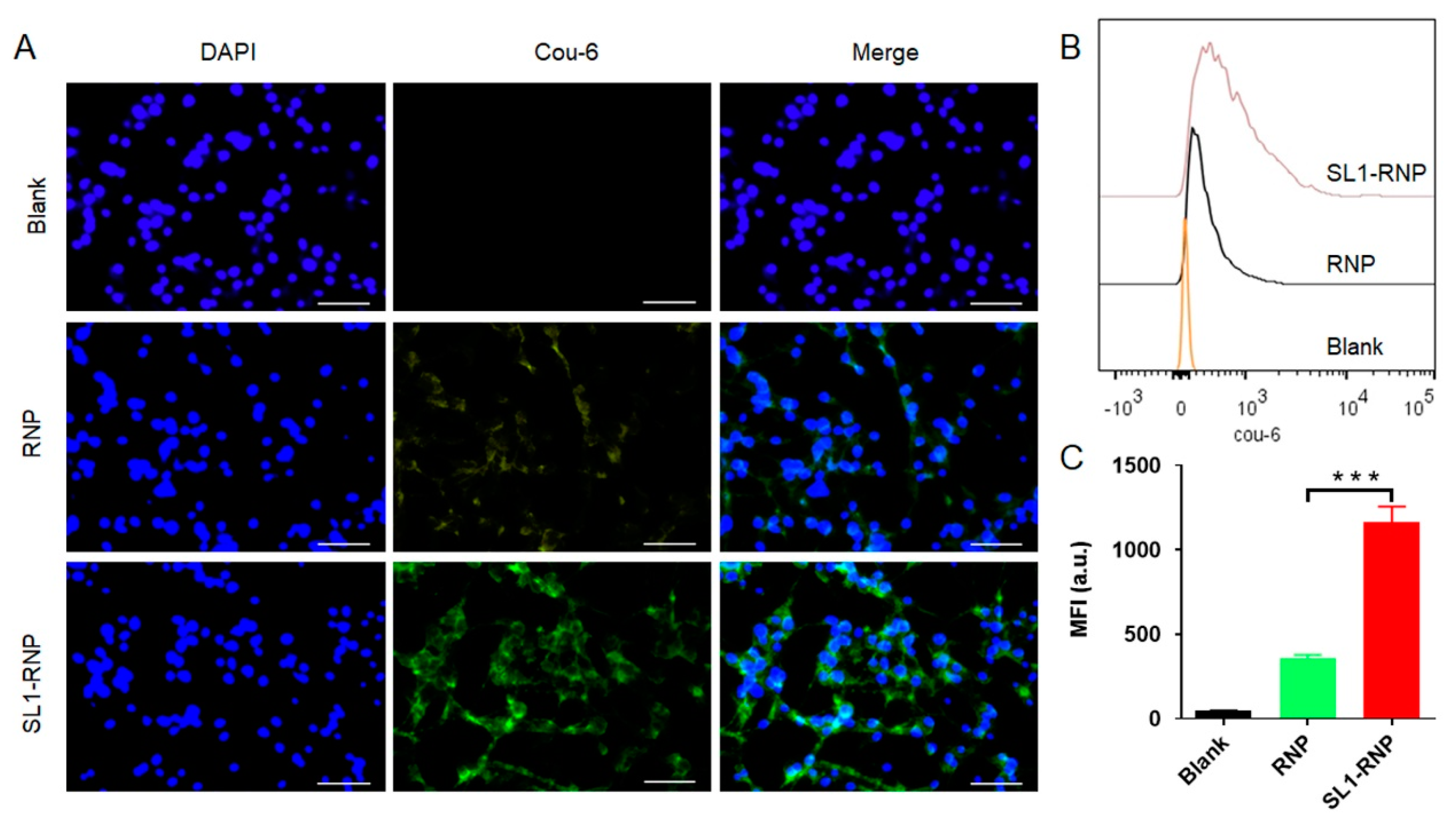
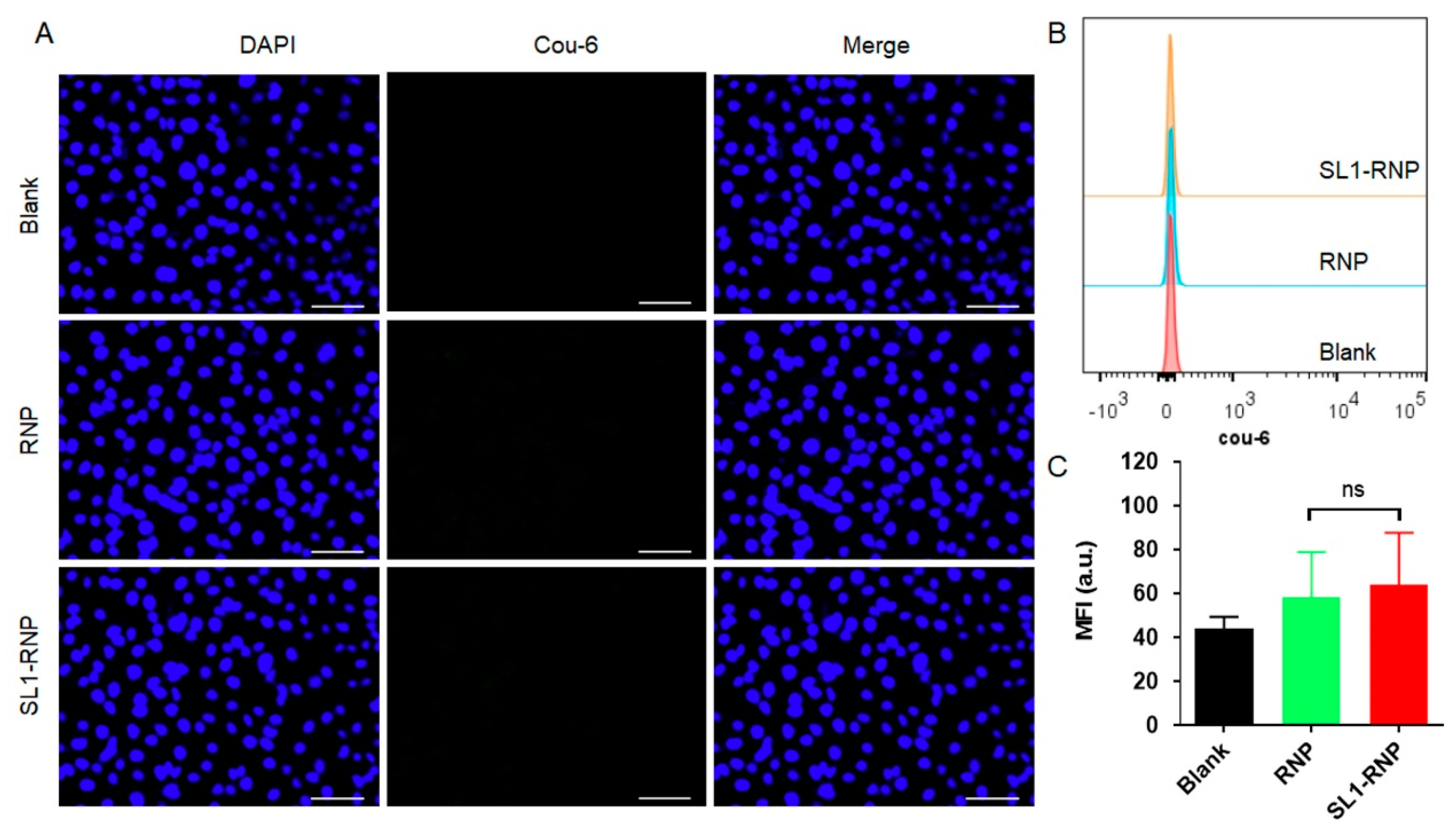
3.3. Cellular Uptake Assay
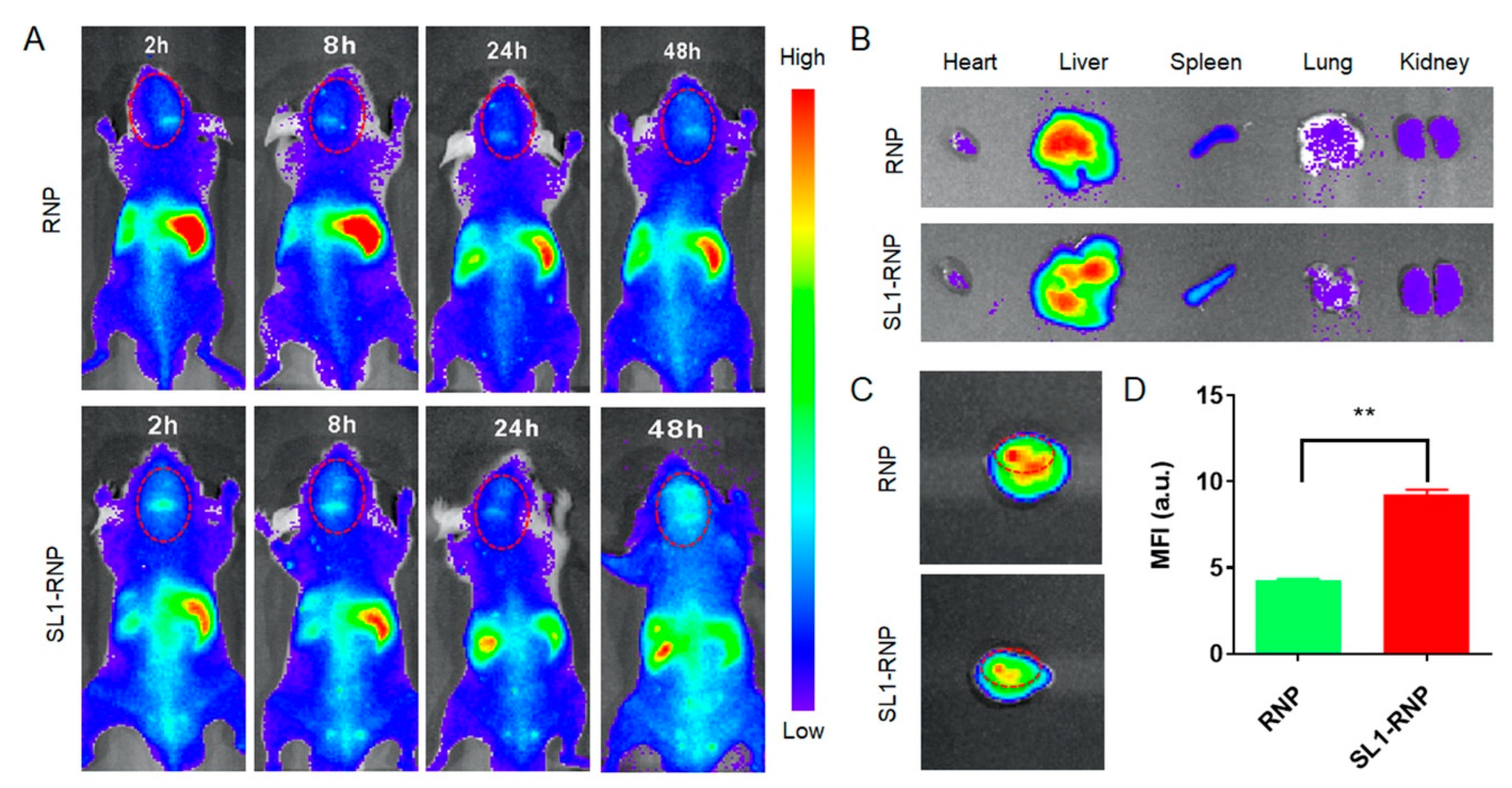
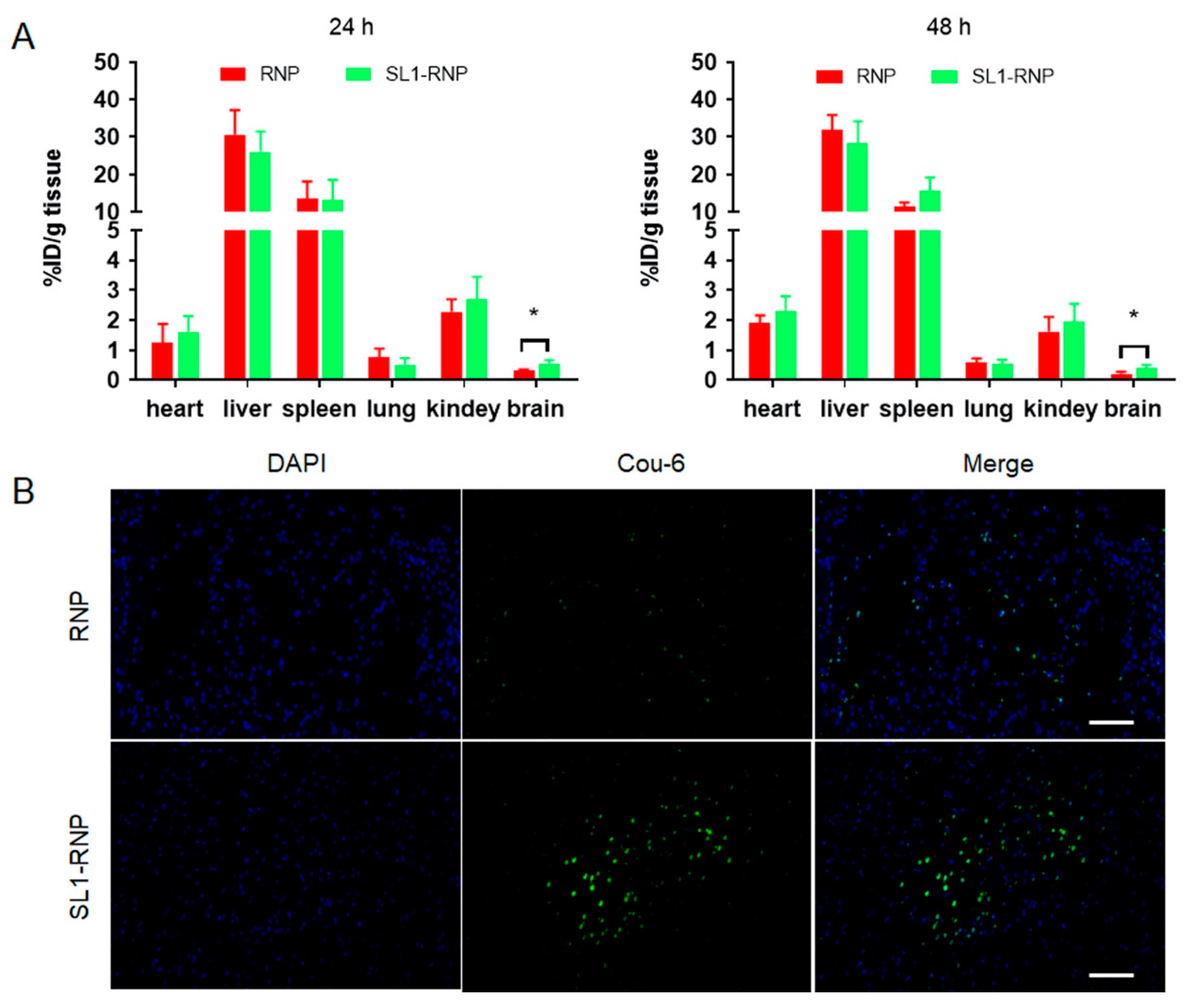
3.4. In Vivo Fluorescence Imaging and Biodistribution
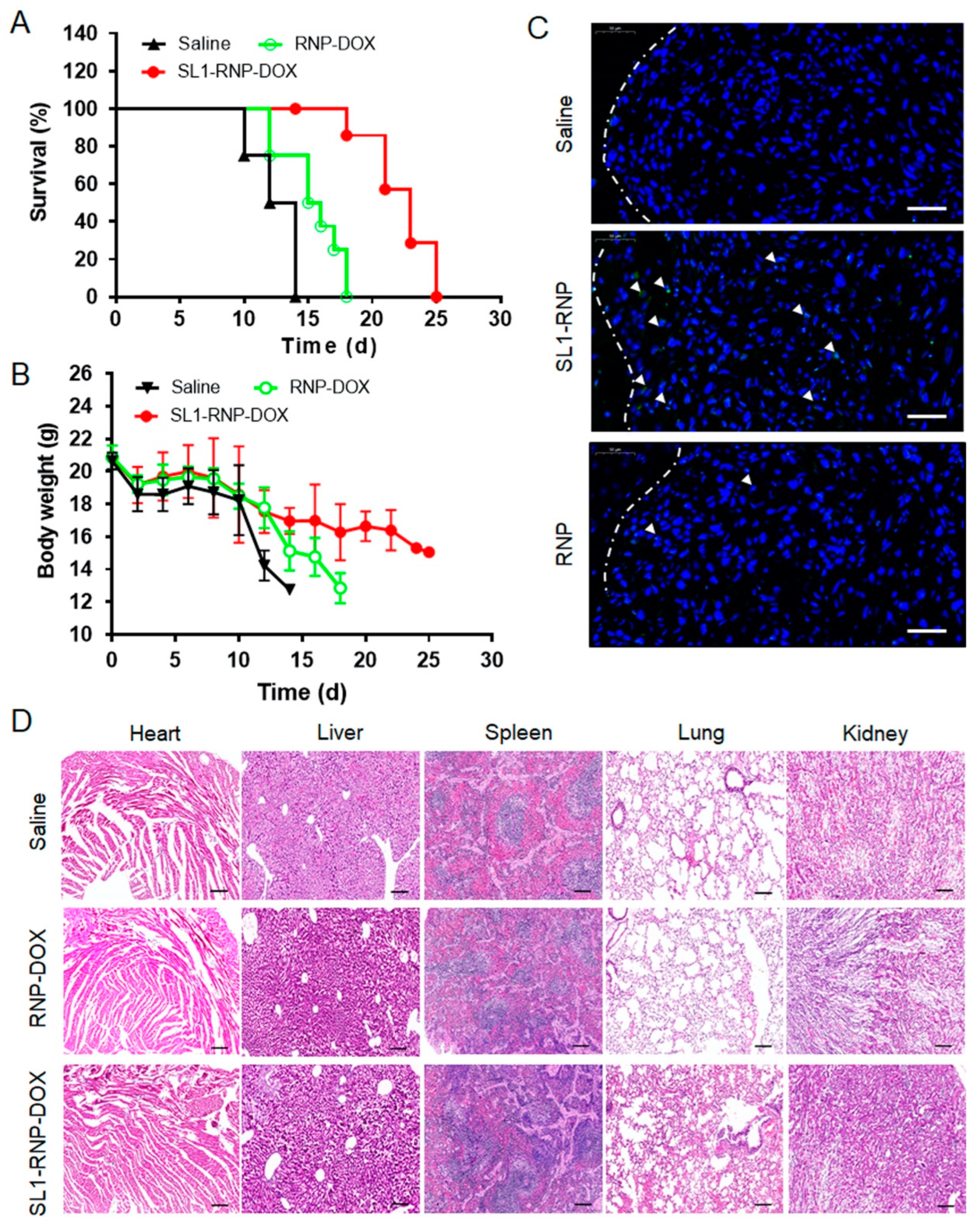
3.5. In Vivo Anti-GBM Efficacy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tan, A.C.; Ashley, D.M.; Lopez, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Klockow, J.L.; Zhang, M.; Lafortune, F.; Chang, E.; Jin, L.; Wu, Y.; Daldrup-Link, H.E. Glioblastoma multiforme (GBM): An overview of current therapies and mechanisms of resistance. Pharmacol. Res. 2021, 171, 105780. [Google Scholar] [CrossRef] [PubMed]
- Da Ros, M.; De Gregorio, V.; Iorio, A.L.; Giunti, L.; Guidi, M.; de Martino, M.; Genitori, L.; Sardi, I. Glioblastoma Chemoresistance: The Double Play by Microenvironment and Blood-Brain Barrier. Int. J. Mol. Sci. 2018, 19, 2879. [Google Scholar] [CrossRef] [Green Version]
- Awad, A.W.; Karsy, M.; Sanai, N.; Spetzler, R.; Zhang, Y.; Xu, Y.; Mahan, M.A. Impact of removed tumor volume and location on patient outcome in glioblastoma. J. Neurooncol. 2017, 135, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Julka, P.K.; Sharma, D.N.; Mallick, S.; Gandhi, A.K.; Joshi, N.; Rath, G.K. Postoperative treatment of glioblastoma multiforme with radiation therapy plus concomitant and adjuvant temozolomide: A mono-institutional experience of 215 patients. J. Cancer Res. Ther. 2013, 9, 381–386. [Google Scholar] [PubMed]
- Wei, X.; Chen, X.; Ying, M.; Lu, W. Brain tumor-targeted drug delivery strategies. Acta Pharm. Sin. B 2014, 4, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alyautdin, R.; Khalin, I.; Nafeeza, M.I.; Haron, M.H.; Kuznetsov, D. Nanoscale drug delivery systems and the blood-brain barrier. Int. J. Nanomed. 2014, 9, 795–811. [Google Scholar]
- Cheng, Y.; Dai, Q.; Morshed, R.A.; Fan, X.; Wegscheid, M.L.; Wainwright, D.A.; Han, Y.; Zhang, L.; Auffinger, B.; Tobias, A.L.; et al. Blood-brain barrier permeable gold nanoparticles: An efficient delivery platform for enhanced malignant glioma therapy and imaging. Small 2014, 10, 5137–5150. [Google Scholar] [CrossRef]
- Liu, X.; Du, C.; Li, H.; Jiang, T.; Luo, Z.; Pang, Z.; Geng, D.; Zhang, J. Engineered superparamagnetic iron oxide nanoparticles (SPIONs) for dual-modality imaging of intracranial glioblastoma via EGFRvIII targeting. Beilstein J. Nanotechnol. 2019, 10, 1860–1872. [Google Scholar] [CrossRef]
- Aryal, S.; Hu, C.M.; Fang, R.H.; Dehaini, D.; Carpenter, C.; Zhang, D.E.; Zhang, L. Erythrocyte membrane-cloaked polymeric nanoparticles for controlled drug loading and release. Nanomedicine 2013, 8, 1271–1280. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.M.; Fang, R.H.; Zhang, L. Erythrocyte-inspired delivery systems. Adv. Healthc. Mater. 2012, 1, 537–547. [Google Scholar] [CrossRef]
- Hu, C.M.; Zhang, L.; Aryal, S.; Cheung, C.; Fang, R.H.; Zhang, L. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc. Natl. Acad. Sci. USA 2011, 108, 10980–10985. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, J.; Chen, W.; Angsantikul, P.; Spiekermann, K.A.; Fang, R.H.; Gao, W.; Zhang, L. Erythrocyte membrane-coated nanogel for combinatorial antivirulence and responsive antimicrobial delivery against Staphylococcus aureus infection. J. Control. Release 2017, 263, 185–191. [Google Scholar] [CrossRef]
- Acharya, S.; Sahoo, S.K. PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv. Drug Deliv. Rev. 2011, 63, 170–183. [Google Scholar] [CrossRef]
- Seiwert, T.Y.; Jagadeeswaran, R.; Faoro, L.; Janamanchi, V.; Nallasura, V.; El Dinali, M.; Yala, S.; Kanteti, R.; Cohen, E.E.; Lingen, M.W.; et al. The MET receptor tyrosine kinase is a potential novel therapeutic target for head and neck squamous cell carcinoma. Cancer Res. 2009, 69, 3021–3031. [Google Scholar] [CrossRef] [Green Version]
- Ponzo, M.G.; Lesurf, R.; Petkiewicz, S.; O’Malley, F.P.; Pinnaduwage, D.; Andrulis, I.L.; Bull, S.B.; Chughtai, N.; Zuo, D.; Souleimanova, M.; et al. Met induces mammary tumors with diverse histologies and is associated with poor outcome and human basal breast cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 12903–12908. [Google Scholar] [CrossRef] [Green Version]
- Pierscianek, D.; Kim, Y.H.; Motomura, K.; Mittelbronn, M.; Paulus, W.; Brokinkel, B.; Keyvani, K.; Wrede, K.; Nakazato, Y.; Tanaka, Y.; et al. MET gain in diffuse astrocytomas is associated with poorer outcome. Brain Pathol. 2013, 23, 13–18. [Google Scholar] [CrossRef]
- Kong, D.S.; Song, S.Y.; Kim, D.H.; Joo, K.M.; Yoo, J.S.; Koh, J.S.; Dong, S.M.; Suh, Y.L.; Lee, J.I.; Park, K.; et al. Prognostic significance of c-Met expression in glioblastomas. Cancer 2009, 115, 140–148. [Google Scholar] [CrossRef]
- Ueki, R.; Sando, S. A DNA aptamer to c-Met inhibits cancer cell migration. Chem. Commun. 2014, 50, 13131–13134. [Google Scholar] [CrossRef]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef]
- Jiang, F.; Liu, B.; Lu, J.; Li, F.; Li, D.; Liang, C.; Dang, L.; Liu, J.; He, B.; Badshah, S.A.; et al. Progress and Challenges in Developing Aptamer-Functionalized Targeted Drug Delivery Systems. Int. J. Mol. Sci. 2015, 16, 23784–23822. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zu, Y. A Highlight of Recent Advances in Aptamer Technology and Its Application. Molecules 2015, 20, 11959–11980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Lao, Y.H.; Phua, K.K.; Leong, K.W. Aptamer nanomedicine for cancer therapeutics: Barriers and potential for translation. ACS Nano 2015, 9, 2235–2254. [Google Scholar] [CrossRef]
- Ueki, R.; Ueki, A.; Kanda, N.; Sando, S. Oligonucleotide-Based Mimetics of Hepatocyte Growth Factor. Angew. Chem. Int. Ed. Engl. 2016, 55, 579–582. [Google Scholar] [CrossRef]
- Luk, B.T.; Fang, R.H.; Hu, C.M.; Copp, J.A.; Thamphiwatana, S.; Dehaini, D.; Gao, W.; Zhang, K.; Li, S.; Zhang, L. Safe and Immunocompatible Nanocarriers Cloaked in RBC Membranes for Drug Delivery to Treat Solid Tumors. Theranostics 2016, 6, 1004–1011. [Google Scholar] [CrossRef] [Green Version]
- Pang, Z.; Gao, H.; Yu, Y.; Guo, L.; Chen, J.; Pan, S.; Ren, J.; Wen, Z.; Jiang, X. Enhanced intracellular delivery and chemotherapy for glioma rats by transferrin-conjugated biodegradable polymersomes loaded with doxorubicin. Bioconjug. Chem. 2011, 22, 1171–1180. [Google Scholar] [CrossRef]
- He, Y.; Li, R.; Li, H.; Zhang, S.; Dai, W.; Wu, Q.; Jiang, L.; Zheng, Z.; Shen, S.; Chen, X.; et al. Erythroliposomes: Integrated Hybrid Nanovesicles Composed of Erythrocyte Membranes and Artificial Lipid Membranes for Pore-Forming Toxin Clearance. ACS Nano 2019, 13, 4148–4159. [Google Scholar] [CrossRef]
- Fang, R.H.; Hu, C.M.; Chen, K.N.; Luk, B.T.; Carpenter, C.W.; Gao, W.; Li, S.; Zhang, D.E.; Lu, W.; Zhang, L. Lipid-insertion enables targeting functionalization of erythrocyte membrane-cloaked nanoparticles. Nanoscale 2013, 5, 8884–8888. [Google Scholar] [CrossRef] [Green Version]
- Copp, J.A.; Fang, R.H.; Luk, B.T.; Hu, C.M.; Gao, W.; Zhang, K.; Zhang, L. Clearance of pathological antibodies using biomimetic nanoparticles. Proc. Natl. Acad. Sci. USA 2014, 111, 13481–13486. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Zhou, Y.; Chen, C.; Zhu, S.; Yan, X. Quantification of Available Ligand Density on the Surface of Targeted Liposomal Nanomedicines at the Single-Particle Level. ACS Nano 2022, 16, 6886–6897. [Google Scholar] [CrossRef]
- Pang, Z.; Lu, W.; Gao, H.; Hu, K.; Chen, J.; Zhang, C.; Gao, X.; Jiang, X.; Zhu, C. Preparation and brain delivery property of biodegradable polymersomes conjugated with OX26. J. Control. Release 2008, 128, 120–127. [Google Scholar] [CrossRef]
- Lu, W.; Zhang, Y.; Tan, Y.Z.; Hu, K.L.; Jiang, X.G.; Fu, S.K. Cationic albumin-conjugated pegylated nanoparticles as novel drug carrier for brain delivery. J. Control. Release 2005, 107, 428–448. [Google Scholar] [CrossRef]
- Alibolandi, M.; Ramezani, M.; Sadeghi, F.; Abnous, K.; Hadizadeh, F. Epithelial cell adhesion molecule aptamer conjugated PEG-PLGA nanopolymersomes for targeted delivery of doxorubicin to human breast adenocarcinoma cell line in vitro. Int. J. Pharm. 2015, 479, 241–251. [Google Scholar] [CrossRef]
- Malinovskaya, Y.; Melnikov, P.; Baklaushev, V.; Gabashvili, A.; Osipova, N.; Mantrov, S.; Ermolenko, Y.; Maksimenko, O.; Gorshkova, M.; Balabanyan, V.; et al. Delivery of doxorubicin-loaded PLGA nanoparticles into U87 human glioblastoma cells. Int. J. Pharm. 2017, 524, 77–90. [Google Scholar] [CrossRef]
- Abd Ellah, N.H.; Abouelmagd, S.A. Surface functionalization of polymeric nanoparticles for tumor drug delivery: Approaches and challenges. Expert Opin. Drug Deliv. 2017, 14, 201–214. [Google Scholar] [CrossRef]
- Zhai, L.; Luo, C.; Gao, H.; Du, S.; Shi, J.; Wang, F. A Dual pH-Responsive DOX-Encapsulated Liposome Combined with Glucose Administration Enhanced Therapeutic Efficacy of Chemotherapy for Cancer. Int. J. Nanomed. 2021, 16, 3185–3199. [Google Scholar] [CrossRef]
- He, H.; Li, Y.; Jia, X.R.; Du, J.; Ying, X.; Lu, W.L.; Lou, J.N.; Wei, Y. PEGylated Poly(amidoamine) dendrimer-based dual-targeting carrier for treating brain tumors. Biomaterials 2011, 32, 478–487. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Chen, Y.; Geng, D.; Li, H.; Jiang, T.; Luo, Z.; Wang, J.; Pang, Z.; Zhang, J. Aptamer-Modified Erythrocyte Membrane-Coated pH-Sensitive Nanoparticles for c-Met-Targeted Therapy of Glioblastoma Multiforme. Membranes 2022, 12, 744. https://doi.org/10.3390/membranes12080744
Liu X, Chen Y, Geng D, Li H, Jiang T, Luo Z, Wang J, Pang Z, Zhang J. Aptamer-Modified Erythrocyte Membrane-Coated pH-Sensitive Nanoparticles for c-Met-Targeted Therapy of Glioblastoma Multiforme. Membranes. 2022; 12(8):744. https://doi.org/10.3390/membranes12080744
Chicago/Turabian StyleLiu, Xianping, Yixin Chen, Daoying Geng, Haichun Li, Ting Jiang, Zimiao Luo, Jianhong Wang, Zhiqing Pang, and Jun Zhang. 2022. "Aptamer-Modified Erythrocyte Membrane-Coated pH-Sensitive Nanoparticles for c-Met-Targeted Therapy of Glioblastoma Multiforme" Membranes 12, no. 8: 744. https://doi.org/10.3390/membranes12080744