

Regulation of Papillary Muscle Contractility by NAD and Ammonia Interplay: Contribution of Ion Channels and Exchangers
 , , and
, , and 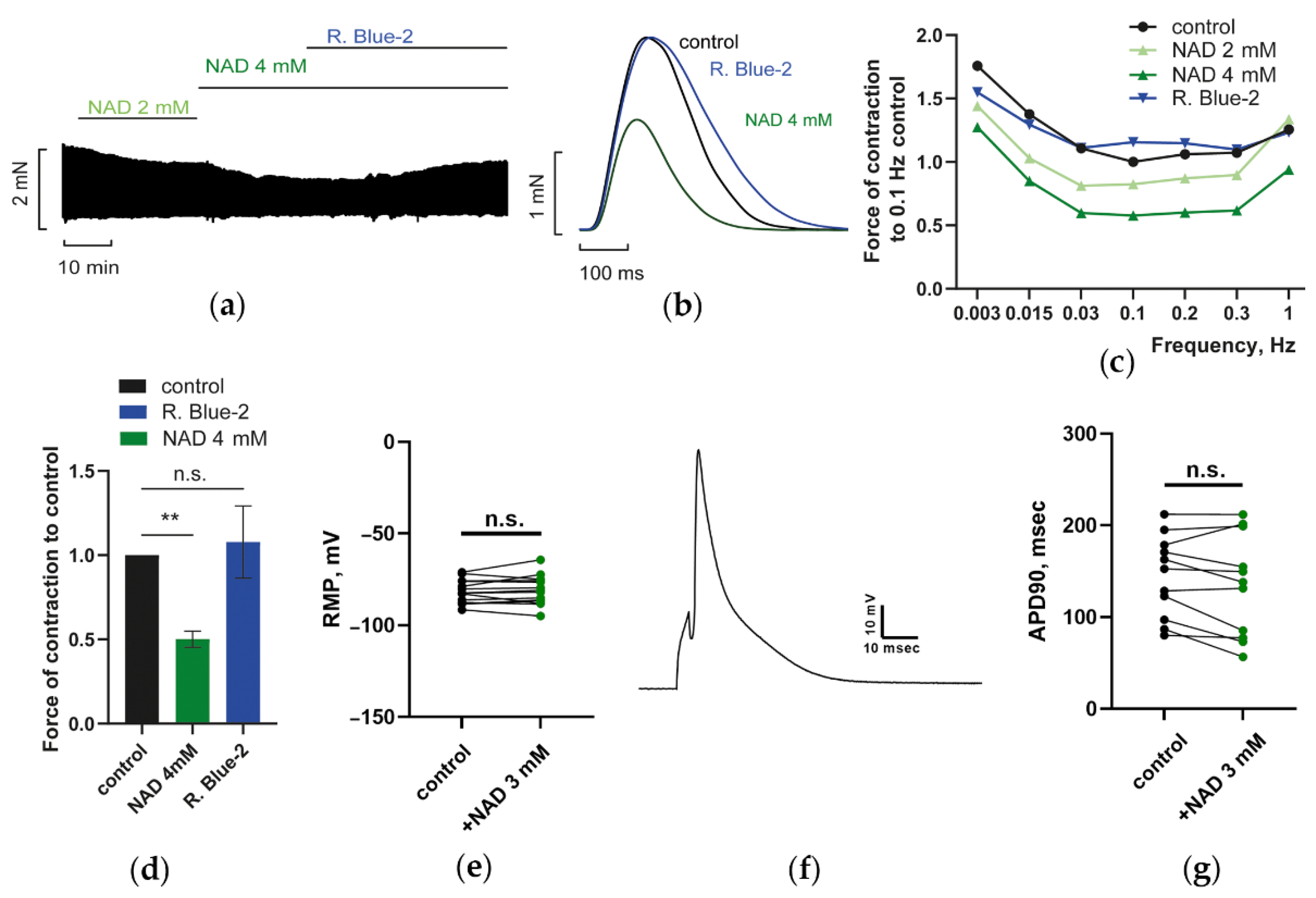{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Handling
2.2. Contractility of Papillary Muscles
2.3. Acute Isolation of Ventricular Cardiomyocytes
2.4. Whole-Cell Patch Clamp Recordings
2.5. Data Analysis and Statistics
3. Results
3.1. NAD and Ammonia Antagonism
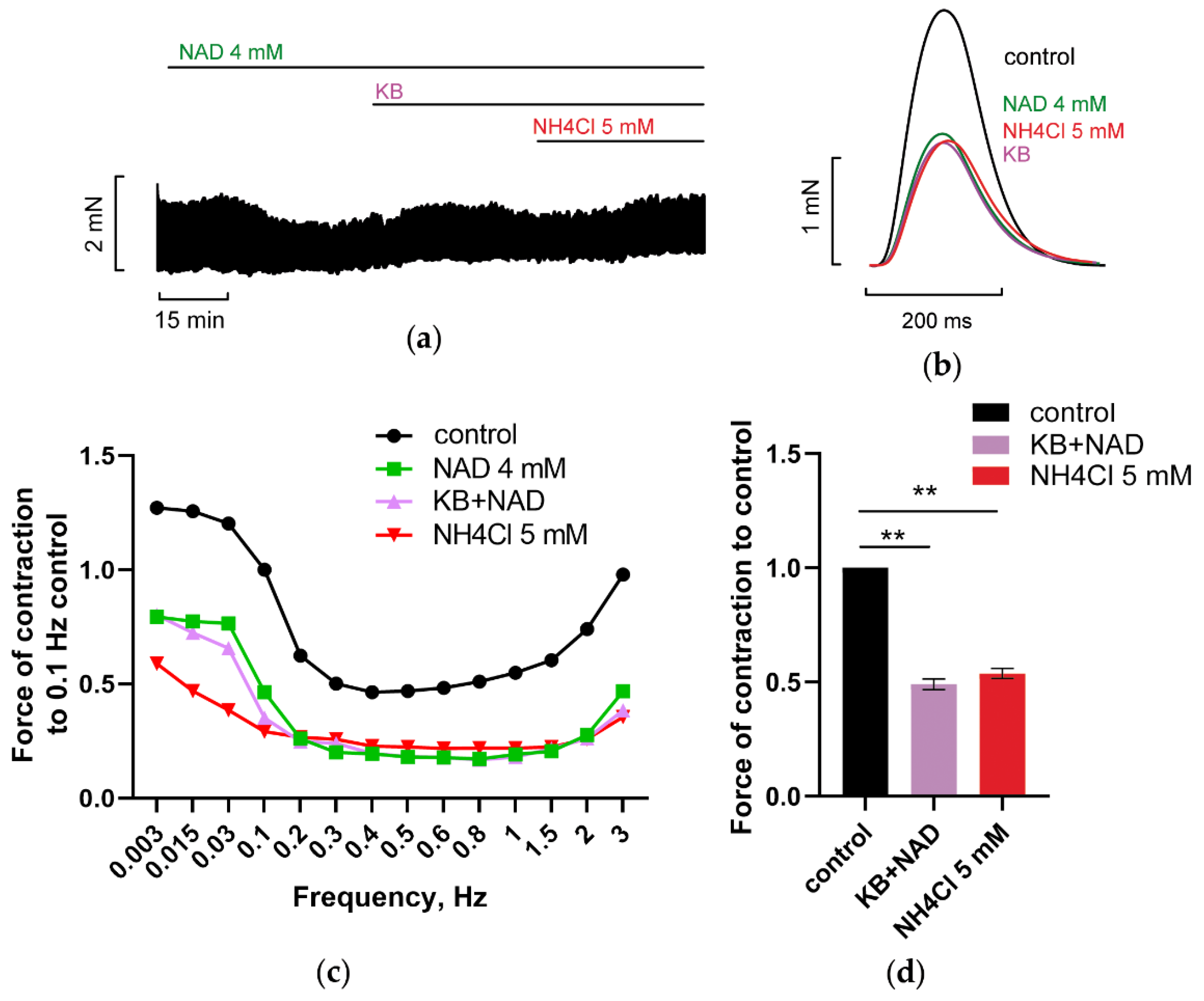
3.1.1. Effects of NAD on PM Contractions, RMP, and AP Duration of CM: P2Y Receptors Antagonist Abrogated Suppression of PM Contractions by NAD
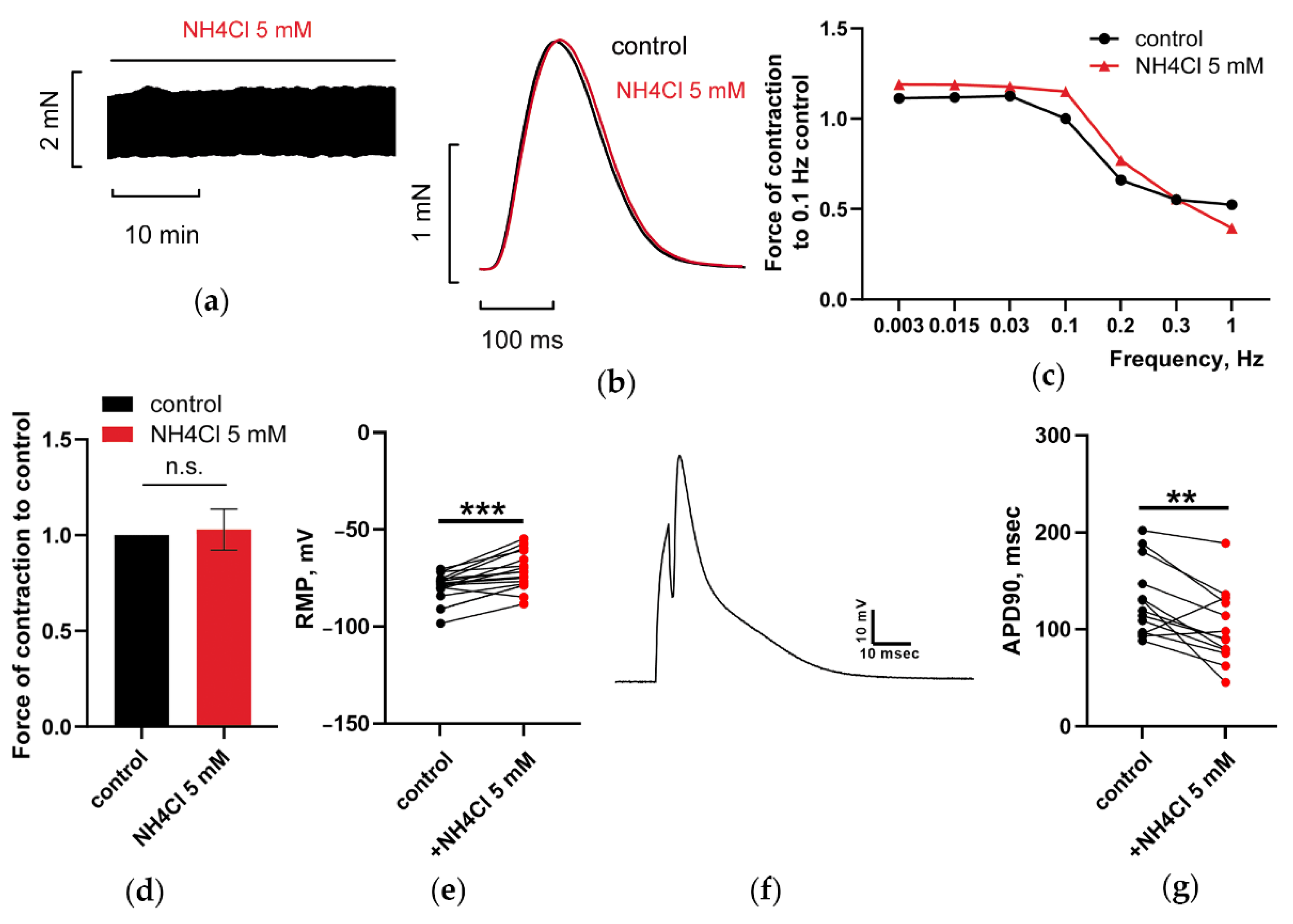
3.1.2. At 5 mM, Ammonia Depolarized RMP and Shortened APD90 in CM, but Does Not Have Any Impact on PM Contractions
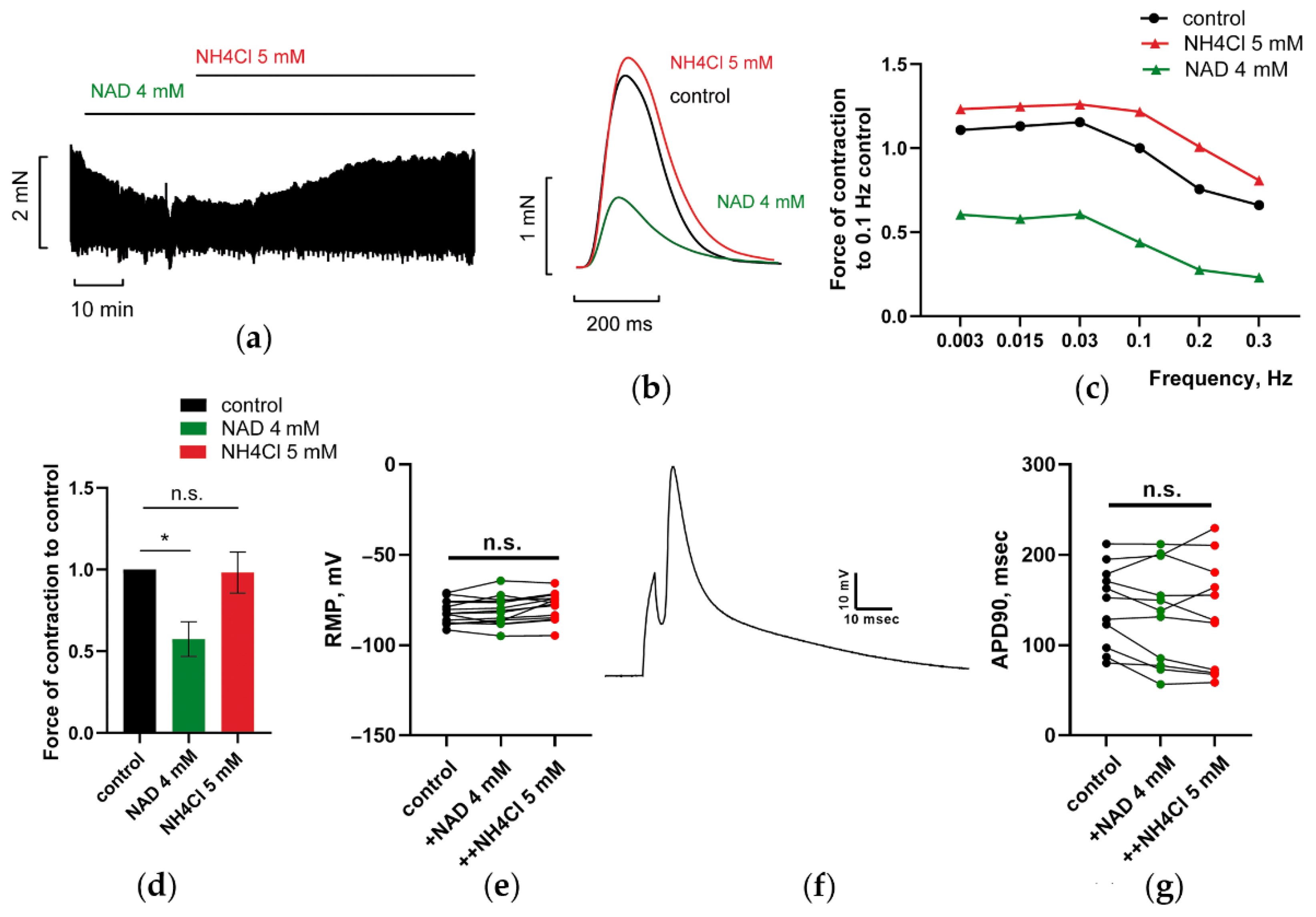
3.1.3. NH4Cl Restored Force of PM Contractions Suppressed by NAD despite Minor Alterations in RMP and APD of CM
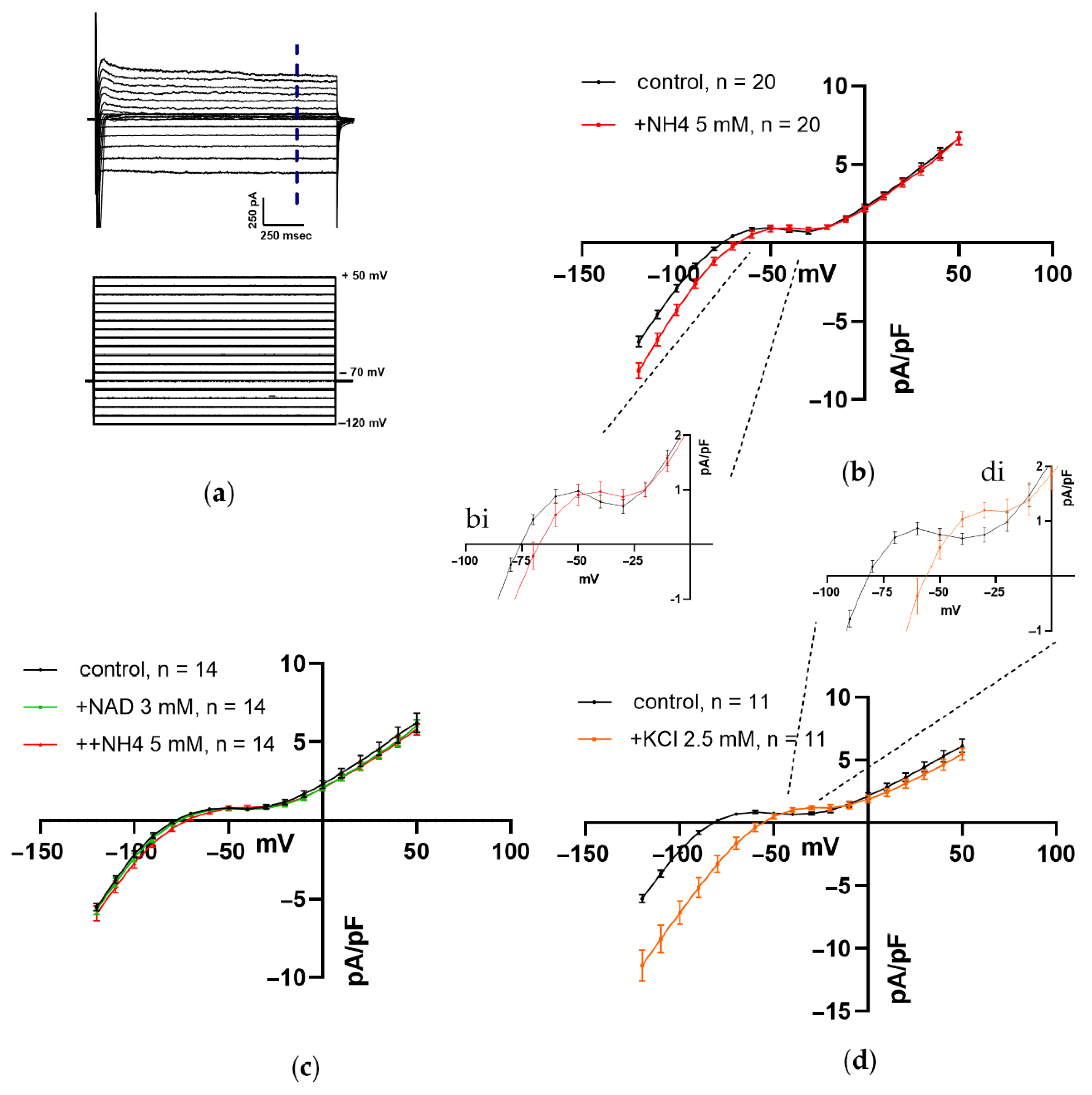
3.1.4. Impact of NH4Cl, NAD, and K+ on Steady-State Current–Voltage (I–V) Relations of Net Current during Repolarizing Voltage Steps in Ventricular CM
3.2. Involvement of Kir2.x and HCN Channels, and Reverse Mode Na+-Ca2+ Exchanger in NAD and Ammonia Antagonism in Paced PM
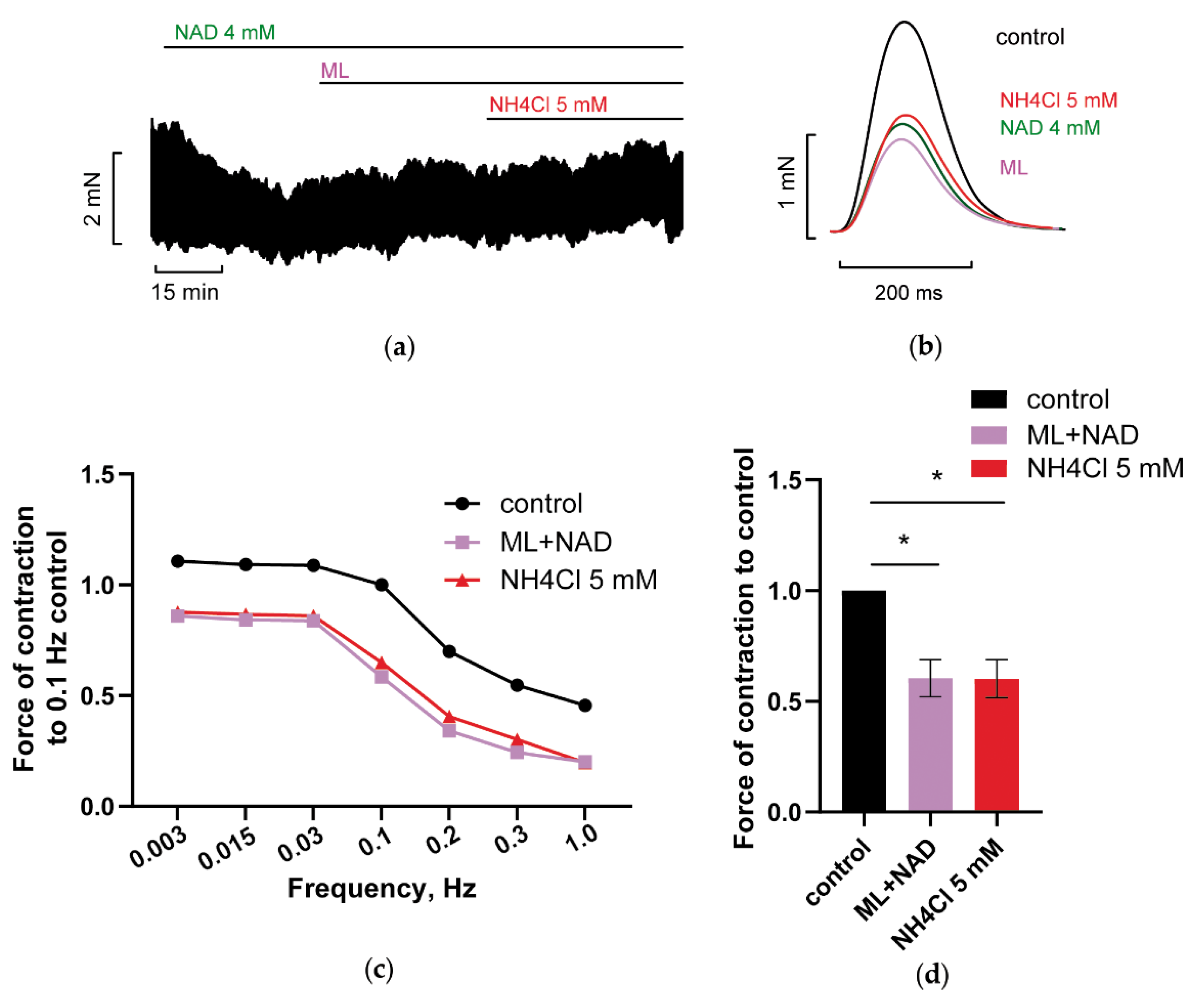
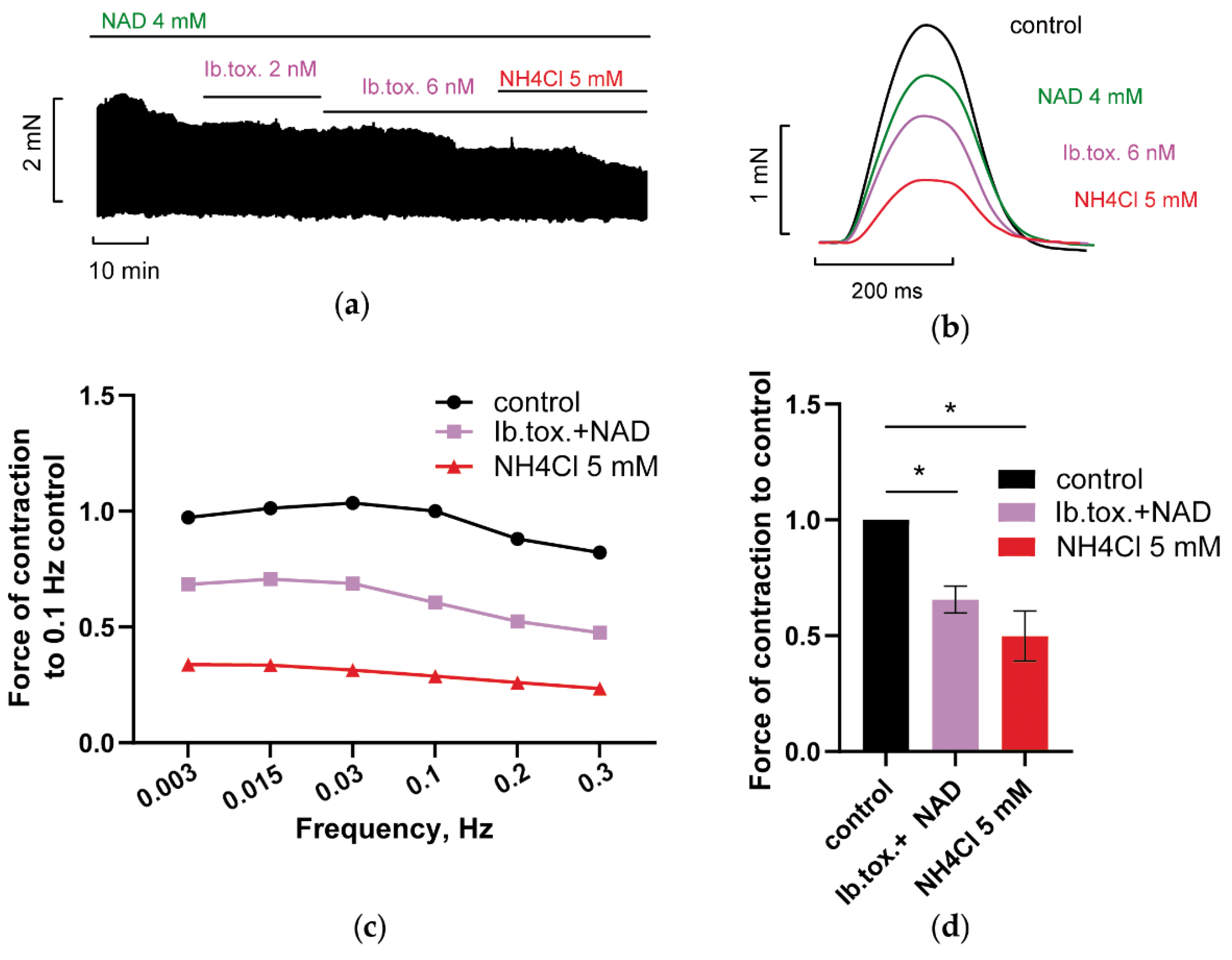
3.2.1. Blockade of Kir2.x Channels Prevents NH4Cl Effect
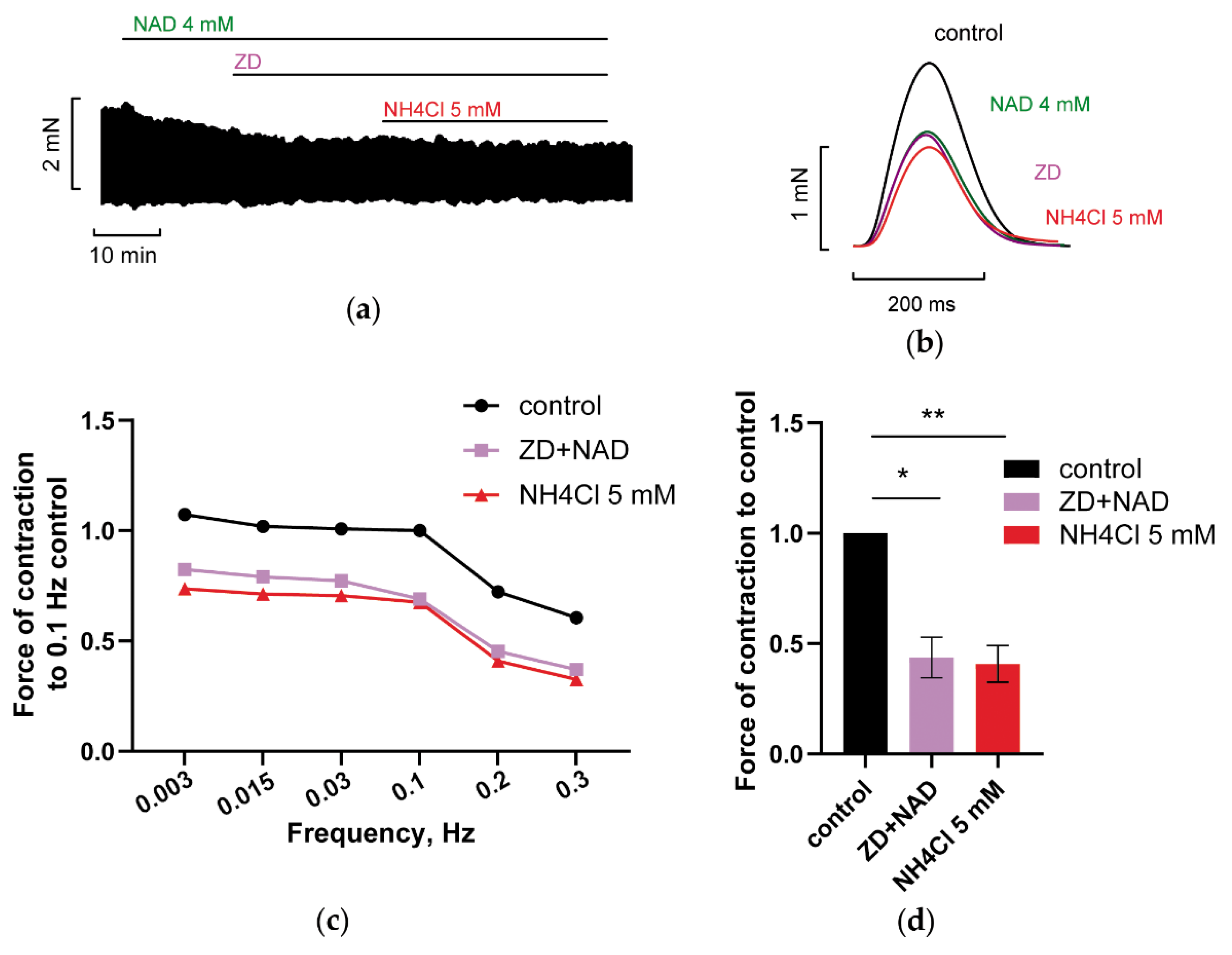
3.2.2. Blockade of HCN Channels Prevents NH4Cl Effect
3.2.3. Inhibition of Reverse Mode Na+-Ca2+ Exchanger (NCX) Abolishes NAD and Ammonia Antagonism
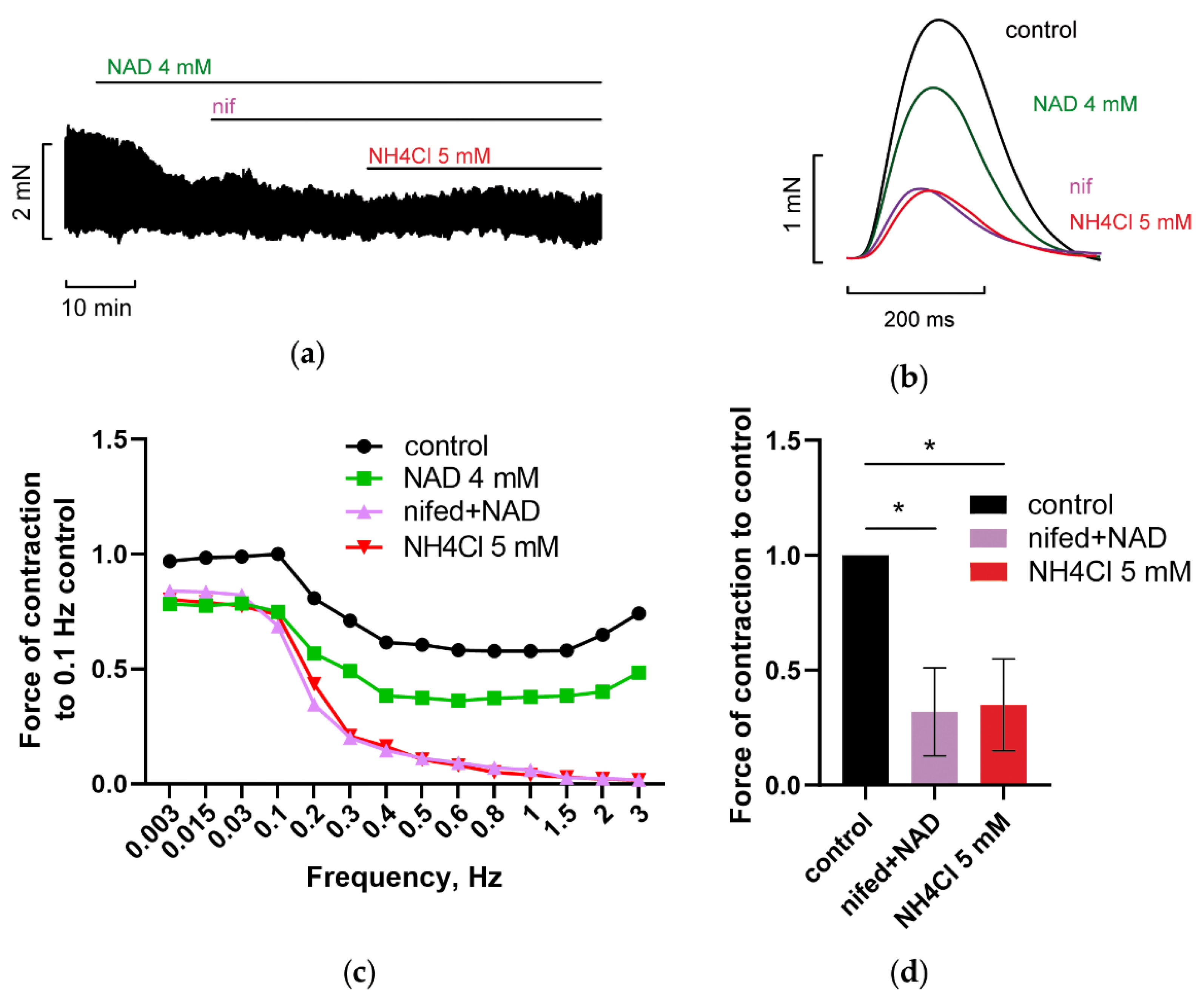
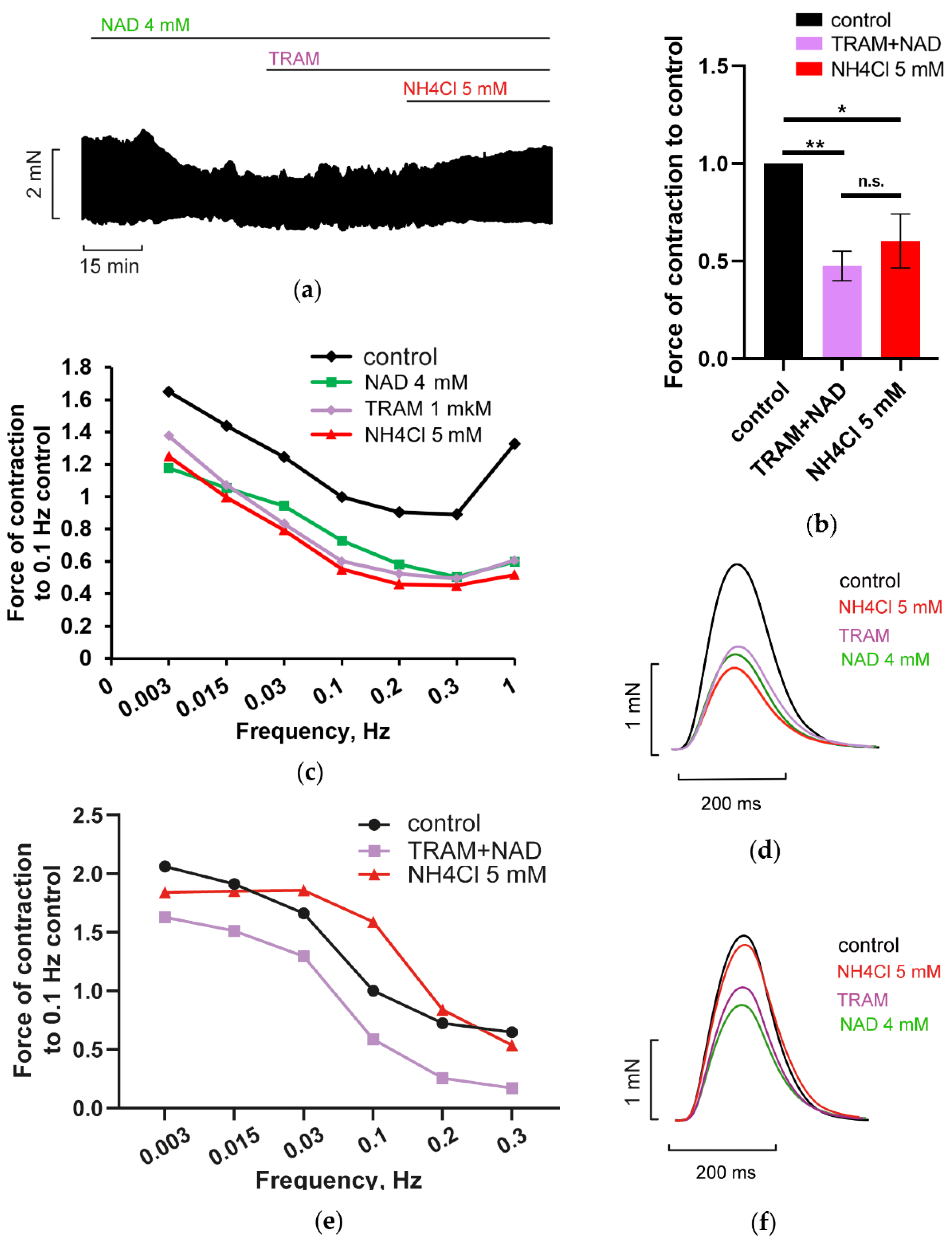
3.3. Blockade of Voltage-Gated L-Type Calcium Channels (LTCC) Abrogated NAD and Ammonia Antagonism
3.4. Implication of Calcium—Activated Potassium Channels in NAD and Ammonia Antagonism
3.4.1. Blockade of BK Channels Prevents NH4Cl Effect
3.4.2. Blockade of IK Channels Prevents NH4Cl Effect
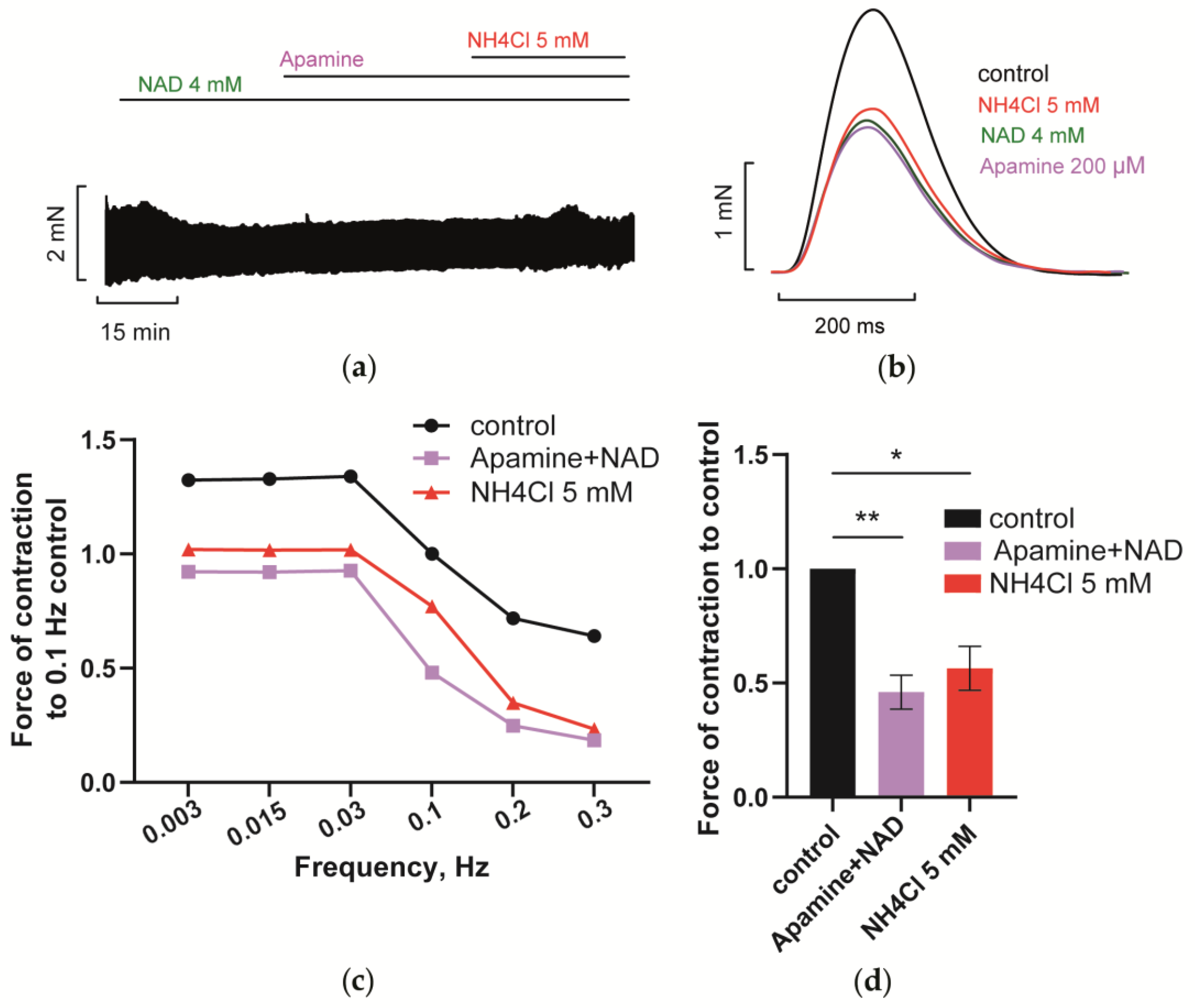
3.4.3. Blockade of SK Channels Prevents NH4Cl Effect
4. Discussion
4.1. NAD and Second Messengers IP3, cAMP and cADPR
4.2. NADo and Ammonia Interplay: Impact of NADo and Ammonia on Membrane Potential and AP Duration in CM and Contractility of PM
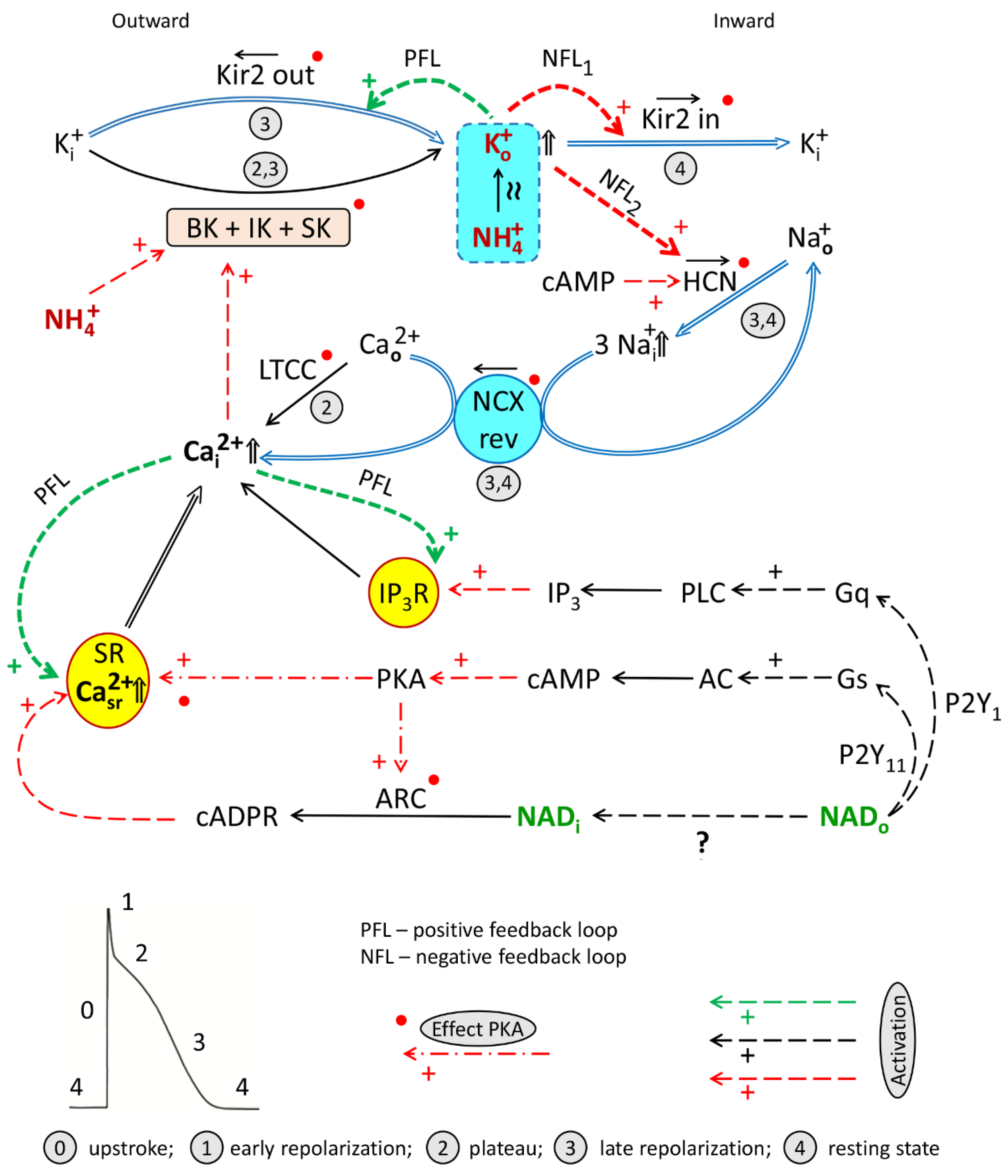
4.2.1. Channels Involved in NADo and Ammonia Antagonism (Interplay)
4.2.2. LTCC, RyR-Encoded CICR, and cADPR Interplay and Signaling Antagonism
4.3. Hypothesis on the Impact of Ammonia on HCN and Kir2.x Channels
4.3.1. Potassium, Kir2.x, and HCN Channels, and Positive/Negative Feedback in the System
4.3.2. Ammonia Evokes Rise of Extracellular Potassium
4.3.3. Suggested “Twin-Brother’s Effect” of Potassium and Ammonia on HCN and Kir2.x Channels Interplay
4.4. Working Hypothesis of NAD/Ammonia Antagonism
- Signaling axes (1) and (3) turned on by NADo and acting through P2Y1 and P2Y11 receptors, respectively, and delivering second messengers IP3, cAMP, required for the activation of HCN channels, and cADPR, involved in LTCC/RyR-dependent CICR/cADPR interplay and CaT rise.
- Cooperation of BK + IK + SK channels. Activation by calcium of BK, IK, and SK channels mediating hyperpolarizing outward K+ current delivering Ko+ required, in turn, for activation of Kir2.x and HCN channels and forming PFL and NFLs;
- PFL/NFLs interplay: sequential turning on of (i) IK1out-dependent hyperpolarizing PFL based on K+o-induced K+o rise and (ii) two NFLs (K+o-induced K+o and Na+o removal) mediating depolarizing If + IK1in net current based on the activation by Ko+ of HCN and Kir2.x channels.
- “Twin-brother’s effect”, based on suggested NH4+-induced rise of [K+]o and overactivation of Kir2.x and HCN channels by both these ions.
- [Na+]i/[Ca2+]i rise: K+o and cAMP-dependent augmentation of the If current providing the rise of [Na+]i and reverse mode operation of NCX (NCXrev) resulting in the accumulation of [Ca2+]i and augmentation of LTCC/cAPDR/RyR interplay determining the magnitude of CaT.
- Activation of BK channels by NH4+.
- Activation of various channels by PKA and CaMKII phosphorylation.
4.5. On the Dormancy of SK, BK, and HCN Channels in Ventricular CM
4.6. Prospects and Clinical Relevance
4.7. Limitations of the Study
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Terentyev, D.; Rochira, J.A.; Terentyeva, R.; Roder, K.; Koren, G.; Li, W. Sarcoplasmic Reticulum Ca2+ Release Is Both Necessary and Sufficient for SK Channel Activation in Ventricular Myocytes. Am. J. Physiol. Circ. Physiol. 2014, 306, H738–H746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, Y.; Zhao, W.; Duan, P.; Chen, Y.; Zhao, W.; Wang, Q.; Tu, H.; Zhang, Q. RyR2 Modulates a Ca2+-Activated K+ Current in Mouse Cardiac Myocytes. PLoS ONE 2014, 9, e94905. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.; Polina, I.; Terentyeva, R.; Bronk, P.; Kim, T.Y.; Roder, K.; Clements, R.T.; Koren, G.; Choi, B.; Terentyev, D. PKA Phosphorylation Underlies Functional Recruitment of Sarcolemmal SK2 Channels in Ventricular Myocytes from Hypertrophic Hearts. J. Physiol. 2020, 598, 2847–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisbrod, D.; Peretz, A.; Ziskind, A.; Menaker, N.; Oz, S.; Barad, L.; Eliyahu, S.; Itskovitz-Eldor, J.; Dascal, N.; Khananshvili, D.; et al. SK4 Ca2+ Activated K+ Channel Is a Critical Player in Cardiac Pacemaker Derived from Human Embryonic Stem Cells. Proc. Natl. Acad. Sci. USA 2013, 110, E1685–E1694. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Yang, M.; Wang, F.; Yang, A.; Zhao, Q.; Wang, X.; Tang, Y.; Wang, T.; Huang, C. Overexpression of the Medium-conductance Calcium-activated Potassium Channel (SK4) and the HCN2 Channel to Generate a Biological Pacemaker. Mol. Med. Rep. 2019, 20, 3406–3414. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Timofeyev, V.; Dennis, A.; Bektik, E.; Wan, X.; Laurita, K.R.; Deschênes, I.; Li, R.A.; Fu, J.-D. A Singular Role of IK1 Promoting the Development of Cardiac Automaticity during Cardiomyocyte Differentiation by IK1 –Induced Activation of Pacemaker Current. Stem Cell Rev. Rep. 2017, 13, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Ponce-Balbuena, D.; Guerrero-Serna, G.; Valdivia, C.R.; Caballero, R.; Diez-Guerra, F.J.; Jiménez-Vázquez, E.N.; Ramírez, R.J.; Monteiro da Rocha, A.; Herron, T.J.; Campbell, K.F.; et al. Cardiac Kir2.1 and Na V 1.5 Channels Traffic Together to the Sarcolemma to Control Excitability. Circ. Res. 2018, 122, 1501–1516. [Google Scholar] [CrossRef]
- Yanagi, K.; Takano, M.; Narazaki, G.; Uosaki, H.; Hoshino, T.; Ishii, T.; Misaki, T.; Yamashita, J.K. Hyperpolarization-Activated Cyclic Nucleotide-Gated Channels and T-Type Calcium Channels Confer Automaticity of Embryonic Stem Cell-Derived Cardiomyocytes. Stem Cells 2007, 25, 2712–2719. [Google Scholar] [CrossRef]
- Zhang, X.; Ai, X.; Nakayama, H.; Chen, B.; Harris, D.M.; Tang, M.; Xie, Y.; Szeto, C.; Li, Y.; Li, Y.; et al. Persistent Increases in Ca2+ Influx through Cav1.2 Shortens Action Potential and Causes Ca2+ Overload-Induced Afterdepolarizations and Arrhythmias. Basic Res. Cardiol. 2016, 111, 4. [Google Scholar] [CrossRef] [Green Version]
- Yampolsky, P.; Koenen, M.; Mosqueira, M.; Geschwill, P.; Nauck, S.; Witzenberger, M.; Seyler, C.; Fink, T.; Kruska, M.; Bruehl, C.; et al. Augmentation of Myocardial If Dysregulates Calcium Homeostasis and Causes Adverse Cardiac Remodeling. Nat. Commun. 2019, 10, 3295. [Google Scholar] [CrossRef]
- Nerbonne, J.M. Molecular Basis of Functional Myocardial Potassium Channel Diversity. Card. Electrophysiol. Clin. 2016, 8, 257–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.L.-H. Murine Electrophysiological Models of Cardiac Arrhythmogenesis. Physiol. Rev. 2017, 97, 283–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisbrod, D. Small and Intermediate Calcium Activated Potassium Channels in the Heart: Role and Strategies in the Treatment of Cardiovascular Diseases. Front. Physiol. 2020, 11, 590534. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.H.; Wu, Y.; Gao, Z.; Anderson, M.E.; Dalziel, J.E.; Meredith, A.L. BK Channels Regulate Sinoatrial Node Firing Rate and Cardiac Pacing in Vivo. Am. J. Physiol. Circ. Physiol. 2014, 307, H1327–H1338. [Google Scholar] [CrossRef] [Green Version]
- Patel, N.H.; Johannesen, J.; Shah, K.; Goswami, S.K.; Patel, N.J.; Ponnalagu, D.; Kohut, A.R.; Singh, H. Inhibition of BK Ca Negatively Alters Cardiovascular Function. Physiol. Rep. 2018, 6, e13748. [Google Scholar] [CrossRef]
- Balderas, E.; Zhang, J.; Stefani, E.; Toro, L. Mitochondrial BKCa Channel. Front. Physiol. 2015, 6, 104. [Google Scholar] [CrossRef] [Green Version]
- DiFrancesco, D. The Role of the Funny Current in Pacemaker Activity. Circ. Res. 2010, 106, 434–446. [Google Scholar] [CrossRef] [Green Version]
- Fenske, S.; Krause, S.; Biel, M.; Wahl-Schott, C. The Role of HCN Channels in Ventricular Repolarization. Trends Cardiovasc. Med. 2011, 21, 216–220. [Google Scholar] [CrossRef]
- Mutafova-Yambolieva, V.N.; Hwang, S.J.; Hao, X.; Chen, H.; Zhu, M.X.; Wood, J.D.; Ward, S.M.; Sanders, K.M. β-Nicotinamide Adenine Dinucleotide Is an Inhibitory Neurotransmitter in Visceral Smooth Muscle. Proc. Natl. Acad. Sci. USA 2007, 104, 16359–16364. [Google Scholar] [CrossRef] [Green Version]
- Sanders, K.M.; Mutafova-Yambolieva, V.N. Neurotransmitters Responsible for Purinergic Motor Neurotransmission and Regulation of GI Motility. Auton. Neurosci. 2021, 234, 102829. [Google Scholar] [CrossRef]
- Alefishat, E.; Alexander, S.P.H.; Ralevic, V. Effects of NAD at Purine Receptors in Isolated Blood Vessels. Purinergic Signal. 2015, 11, 47–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umapathy, N.S.; Zemskov, E.A.; Gonzales, J.; Gorshkov, B.A.; Sridhar, S.; Chakraborty, T.; Lucas, R.; Verin, A.D. Extracellular Beta-Nicotinamide Adenine Dinucleotide (Beta-NAD) Promotes the Endothelial Cell Barrier Integrity via PKA- and EPAC1/Rac1-Dependent Actin Cytoskeleton Rearrangement. J. Cell. Physiol. 2010, 223, 215–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreschi, I.; Bruzzone, S.; Nicholas, R.A.; Fruscione, F.; Sturla, L.; Benvenuto, F.; Usai, C.; Meis, S.; Kassack, M.U.; Zocchi, E.; et al. Extracellular NAD+ Is an Agonist of the Human P2Y 11 Purinergic Receptor in Human Granulocytes. J. Biol. Chem. 2006, 281, 31419–31429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, C.; Grahnert, A.; Abdelrahman, A.; Müller, C.E.; Hauschildt, S. Extracellular NAD+ Induces a Rise in [Ca2+]i in Activated Human Monocytes via Engagement of P2Y1 and P2Y11 Receptors. Cell Calcium 2009, 46, 263–272. [Google Scholar] [CrossRef]
- Djerada, Z.; Millart, H. Intracellular NAADP Increase Induced by Extracellular NAADP via the P2Y11-like Receptor. Biochem. Biophys. Res. Commun. 2013, 436, 199–203. [Google Scholar] [CrossRef]
- Coppi, E.; Pedata, F.; Gibb, A.J. P2Y 1 Receptor Modulation of Ca2+-Activated K+ Currents in Medium-Sized Neurons from Neonatal Rat Striatal Slices. J. Neurophysiol. 2012, 107, 1009–1021. [Google Scholar] [CrossRef] [Green Version]
- Pustovit, K.B.; Abramochkin, D.V. Effects of Nicotinamide Adenine Dinucleotide (NAD+) and Diadenosine Tetraphosphate (Ap4A) on Electrical Activity of Working and Pacemaker Atrial Myocardium in Guinea Pigs. Bull. Exp. Biol. Med. 2016, 160, 733–736. [Google Scholar] [CrossRef]
- Pustovit, K.B.; Potekhina, V.M.; Ivanova, A.D.; Petrov, A.M.; Abramochkin, D.V.; Kuzmin, V.S. Extracellular ATP and β-NAD Alter Electrical Properties and Cholinergic Effects in the Rat Heart in Age-Specific Manner. Purinergic Signal. 2019, 15, 107–117. [Google Scholar] [CrossRef]
- Cooper, A.J.L.; Lai, J.C.K. Cerebral Ammonia Metabolism in Normal and Hyperammonemic Rats. Neurochem. Pathol. 1987, 6, 67–95. [Google Scholar] [CrossRef]
- Hertz, L.; Song, D.; Peng, L.; Chen, Y. Multifactorial Effects on Different Types of Brain Cells Contribute to Ammonia Toxicity. Neurochem. Res. 2017, 42, 721–736. [Google Scholar] [CrossRef]
- Dasarathy, S.; Mookerjee, R.P.; Rackayova, V.; Rangroo Thrane, V.; Vairappan, B.; Ott, P.; Rose, C.F. Ammonia Toxicity: From Head to Toe? Metab. Brain Dis. 2017, 32, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, A.R.; Norenberg, M.D. Hyperammonemia in Hepatic Encephalopathy. J. Clin. Exp. Hepatol. 2018, 8, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Knepper, M.A.; Packer, R.; Good, D.W. Ammonium Transport in the Kidney. Physiol. Rev. 1989, 69, 179–249. [Google Scholar] [CrossRef]
- Dahlmann, A.; Li, M.; Gao, Z.; McGarrigle, D.; Sackin, H.; Palmer, L.G. Regulation of Kir Channels by Intracellular PH and Extracellular K+. J. Gen. Physiol. 2004, 123, 441–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, R.L.; Morad, M. Permeance of Cs+ and Rb+ through the Inwardly Rectifying K+ Channel in Guinea Pig Ventricular Myocytes. J. Membr. Biol. 1991, 122, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Edvinsson, J.M.; Shah, A.J.; Palmer, L.G. Kir4.1 K+ Channels Are Regulated by External Cations. Channels 2011, 5, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Stephan, J.; Haack, N.; Kafitz, K.W.; Durry, S.; Koch, D.; Hochstrate, P.; Seifert, G.; Steinhäuser, C.; Rose, C.R. Kir4.1 Channels Mediate a Depolarization of Hippocampal Astrocytes under Hyperammonemic Conditions in Situ. Glia 2012, 60, 965–978. [Google Scholar] [CrossRef]
- Blatz, A.L.; Magleby, K.L. Ion Conductance and Selectivity of Single Calcium-Activated Potassium Channels in Cultured Rat Muscle. J. Gen. Physiol. 1984, 84, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Hadley, R.W.; Hume, J.R. Permeability of Time-Dependent K+ Channel in Guinea Pig Ventricular Myocytes to Cs+, Na+, NH4+, and Rb+. Am. J. Physiol. Circ. Physiol. 1990, 259, H1448–H1454. [Google Scholar] [CrossRef]
- Wollmuth, L.P.; Hille, B. Ionic Selectivity of Ih Channels of Rod Photoreceptors in Tiger Salamanders. J. Gen. Physiol. 1992, 100, 749–765. [Google Scholar] [CrossRef]
- Carrisoza-Gaytán, R.; Rangel, C.; Salvador, C.; Saldaña-Meyer, R.; Escalona, C.; Satlin, L.M.; Liu, W.; Zavilowitz, B.; Trujillo, J.; Bobadilla, N.A.; et al. The Hyperpolarization-Activated Cyclic Nucleotide-Gated HCN2 Channel Transports Ammonium in the Distal Nephron. Kidney Int. 2011, 80, 832–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehsenfeld, S.; Wood, C.M. A Potential Role for Hyperpolarization-Activated Cyclic Nucleotide-Gated Sodium/Potassium Channels (HCNs) in Teleost Acid-Base and Ammonia Regulation. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2020, 248–249, 110469. [Google Scholar] [CrossRef] [PubMed]
- Marcaggi, P.; Coles, J.A. Ammonium in Nervous Tissue: Transport across Cell Membranes, Fluxes from Neurons to Glial Cells, and Role in Signalling. Prog. Neurobiol. 2001, 64, 157–183. [Google Scholar] [CrossRef] [PubMed]
- Weiner, I.D.; Hamm, L.L. Molecular Mechanisms of Renal Ammonia Transport. Annu. Rev. Physiol. 2007, 69, 317–340. [Google Scholar] [CrossRef] [Green Version]
- Braun, A.P. Ammonium Ion Enhances the Calcium-Dependent Gating of a Mammalian Large Conductance, Calcium-Sensitive K+ Channel. Can. J. Physiol. Pharmacol. 2001, 79, 919–923. [Google Scholar] [CrossRef]
- Fan, P.; Szerb, J.C. Effects of Ammonium Ions on Synaptic Transmission and on Responses to Quisqualate and N-Methyl-d-Aspartate in Hippocampal CA1 Pyramidal Neurons in Vitro. Brain Res. 1993, 632, 225–231. [Google Scholar] [CrossRef]
- Kelly, T.; Rose, C.R. Ammonium Influx Pathways into Astrocytes and Neurones of Hippocampal Slices. J. Neurochem. 2010, 115, 1123–1136. [Google Scholar] [CrossRef]
- Allert, N.; Köller, H.; Siebler, M. Ammonia-Induced Depolarization of Cultured Rat Cortical Astrocytes. Brain Res. 1998, 782, 261–270. [Google Scholar] [CrossRef]
- Schwarz, C.-S.; Ferrea, S.; Quasthoff, K.; Walter, J.; Görg, B.; Häussinger, D.; Schnitzler, A.; Hartung, H.-P.; Dihné, M. Ammonium Chloride Influences in Vitro-Neuronal Network Activity. Exp. Neurol. 2012, 235, 368–373. [Google Scholar] [CrossRef]
- Dynnik, V.V.; Kononov, A.V.; Sergeev, A.I.; Teplov, I.Y.; Tankanag, A.V.; Zinchenko, V.P. To Break or to Brake Neuronal Network Accelerated by Ammonium Ions? PLoS ONE 2015, 10, e0134145. [Google Scholar] [CrossRef]
- Kononov, A.V.; Galimova, M.H.; Dynnik, V.V. Impact of Methyl L-Methionine, Coenzyme NAD+, Certain Blockers of Cation Channels and Kinase G on Hyperactivation of Neuronal Networks by Ammonium Ions. MEDLINE.RU 2015, 16, 1062–1076. [Google Scholar]
- Stull, L.B.; Leppo, M.K.; Marbán, E.; Janssen, P.M.L. Physiological Determinants of Contractile Force Generation and Calcium Handling in Mouse Myocardium. J. Mol. Cell. Cardiol. 2002, 34, 1367–1376. [Google Scholar] [CrossRef]
- Taylor, D. Quantification of the Rat Left Ventricle Force and Ca2+-Frequency Relationships: Similarities to Dog and Human. Cardiovasc. Res. 2004, 61, 77–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez-Correa, G.A.; Cortassa, S.; Stanley, B.; Gao, W.D.; Murphy, A.M. Calcium Sensitivity, Force Frequency Relationship and Cardiac Troponin I: Critical Role of PKA and PKC Phosphorylation Sites. J. Mol. Cell. Cardiol. 2010, 48, 943–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Averin, A.S.; Nenov, M.N.; Starkov, V.G.; Tsetlin, V.I.; Utkin, Y.N. Effects of Cardiotoxins from Naja Oxiana Cobra Venom on Rat Heart Muscle and Aorta: A Comparative Study of Toxin-Induced Contraction Mechanisms. Toxins 2022, 14, 88. [Google Scholar] [CrossRef] [PubMed]
- Khokhlova, A.; Solovyova, O.; Kohl, P.; Peyronnet, R. Single Cardiomyocytes from Papillary Muscles Show Lower Preload-Dependent Activation of Force Compared to Cardiomyocytes from the Left Ventricular Free Wall. J. Mol. Cell. Cardiol. 2022, 166, 127–136. [Google Scholar] [CrossRef]
- Yamashita, T.; Horio, Y.; Yamada, M.; Takahashi, N.; Kondo, C.; Kurachi, Y. Competition between Mg2+ and Spermine for a Cloned IRK2 Channel Expressed in a Human Cell Line. J. Physiol. 1996, 493, 143–156. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, K.; Ehara, T. A Repolarization-Induced Transient Increase in the Outward Current of the Inward Rectifier K+ Channel in Guinea-Pig Cardiac Myocytes. J. Physiol. 1998, 510, 755–771. [Google Scholar] [CrossRef]
- Dhamoon, A.S.; Pandit, S.V.; Sarmast, F.; Parisian, K.R.; Guha, P.; Li, Y.; Bagwe, S.; Taffet, S.M.; Anumonwo, J.M.B. Unique Kir2.x Properties Determine Regional and Species Differences in the Cardiac Inward Rectifier K+ Current. Circ. Res. 2004, 94, 1332–1339. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Zuo, D.; Liu, Z.; Chen, H. Kir2.1 Channels Set Two Levels of Resting Membrane Potential with Inward Rectification. Pflügers Arch.-Eur. J. Physiol. 2018, 470, 599–611. [Google Scholar] [CrossRef]
- Anumonwo, J.M.B.; Lopatin, A.N. Cardiac Strong Inward Rectifier Potassium Channels. J. Mol. Cell. Cardiol. 2010, 48, 45–54. [Google Scholar] [CrossRef] [PubMed]
- McCormick, D.A.; Pape, H.C. Properties of a Hyperpolarization-Activated Cation Current and Its Role in Rhythmic Oscillation in Thalamic Relay Neurones. J. Physiol. 1990, 431, 291–318. [Google Scholar] [CrossRef] [PubMed]
- Azene, E.; Xue, T.; Li, R.A. Molecular Basis of the Effect of Potassium on Heterologously Expressed Pacemaker (HCN) Channels. J. Physiol. 2003, 547, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Carlen, P.L. Enhanced I h Depresses Rat Entopeduncular Nucleus Neuronal Activity From High-Frequency Stimulation or Raised Ke+. J. Neurophysiol. 2008, 99, 2203–2219. [Google Scholar] [CrossRef]
- Wang, L.; Dufour, S.; Valiante, T.A.; Carlen, P.L. Extracellular Potassium and Seizures: Excitation, Inhibition and the Role of Ih. Int. J. Neural Syst. 2016, 26, 1650044. [Google Scholar] [CrossRef]
- Armoundas, A.A.; Hobai, I.A.; Tomaselli, G.F.; Winslow, R.L.; O’Rourke, B. Role of Sodium-Calcium Exchanger in Modulating the Action Potential of Ventricular Myocytes From Normal and Failing Hearts. Circ. Res. 2003, 93, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Sher, A.A.; Noble, P.J.; Hinch, R.; Gavaghan, D.J.; Noble, D. The Role of the Na+/Ca2+ Exchangers in Ca2+ Dynamics in Ventricular Myocytes. Prog. Biophys. Mol. Biol. 2008, 96, 377–398. [Google Scholar] [CrossRef]
- Lyashkov, A.E.; Behar, J.; Lakatta, E.G.; Yaniv, Y.; Maltsev, V.A. Positive Feedback Mechanisms among Local Ca Releases, NCX, and ICaL Ignite Pacemaker Action Potentials. Biophys. J. 2018, 114, 1176–1189. [Google Scholar] [CrossRef]
- Baartscheer, A. [Na+]i and the Driving Force of the Na+/Ca2+-Exchanger in Heart Failure. Cardiovasc. Res. 2003, 57, 986–995. [Google Scholar] [CrossRef] [Green Version]
- Bers, D.M.; Despa, S. Cardiac Myocytes Ca2+ and Na+ Regulation in Normal and Failing Hearts. J. Pharmacol. Sci. 2006, 100, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.S. (Ed.) Calcium Signaling; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2020; Volume 1131, Chapter 16; pp. 344–395. ISBN 978-3-030-12456-4. [Google Scholar]
- Guse, A.H. Second Messenger Function and the Structure-Activity Relationship of Cyclic Adenosine Diphosphoribose (CADPR). FEBS J. 2005, 272, 4590–4597. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Aarhus, R. ADP-Ribosyl Cyclase: An Enzyme That Cyclizes NAD+ into a Calcium-Mobilizing Metabolite. Cell Regul. 1991, 2, 203–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galione, A.; Lee, H.C.; Busa, W.B. Ca2+-Induced Ca2+ Release in Sea Urchin Egg Homogenates: Modulation by Cyclic ADP-Ribose. Science 1991, 253, 1143–1146. [Google Scholar] [CrossRef] [PubMed]
- Verderio, C.; Bruzzone, S.; Zocchi, E.; Fedele, E.; Schenk, U.; De Flora, A.; Matteoli, M. Evidence of a Role for Cyclic ADP-Ribose in Calcium Signalling and Neurotransmitter Release in Cultured Astrocytes. J. Neurochem. 2001, 78, 646–657. [Google Scholar] [CrossRef] [PubMed]
- De Flora, A.; Zocchi, E.; Guida, L.; Franco, L.; Bruzzone, S. Autocrine and Paracrine Calcium Signaling by the CD38/NAD+/Cyclic ADP-Ribose System. Ann. N. Y. Acad. Sci. 2004, 1028, 176–191. [Google Scholar] [CrossRef]
- Higashida, H.; Salmina, A.B.; Olovyannikova, R.Y.; Hashii, M.; Yokoyama, S.; Koizumi, K.; Jin, D.; Liu, H.-X.; Lopatina, O.; Amina, S.; et al. Cyclic ADP-Ribose as a Universal Calcium Signal Molecule in the Nervous System. Neurochem. Int. 2007, 51, 192–199. [Google Scholar] [CrossRef] [Green Version]
- Capel, R.A.; Bose, S.J.; Collins, T.P.; Rajasundaram, S.; Ayagama, T.; Zaccolo, M.; Burton, R.-A.B.; Terrar, D.A. IP3-Mediated Ca2+ Release Regulates Atrial Ca2+ Transients and Pacemaker Function by Stimulation of Adenylyl Cyclases. Am. J. Physiol. Circ. Physiol. 2021, 320, H95–H107. [Google Scholar] [CrossRef]
- Balogh, J.; Wihlborg, A.; Isackson, H.; Joshi, B.; Jacobson, K.; Arner, A.; Erlinge, D. Phospholipase C and cAMP-Dependent Positive Inotropic Effects of ATP in Mouse Cardiomyocytes via P2Y-like Receptors. J. Mol. Cell. Cardiol. 2005, 39, 223–230. [Google Scholar] [CrossRef] [Green Version]
- King, B.F.; Townsend-Nicholson, A. Involvement of P2Y 1 and P2Y 11 Purinoceptors in Parasympathetic Inhibition of Colonic Smooth Muscle. J. Pharmacol. Exp. Ther. 2008, 324, 1055–1063. [Google Scholar] [CrossRef] [Green Version]
- Gul, R.; Park, D.-R.; Shawl, A.I.; Im, S.-Y.; Nam, T.-S.; Lee, S.-H.; Ko, J.-K.; Jang, K.Y.; Kim, D.; Kim, U.-H. Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) and Cyclic ADP-Ribose (CADPR) Mediate Ca2+ Signaling in Cardiac Hypertrophy Induced by β-Adrenergic Stimulation. PLoS ONE 2016, 11, e0149125. [Google Scholar] [CrossRef]
- Lewis, A.M.; Aley, P.K.; Roomi, A.; Thomas, J.M.; Masgrau, R.; Garnham, C.; Shipman, K.; Paramore, C.; Bloor-Young, D.; Sanders, L.E.L.; et al. SS-Adrenergic Receptor Signaling Increases NAADP and CADPR Levels in the Heart. Biochem. Biophys. Res. Commun. 2012, 427, 326–329. [Google Scholar] [CrossRef]
- Lin, W.K.; Bolton, E.L.; Cortopassi, W.A.; Wang, Y.; O’Brien, F.; Maciejewska, M.; Jacobson, M.P.; Garnham, C.; Ruas, M.; Parrington, J.; et al. Synthesis of the Ca2+-Mobilizing Messengers NAADP and CADPR by Intracellular CD38 Enzyme in the Mouse Heart: Role in β-Adrenoceptor Signaling. J. Biol. Chem. 2017, 292, 13243–13257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willmott, N.; Sethi, J.K.; Walseth, T.F.; Lee, H.C.; White, A.M.; Galione, A. Nitric Oxide-Induced Mobilization of Intracellular Calcium via the Cyclic ADP-Ribose Signaling Pathway. J. Biol. Chem. 1996, 271, 3699–3705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rah, S.-Y.; Park, K.-H.; Nam, T.-S.; Kim, S.-J.; Kim, H.; Im, M.-J.; Kim, U.-H. Association of CD38 with Nonmuscle Myosin Heavy Chain IIA and Lck Is Essential for the Internalization and Activation of CD38. J. Biol. Chem. 2007, 282, 5653–5660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashii, M.; Minabe, Y.; Higashida, H. CADP-Ribose Potentiates Cytosolic Ca2+ Elevation and Ca2+ Entry via L-Type Voltage-Activated Ca2+ Channels in NG108-15 Neuronal Cells. Biochem. J. 2000, 345 Pt 2, 207–215. [Google Scholar] [CrossRef]
- Clark, R.B.; Tremblay, A.; Melnyk, P.; Allen, B.G.; Giles, W.R.; Fiset, C. T-Tubule Localization of the Inward-Rectifier K+ Channel in Mouse Ventricular Myocytes: A Role in K+ Accumulation. J. Physiol. 2001, 537, 979–992. [Google Scholar] [CrossRef]
- Reilly, L.; Eckhardt, L.L. Cardiac Potassium Inward Rectifier Kir2: Review of Structure, Regulation, Pharmacology, and Arrhythmogenesis. Hear. Rhythm 2021, 18, 1423–1434. [Google Scholar] [CrossRef]
- Gelens, L.; Anderson, G.A.; Ferrell, J.E. Spatial Trigger Waves: Positive Feedback Gets You a Long Way. Mol. Biol. Cell 2014, 25, 3486–3493. [Google Scholar] [CrossRef] [Green Version]
- Raimondo, J.V.; Burman, R.J.; Katz, A.A.; Akerman, C.J. Ion Dynamics during Seizures. Front. Cell. Neurosci. 2015, 9, 419. [Google Scholar] [CrossRef] [Green Version]
- Rangroo Thrane, V.; Thrane, A.S.; Wang, F.; Cotrina, M.L.; Smith, N.A.; Chen, M.; Xu, Q.; Kang, N.; Fujita, T.; Nagelhus, E.A.; et al. Ammonia Triggers Neuronal Disinhibition and Seizures by Impairing Astrocyte Potassium Buffering. Nat. Med. 2013, 19, 1643–1648. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, H.; Koehler, R.C.; Wilson, D.A.; Brusilow, S.W.; Traystman, R.J. Methionine Sulfoximine, a Glutamine Synthetase Inhibitor, Attenuates Increased Extracellular Potassium Activity during Acute Hyperammonemia. J. Cereb. Blood Flow Metab. 1997, 17, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Grishina, E.V.; Kravtchenko, I.N.; Lobanov, A.V.; Sergeev, A.I.; Sadovnikova, E.S.; Dynnik, V.V. Selection of Complex Compositions of Protectors of Hyperammonemia and Acute Hepatic Encephalopathy. MEDLINE.RU 2015, 16, 1077–1098. [Google Scholar]
- Tuteja, D.; Xu, D.; Timofeyev, V.; Lu, L.; Sharma, D.; Zhang, Z.; Xu, Y.; Nie, L.; Vázquez, A.E.; Young, J.N.; et al. Differential Expression of Small-Conductance Ca2+-Activated K+ Channels SK1, SK2, and SK3 in Mouse Atrial and Ventricular Myocytes. Am. J. Physiol. Circ. Physiol. 2005, 289, H2714–H2723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonilla, I.M.; Long, V.P.; Vargas-Pinto, P.; Wright, P.; Belevych, A.; Lou, Q.; Mowrey, K.; Yoo, J.; Binkley, P.F.; Fedorov, V.V.; et al. Calcium-Activated Potassium Current Modulates Ventricular Repolarization in Chronic Heart Failure. PLoS ONE 2014, 9, e108824. [Google Scholar] [CrossRef]
- Pineda, S.; Nikolova-Krstevski, V.; Leimena, C.; Atkinson, A.J.; Altekoester, A.-K.; Cox, C.D.; Jacoby, A.; Huttner, I.G.; Ju, Y.-K.; Soka, M.; et al. Conserved Role of the Large Conductance Calcium-Activated Potassium Channel, K Ca 1.1, in Sinus Node Function and Arrhythmia Risk. Circ. Genomic Precis. Med. 2021, 14, e003144. [Google Scholar] [CrossRef]
- Sanghvi, S.; Szteyn, K.; Ponnalagu, D.; Sridharan, D.; Lam, A.; Hansra, I.; Chaudhury, A.; Majumdar, U.; Kohut, A.R.; Gururaja Rao, S.; et al. Inhibition of BKCa Channels Protects Neonatal Hearts against Myocardial Ischemia and Reperfusion Injury. Cell Death Discov. 2022, 8, 175. [Google Scholar] [CrossRef]
- Herrmann, S.; Layh, B.; Ludwig, A. Novel Insights into the Distribution of Cardiac HCN Channels: An Expression Study in the Mouse Heart. J. Mol. Cell. Cardiol. 2011, 51, 997–1006. [Google Scholar] [CrossRef]
- Hofmann, F.; Fabritz, L.; Stieber, J.; Schmitt, J.; Kirchhof, P.; Ludwig, A.; Herrmann, S. Ventricular HCN Channels Decrease the Repolarization Reserve in the Hypertrophic Heart. Cardiovasc. Res. 2012, 95, 317–326. [Google Scholar] [CrossRef] [Green Version]
- Hadova, K.; Kralova, E.; Doka, G.; Bies Pivackova, L.; Kmecova, Z.; Krenek, P.; Klimas, J. Isolated Downregulation of HCN2 in Ventricles of Rats with Streptozotocin-Induced Diabetic Cardiomyopathy. BMC Cardiovasc. Disord. 2021, 21, 118. [Google Scholar] [CrossRef]
- Eren-Koçak, E.; Dalkara, T. Ion Channel Dysfunction and Neuroinflammation in Migraine and Depression. Front. Pharmacol. 2021, 12, 777607. [Google Scholar] [CrossRef]
- Yu, F.-X.; Ke, J.-J.; Fu, Y.; Liao, B.; Shi, Y.-K. Protective Effects of Adenosine in Rabbit Sinoatrial Node Ischemia–Reperfusion Model in Vivo: Control of Arrhythmia by Hyperpolarization-Activated Cyclic Nucleotide-Gated (HCN)4 Channels. Mol. Biol. Rep. 2011, 38, 1723–1731. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Sun, J.; Zhang, L.; Xu, Y.; Wu, B.; Cao, J. The Agonist of Inward Rectifier Potassium Channel (IK1) Attenuates Rat Reperfusion Arrhythmias Linked to CaMKII Signaling. Int. Heart J. 2021, 62, 21–379. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Qiao, X.; Zhang, L.; Wang, D.; Zhang, L.; Feng, Q.; Wu, B.; Cao, J.; Liu, Q. IK1 Channel Agonist Zacopride Suppresses Ventricular Arrhythmias in Conscious Rats with Healing Myocardial Infarction. Life Sci. 2019, 239, 117075. [Google Scholar] [CrossRef]
- Mohamed, M.S.A. Calcium-Activated Potassium Channels in Ischemia–Reperfusion: Learning for the Clinical Application. Front. Med. 2015, 2, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankenreiter, S.; Bednarczyk, P.; Kniess, A.; Bork, N.I.; Straubinger, J.; Koprowski, P.; Wrzosek, A.; Mohr, E.; Logan, A.; Murphy, M.P.; et al. CGMP-Elevating Compounds and Ischemic Conditioning Provide Cardioprotection Against Ischemia and Reperfusion Injury via Cardiomyocyte-Specific BK Channels. Circulation 2017, 136, 2337–2355. [Google Scholar] [CrossRef] [Green Version]
- Nuss, H.B.; Marbán, E.; Johns, D.C. Overexpression of a Human Potassium Channel Suppresses Cardiac Hyperexcitability in Rabbit Ventricular Myocytes. J. Clin. Investig. 1999, 103, 889–896. [Google Scholar] [CrossRef]
- Sturm, P.; Wimmers, S.; Schwarz, J.R.; Bauer, C.K. Extracellular Potassium Effects Are Conserved within the Rat Erg K+ Channel Family. J. Physiol. 2005, 564, 329–345. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Averin, A.S.; Konakov, M.V.; Pimenov, O.Y.; Galimova, M.H.; Berezhnov, A.V.; Nenov, M.N.; Dynnik, V.V. Regulation of Papillary Muscle Contractility by NAD and Ammonia Interplay: Contribution of Ion Channels and Exchangers. Membranes 2022, 12, 1239. https://doi.org/10.3390/membranes12121239
Averin AS, Konakov MV, Pimenov OY, Galimova MH, Berezhnov AV, Nenov MN, Dynnik VV. Regulation of Papillary Muscle Contractility by NAD and Ammonia Interplay: Contribution of Ion Channels and Exchangers. Membranes. 2022; 12(12):1239. https://doi.org/10.3390/membranes12121239
Chicago/Turabian StyleAverin, Alexey S., Maxim V. Konakov, Oleg Y. Pimenov, Miliausha H. Galimova, Alexey V. Berezhnov, Miroslav N. Nenov, and Vladimir V. Dynnik. 2022. "Regulation of Papillary Muscle Contractility by NAD and Ammonia Interplay: Contribution of Ion Channels and Exchangers" Membranes 12, no. 12: 1239. https://doi.org/10.3390/membranes12121239