
Membrane Contact Sites in Yeast: Control Hubs of Sphingolipid Homeostasis


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
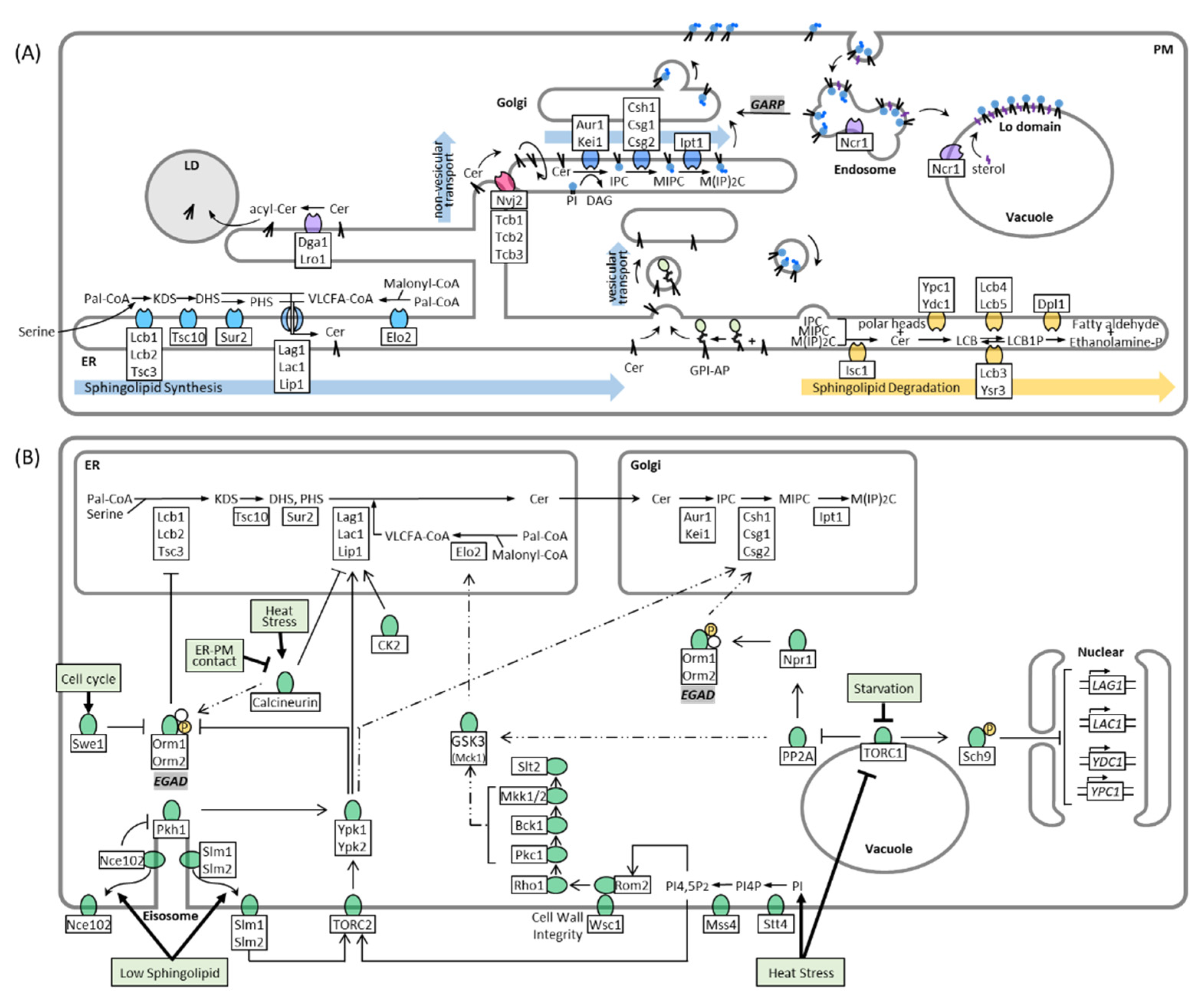
2. Sphingolipid Synthesis, Catabolism, and Trafficking in Yeast
3. Regulation of Sphingolipid Metabolism
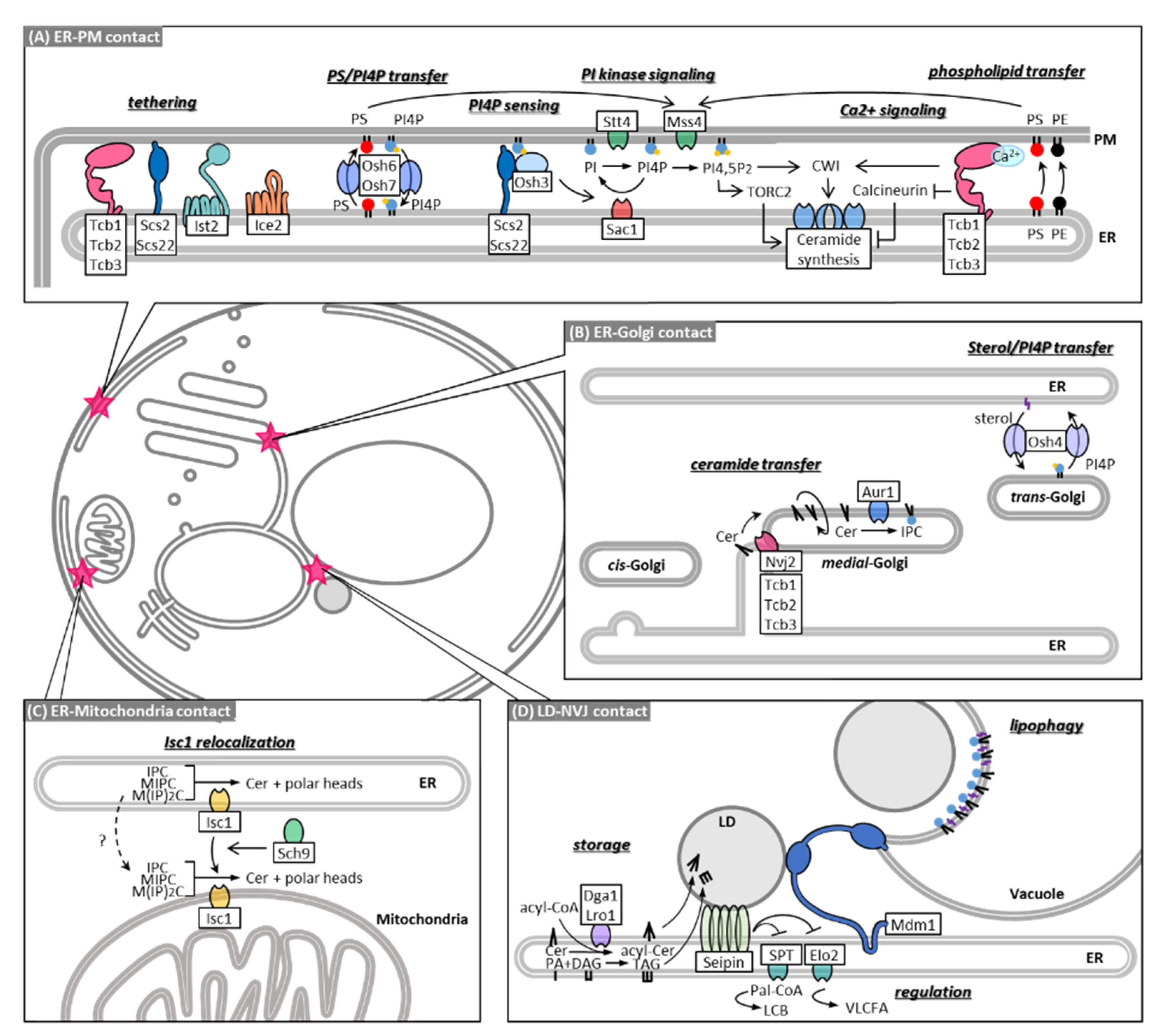
4. MCSs in Sphingolipid Metabolism
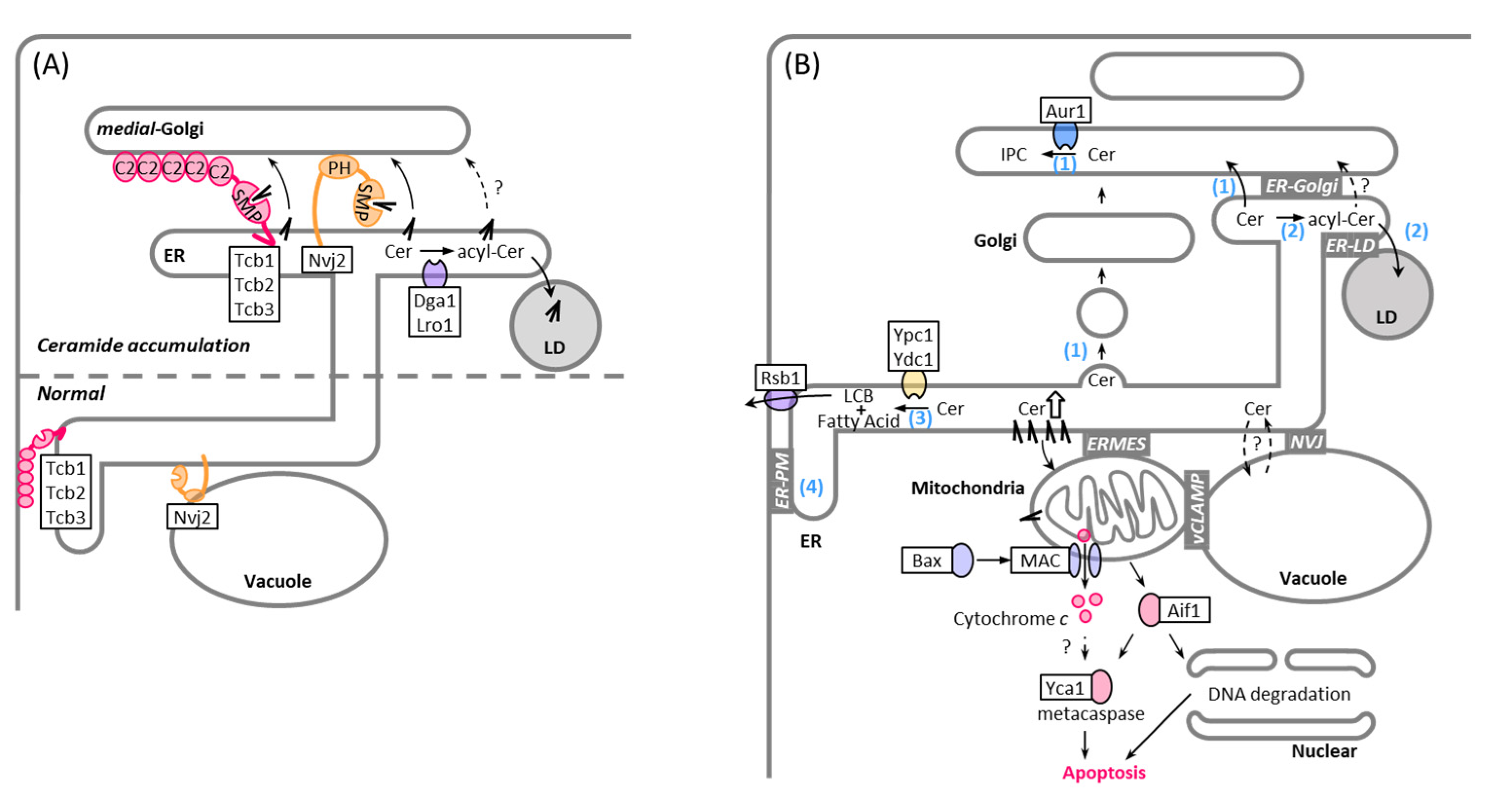
5. MCSs in Non-Vesicular Transport of Ceramide
6. MCSs and LDs in Ceramide Lipotoxicity
7. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Ziółkowska, N.E.; Christiano, R.; Walther, T.C. Organized living: Formation mechanisms and functions of plasma membrane domains in yeast. Trends Cell Biol. 2012, 22, 151–158. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Pettus, B.J.; Chalfant, C.E.; Hannun, Y.A. Ceramide in apoptosis: An overview and current perspectives. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2002, 1585, 114–125. [Google Scholar] [CrossRef]
- Mandala, S.M.; Thornton, R.; Tu, Z.; Kurtz, M.B.; Nickels, J.; Broach, J.; Menzeleev, R.; Spiegel, S. Sphingoid base 1-phosphate phosphatase: A key regulator of sphingolipid metabolism and stress response. Proc Natl. Acad. Sci. USA 1998, 95, 150–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morad, S.A.; Cabot, M.C. Ceramide-orchestrated signalling in cancer cells. Nat. Rev. Cancer 2013, 13, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Shimobayashi, M.; Oppliger, W.; Moes, S.; Jenö, P.; Hall, M.N. TORC1-regulated protein kinase Npr1 phosphorylates Orm to stimulate complex sphingolipid synthesis. Mol. Biol. Cell 2013, 24, 870–881. [Google Scholar] [CrossRef] [Green Version]
- Berchtold, D.; Piccolis, M.; Chiaruttini, N.; Riezman, I.; Riezman, H.; Roux, A.; Walther, T.; Loewith, R. Plasma membrane stress induces relocalization of Slm proteins and activation of TORC2 to promote sphingolipid synthesis. Nat. Cell Biol. 2012, 14, 542–547. [Google Scholar] [CrossRef]
- Gururaj, C.; Federman, R.; Chang, A. Orm proteins integrate multiple signals to maintain sphingolipid homeostasis. J. Biol. Chem. 2013, 288, 20453–20463. [Google Scholar] [CrossRef] [Green Version]
- Olson, D.K.; Fröhlich, F.; Farese, R.V., Jr.; Walther, T.C. Taming the sphinx: Mechanisms of cellular sphingolipid homeostasis. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2016, 1861, 784–792. [Google Scholar]
- Senkal, C.E.; Salama, M.F.; Snider, A.J.; Allopenna, J.J.; Rana, N.A.; Koller, A.; Hannun, Y.A.; Obeid, L.M. Ceramide is metabolized to acylceramide and stored in lipid droplets. Cell Metab. 2017, 25, 686–697. [Google Scholar] [CrossRef] [Green Version]
- Voynova, N.S.; Vionnet, C.; Ejsing, C.S.; Conzelmann, A. A novel pathway of ceramide metabolism in Saccharomyces cerevisiae. Biochem. J. 2012, 447, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, A.; Schlarmann, P.; Kurokawa, K.; Nakano, A.; Riezman, H.; Funato, K. Tricalbins are required for non-vesicular ceramide transport at ER-Golgi contacts and modulate lipid droplet biogenesis. Iscience 2020, 23, 101603. [Google Scholar] [CrossRef]
- Mandon, E.C.; Ehses, I.; Rother, J.; van Echten, G.; Sandhoff, K. Subcellular localization and membrane topology of serine palmitoyltransferase, 3-dehydrosphinganine reductase, and sphinganine N-acyltransferase in mouse liver. J. Biol. Chem. 1992, 267, 11144–11148. [Google Scholar] [CrossRef]
- Han, G.; Gable, K.; Yan, L.; Natarajan, M.; Krishnamurthy, J.; Gupta, S.D.; Borovitskaya, A.; Harmon, J.M.; Dunn, T.M. The topology of the Lcb1p subunit of yeast serine palmitoyltransferase. J. Biol. Chem. 2004, 279, 53707–53716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gable, K.; Han, G.; Monaghan, E.; Bacikova, D.; Natarajan, M.; Williams, R.; Dunn, T.M. Mutations in the yeast LCB1 and LCB2Genes, including those corresponding to the hereditary sensory neuropathy type I mutations, dominantly inactivate serine palmitoyltransferase. J. Biol. Chem. 2002, 277, 10194–10200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kihara, A.; Igarashi, Y. FVT-1 is a mammalian 3-ketodihydrosphingosine reductase with an active site that faces the cytosolic side of the endoplasmic reticulum membrane. J. Biol. Chem. 2004, 279, 49243–49250. [Google Scholar] [CrossRef] [Green Version]
- Kageyama-Yahara, N.; Riezman, H. Transmembrane topology of ceramide synthase in yeast. Biochem. J. 2006, 398, 585–593. [Google Scholar] [CrossRef] [Green Version]
- Megyeri, M.; Prasad, R.; Volpert, G.; Sliwa-Gonzalez, A.; Haribowo, A.G.; Aguilera-Romero, A.; Riezman, H.; Barral, Y.; Futerman, A.; Schuldiner, M. Yeast ceramide synthases, Lag1 and Lac1, have distinct substrate specificity. J. Cell Sci. 2019, 132, jcs228411. [Google Scholar] [CrossRef] [Green Version]
- Vallée, B.; Riezman, H. Lip1p: A novel subunit of acyl-CoA ceramide synthase. EMBO J. 2005, 24, 730–741. [Google Scholar] [CrossRef] [Green Version]
- Funato, K.; Lombardi, R.; Vallée, B.; Riezman, H. Lcb4p is a key regulator of ceramide synthesis from exogenous long chain sphingoid base in Saccharomyces cerevisiae. J. Biol. Chem. 2003, 278, 7325–7334. [Google Scholar] [CrossRef] [Green Version]
- Kihara, A.; Sano, T.; Iwaki, S.; Igarashi, Y. Transmembrane topology of sphingoid long-chain base-1-phosphate phosphatase, Lcb3p. Genes Cells 2003, 8, 525–535. [Google Scholar] [CrossRef]
- Funato, K.; Vallée, B.; Riezman, H. Biosynthesis and trafficking of sphingolipids in the yeast Saccharomyces cerevisiae. Biochemistry 2002, 41, 15105–15114. [Google Scholar] [CrossRef]
- Kondo, N.; Ohno, Y.; Yamagata, M.; Obara, T.; Seki, N.; Kitamura, T.; Naganuma, T.; Kihara, A. Identification of the phytosphingosine metabolic pathway leading to odd-numbered fatty acids. Nat. Commun. 2014, 5, 5338. [Google Scholar] [CrossRef] [Green Version]
- Mayor, S.; Riezman, H. Sorting GPI-anchored proteins. Nat. Rev. Mol. Cell Biol. 2004, 5, 110–120. [Google Scholar] [CrossRef]
- Bosson, R.; Guillas, I.; Vionnet, C.; Roubaty, C.; Conzelmann, A. Incorporation of ceramides into Saccharomyces cerevisiae glycosylphosphatidylinositol-anchored proteins can be monitored in vitro. Eukaryot. Cell 2009, 8, 306–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swain, E.; Stukey, J.; McDonough, V.; Germann, M.; Liu, Y.; Sturley, S.L.; Nickels, J.T. Yeast cells lacking the ARV1 gene harbor defects in sphingolipid metabolism: Complementation by human ARV1. J. Biol. Chem. 2002, 277, 36152–36160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajiwara, K.; Watanabe, R.; Pichler, H.; Ihara, K.; Murakami, S.; Riezman, H.; Funato, K. Yeast ARV1 is required for efficient delivery of an early GPI intermediate to the first mannosyltransferase during GPI assembly and controls lipid flow from the endoplasmic reticulum. Mol. Biol. Cell 2008, 19, 2069–2082. [Google Scholar] [CrossRef] [Green Version]
- Sutterlin, C.; Doering, T.L.; Schimmoller, F.; Schroder, S.; Riezman, H. Specific requirements for the ER to Golgi transport of GPI-anchored proteins in yeast. J. Cell Sci. 1997, 110, 2703–2714. [Google Scholar] [CrossRef] [PubMed]
- Bagnat, M.; Keränen, S.; Shevchenko, A.; Shevchenko, A.; Simons, K. Lipid rafts function in biosynthetic delivery of proteins to the cell surface in yeast. Proc Natl. Acad. Sci. USA 2000, 97, 3254–3259. [Google Scholar] [CrossRef]
- Rodriguez-Gallardo, S.; Kurokawa, K.; Sabido-Bozo, S.; Cortes-Gomez, A.; Ikeda, A.; Zoni, V.; Aguilera-Romero, A.; Perez-Linero, A.M.; Lopez, S.; Waga, M.; et al. Ceramide chain length–dependent protein sorting into selective endoplasmic reticulum exit sites. Sci. Adv. 2020, 6, eaba8237. [Google Scholar] [CrossRef] [PubMed]
- Funato, K.; Riezman, H. Vesicular and nonvesicular transport of ceramide from ER to the Golgi apparatus in yeast. J. Cell Biol. 2001, 155, 949–960. [Google Scholar] [CrossRef] [Green Version]
- Levine, T.P.; Wiggins, C.A.; Munro, S. Inositol phosphorylceramide synthase is located in the Golgi apparatus of Saccharomyces cerevisiae. Mol. Biol. Cell 2000, 11, 2267–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, K.; Noda, Y.; Yoda, K. Kei1: A novel subunit of inositolphosphorylceramide synthase, essential for its enzyme activity and Golgi localization. Mol. Biol. Cell 2009, 20, 4444–4457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisman, Q.; Pomorski, T.; Vogelzangs, C.; Urli-Stam, D.; van Delwijnen, W.D.C.; Holthuis, J.C. Protein sorting in the late Golgi of Saccharomyces cerevisiae does not require mannosylated sphingolipids. J. Biol. Chem. 2004, 279, 1020–1029. [Google Scholar] [CrossRef] [Green Version]
- Uemura, S.; Kihara, A.; Inokuchi, J.I.; Igarashi, Y. Csg1p and newly identified Csh1p function in mannosylinositol phosphorylceramide synthesis by interacting with Csg2p. J. Biol. Chem. 2003, 278, 45049–45055. [Google Scholar] [CrossRef] [Green Version]
- Dickson, R.C.; Nagiec, E.E.; Wells, G.B.; Nagiec, M.M.; Lester, R.L. Synthesis of mannose-(inositol-P) 2-ceramide, the major sphingolipid in Saccharomyces cerevisiae, requires the IPT1 (YDR072c) gene. J. Biol. Chem. 1997, 272, 29620–29625. [Google Scholar] [CrossRef] [Green Version]
- Schnabl, M.; Daum, G.; Pichler, H. Multiple lipid transport pathways to the plasma membrane in yeast. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2005, 1687, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Sawai, H.; Okamoto, Y.; Luberto, C.; Mao, C.; Bielawska, A.; Domae, N.; Hannun, Y.A. Identification of ISC1 (YER019w) as inositol phosphosphingolipid phospholipase C in Saccharomyces cerevisiae. J. Biol. Chem. 2000, 275, 39793–39798. [Google Scholar] [CrossRef] [Green Version]
- De Avalos, S.V.; Okamoto, Y.; Hannun, Y.A. Activation and localization of inositol phosphosphingolipid phospholipase C, Isc1p, to the mitochondria during growth of Saccharomyces cerevisiae. J. Biol. Chem. 2004, 279, 11537–11545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, Y.; Kamiyama, N.; Nakamichi, S.; Kihara, A. PNPLA1 is a transacylase essential for the generation of the skin barrier lipid ω-O-acylceramide. Nat. Commun. 2017, 8, 14610. [Google Scholar] [CrossRef] [PubMed]
- Klug, L.; Daum, G. Yeast lipid metabolism at a glance. FEMS Yeast Res. 2014, 14, 369–388. [Google Scholar] [CrossRef] [Green Version]
- Breslow, D.K. Sphingolipid homeostasis in the endoplasmic reticulum and beyond. Cold Spring Harb. Perspect. Biol. 2013, 5, a013326. [Google Scholar] [CrossRef] [Green Version]
- Platt, F.M. Sphingolipid lysosomal storage disorders. Nature 2014, 510, 68–75. [Google Scholar] [CrossRef]
- Teixeira, V.; Medeiros, T.C.; Vilaça, R.; Ferreira, J.; Moradas-Ferreira, P.; Costa, V. Ceramide signaling targets the PP2A-like protein phosphatase Sit4p to impair vacuolar function, vesicular trafficking and autophagy in Isc1p deficient cells. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2016, 1861, 21–33. [Google Scholar] [CrossRef]
- Hurst, L.R.; Fratti, R.A. Lipid rafts, sphingolipids, and ergosterol in yeast vacuole fusion and maturation. Front. Cell Dev. Biol. 2020, 8, 539. [Google Scholar] [CrossRef]
- Barbosa, A.D.; Osório, H.; Sims, K.J.; Almeida, T.; Alves, M.; Bielawski, J.; Amorim, M.A.; Moradas-Ferreira, P.; Hannun, Y.A.; Costa, V. Role for Sit4p-dependent mitochondrial dysfunction in mediating the shortened chronological lifespan and oxidative stress sensitivity of Isc1p-deficient cells. Mol. Microbiol. 2011, 81, 515–527. [Google Scholar] [CrossRef] [Green Version]
- Malathi, K.; Higaki, K.; Tinkelenberg, A.H.; Balderes, D.A.; Almanzar-Paramio, D.; Wilcox, L.J.; Erdeniz, N.; Redican, F.; Padamsee, M.; Liu, Y.; et al. Mutagenesis of the putative sterol-sensing domain of yeast Niemann Pick C–related protein reveals a primordial role in subcellular sphingolipid distribution. J. Cell Biol. 2004, 164, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Conibear, E.; Cleck, J.N.; Stevens, T.H. Vps51p mediates the association of the GARP (Vps52/53/54) complex with the late Golgi t-SNARE Tlg1p. Mol. Biol. Cell 2003, 14, 1610–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fröhlich, F.; Petit, C.; Kory, N.; Christiano, R.; Hannibal-Bach, H.K.; Graham, M.; Liu, X.; Ejsing, C.S.; Farese, R.V.; Walther, T.C. The GARP complex is required for cellular sphingolipid homeostasis. eLife 2015, 4, e08712. [Google Scholar] [CrossRef]
- Tsuji, T.; Fujimoto, M.; Tatematsu, T.; Cheng, J.; Orii, M.; Takatori, S.; Fujimoto, T. Niemann-Pick type C proteins promote microautophagy by expanding raft-like membrane domains in the yeast vacuole. eLife 2017, 6, e25960. [Google Scholar] [CrossRef] [PubMed]
- Esch, B.M.; Limar, S.; Bogdanowski, A.; Gournas, C.; More, T.; Sundag, C.; Walter, S.; Heinisch, J.J.; Ejsing, C.S.; André, B.; et al. Uptake of exogenous serine is important to maintain sphingolipid homeostasis in Saccharomyces cerevisiae. PLoS Genet. 2020, 16, e1008745. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Lone, M.A.; Schneiter, R.; Chang, A. Orm1 and Orm2 are conserved endoplasmic reticulum membrane proteins regulating lipid homeostasis and protein quality control. Proc Natl. Acad. Sci. USA 2010, 107, 5851–5856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breslow, D.K.; Collins, S.R.; Bodenmiller, B.; Aebersold, R.; Simons, K.; Shevchenko, A.; Ejsing, C.S.; Weissman, J.S. Orm family proteins mediate sphingolipid homeostasis. Nature 2010, 463, 1048–1053. [Google Scholar] [CrossRef] [Green Version]
- Niles, B.J.; Powers, T. Plasma membrane proteins Slm1 and Slm2 mediate activation of the AGC kinase Ypk1 by TORC2 and sphingolipids in S. cerevisiae. Cell Cycle 2012, 11, 3745–3749. [Google Scholar] [CrossRef] [Green Version]
- Fröhlich, F.; Moreira, K.; Aguilar, P.S.; Hubner, N.C.; Mann, M.; Walter, P.; Walther, T.C. A genome-wide screen for genes affecting eisosomes reveals Nce102 function in sphingolipid signaling. J. Cell Biol. 2009, 185, 1227–1242. [Google Scholar] [CrossRef] [Green Version]
- Roelants, F.M.; Breslow, D.K.; Muir, A.; Weissman, J.S.; Thorner, J. Protein kinase Ypk1 phosphorylates regulatory proteins Orm1 and Orm2 to control sphingolipid homeostasis in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2011, 108, 19222–19227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, O.; Weyer, Y.; Baumann, V.; Widerin, M.A.; Eising, S.; Angelova, M.; Schleiffer, A.; Kremser, L.; Lindner, H.; Peter, M.; et al. Endosome and Golgi-associated degradation (EGAD) of membrane proteins regulates sphingolipid metabolism. EMBO J. 2019, 38, e101433. [Google Scholar] [CrossRef]
- Muir, A.; Ramachandran, S.; Roelants, F.M.; Timmons, G.; Thorner, J. TORC2-dependent protein kinase Ypk1 phosphorylates ceramide synthase to stimulate synthesis of complex sphingolipids. eLife 2014, 3, e03779. [Google Scholar] [CrossRef]
- Aronova, S.; Wedaman, K.; Aronov, P.A.; Fontes, K.; Ramos, K.; Hammock, B.D.; Powers, T. Regulation of ceramide biosynthesis by TOR complex 2. Cell Metab. 2008, 7, 148–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fröhlich, F.; Olson, D.K.; Christiano, R.; Farese, R.V., Jr.; Walther, T.C. Proteomic and phosphoproteomic analyses of yeast reveal the global cellular response to sphingolipid depletion. Proteomics 2016, 16, 2759–2763. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, N.; Han, G.; Somashekarappa, N.; Gable, K.; Dunn, T.; Kohlwein, S.D. Regulation of sphingolipid biosynthesis by the morphogenesis checkpoint kinase Swe1. J. Biol. Chem. 2016, 291, 2524–2534. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, N.; Visram, M.; Cristobal-Sarramian, A.; Sarkleti, F.; Kohlwein, S.D. Morphogenesis checkpoint kinase Swe1 is the executor of lipolysis-dependent cell-cycle progression. Proc Natl. Acad. Sci. USA 2015, 112, E1077–E1085. [Google Scholar] [CrossRef] [Green Version]
- Meggio, F.; Pinna, L.A. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003, 17, 349–368. [Google Scholar] [CrossRef]
- Trembley, J.H.; Wang, G.; Unger, G.; Slaton, J.; Ahmed, K. Protein kinase CK2 in health and disease. Cell. Mol. Life Sci. 2009, 66, 1858–1867. [Google Scholar] [CrossRef] [Green Version]
- Fresques, T.; Niles, B.; Aronova, S.; Mogri, H.; Rakhshandehroo, T.; Powers, T. Regulation of ceramide synthase by casein kinase 2-dependent phosphorylation in Saccharomyces cerevisiae. J. Biol. Chem. 2015, 290, 1395–1403. [Google Scholar] [CrossRef] [Green Version]
- González, A.; Hall, M.N. Nutrient sensing and TOR signaling in yeast and mammals. EMBO J. 2017, 36, 397–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, G.; Shen, X.; Jiang, Y. Rapamycin activates Tap42-associated phosphatases by abrogating their association with Tor complex 1. EMBO J. 2006, 25, 3546–3555. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, C.; Santos, A.; Gable, K.; Epstein, S.; Gururaj, C.; Chymkowitch, P.; Pultz, D.; Rødkær, S.V.; Clay, L.; Bjørås, M.; et al. TORC1 inhibits GSK3-mediated Elo2 phosphorylation to regulate very long chain fatty acid synthesis and autophagy. Cell Rep. 2013, 5, 1036–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, D.K.; Fröhlich, F.; Christiano, R.; Hannibal-Bach, H.K.; Ejsing, C.S.; Walther, T.C. Rom2-dependent phosphorylation of Elo2 controls the abundance of very long-chain fatty acids. J. Biol. Chem. 2015, 290, 4238–4247. [Google Scholar] [CrossRef] [Green Version]
- Swinnen, E.; Wilms, T.; Idkowiak-Baldys, J.; Smets, B.; De Snijder, P.; Accardo, S.; Ghillebert, R.; Thevissen, K.; Cammue, B.; De Vos, D.; et al. The protein kinase Sch9 is a key regulator of sphingolipid metabolism in Saccharomyces cerevisiae. Mol. Biol. Cell 2014, 25, 196–211. [Google Scholar] [CrossRef]
- Urban, J.; Soulard, A.; Huber, A.; Lippman, S.; Mukhopadhyay, D.; Deloche, O.; Wanke, V.; Anrather, D.; Ammerer, G.; Riezman, H.; et al. Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol. Cell 2007, 26, 663–674. [Google Scholar] [CrossRef]
- Loewith, R. TORC1 signaling in budding yeast. In The Enzymes; Academic Press: Cambridge, MA, USA, 2010; Volume 27, pp. 147–175. [Google Scholar]
- Takahara, T.; Maeda, T. Transient sequestration of TORC1 into stress granules during heat stress. Mol. Cell 2012, 47, 242–252. [Google Scholar] [CrossRef] [Green Version]
- De Craene, J.O.; Bertazzi, D.L.; Bär, S.; Friant, S. Phosphoinositides, major actors in membrane trafficking and lipid signaling pathways. Int. J. Mol. Sci. 2017, 18, 634. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Takematsu, H.; Yamaji, T.; Hiramoto, S.; Kozutsumi, Y. Disturbance of sphingolipid biosynthesis abrogates the signaling of Mss4, phosphatidylinositol-4-phosphate 5-kinase, in yeast. J. Biol. Chem. 2005, 280, 18087–18094. [Google Scholar] [CrossRef] [Green Version]
- Gallego, O.; Betts, M.J.; Gvozdenovic-Jeremic, J.; Maeda, K.; Matetzki, C.; Aguilar-Gurrieri, C.; Beltran-Alvarez, P.; Bonn, S.; Fernández-Tornero, C.; Jensen, L.J.; et al. A systematic screen for protein–lipid interactions in Saccharomyces cerevisiae. Mol. Syst. Biol. 2010, 6, 430. [Google Scholar] [CrossRef] [Green Version]
- Jesch, S.A.; Gaspar, M.L.; Stefan, C.J.; Aregullin, M.A.; Henry, S.A. Interruption of inositol sphingolipid synthesis triggers Stt4p-dependent protein kinase C signaling. J. Biol. Chem. 2010, 285, 41947–41960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desrivieres, S.; Cooke, F.T.; Parker, P.J.; Hall, M.N. MSS4, a phosphatidylinositol-4-phosphate 5-kinase required for organization of the actin cytoskeleton in Saccharomyces cerevisiae. J. Biol. Chem. 1998, 273, 15787–15793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audhya, A.; Emr, S.D. Stt4 PI 4-kinase localizes to the plasma membrane and functions in the Pkc1-mediated MAP kinase cascade. Dev. Cell 2002, 2, 593–605. [Google Scholar] [CrossRef] [Green Version]
- Daquinag, A.; Fadri, M.; Jung, S.Y.; Qin, J.; Kunz, J. The yeast PH domain proteins Slm1 and Slm2 are targets of sphingolipid signaling during the response to heat stress. Mol. Cell. Biol. 2007, 27, 633–650. [Google Scholar] [CrossRef] [Green Version]
- Levin, D.E. Regulation of cell wall biogenesis in Saccharomyces cerevisiae: The cell wall integrity signaling pathway. Genetics 2011, 189, 1145–1175. [Google Scholar] [CrossRef] [Green Version]
- Dunayevich, P.; Baltanás, R.; Clemente, J.A.; Couto, A.; Sapochnik, D.; Vasen, G.; Colman-Lerner, A. Heat-stress triggers MAPK crosstalk to turn on the hyperosmotic response pathway. Sci. Rep. 2018, 8, 15168. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, Y.; Katsuki, Y.; Tanaka, S.; Kawaguchi, R.; Denda, H.; Ikeda, T.; Funato, K.; Tani, M. Protective role of the HOG pathway against the growth defect caused by impaired biosynthesis of complex sphingolipids in yeast Saccharomyces cerevisiae. Mol. Microbiol. 2018, 107, 363–386. [Google Scholar] [CrossRef] [Green Version]
- Tani, M.; Funato, K. Protection mechanisms against aberrant metabolism of sphingolipids in budding yeast. Curr. Genet. 2018, 64, 1021–1028. [Google Scholar] [CrossRef]
- Jenkins, G.M.; Richards, A.; Wahl, T.; Mao, C.; Obeid, L.; Hannun, Y. Involvement of yeast sphingolipids in the heat stress response of Saccharomyces cerevisiae. J. Biol. Chem. 1997, 272, 32566–32572. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Miao, Y.; Yamane, Y.; Zhang, C.; Shokat, K.M.; Takematsu, H.; Kozutsumi, Y.; Drubin, D.G. Orm protein phosphoregulation mediates transient sphingolipid biosynthesis response to heat stress via the Pkh-Ypk and Cdc55-PP2A pathways. Mol. Biol. Cell 2012, 23, 2388–2398. [Google Scholar] [CrossRef] [PubMed]
- Friant, S.; Lombardi, R.; Schmelzle, T.; Hall, M.N.; Riezman, H. Sphingoid base signaling via Pkh kinases is required for endocytosis in yeast. EMBO J. 2001, 20, 6783–6792. [Google Scholar] [CrossRef] [Green Version]
- Prinz, W.A.; Toulmay, A.; Balla, T. The functional universe of membrane contact sites. Nat. Rev. Mol. Cell Biol. 2020, 21, 7–24. [Google Scholar] [CrossRef]
- Scorrano, L.; De Matteis, M.A.; Emr, S.; Giordano, F.; Hajnóczky, G.; Kornmann, B.; Lackner, L.L.; Pellegrini, L.; Reinisch, K.; Rizzuto, R.; et al. Coming together to define membrane contact sites. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Manford, A.G.; Stefan, C.J.; Yuan, H.L.; MacGurn, J.A.; Emr, S.D. ER-to-plasma membrane tethering proteins regulate cell signaling and ER morphology. Dev. Cell 2012, 23, 1129–1140. [Google Scholar] [CrossRef] [Green Version]
- West, M.; Zurek, N.; Hoenger, A.; Voeltz, G.K. A 3D analysis of yeast ER structure reveals how ER domains are organized by membrane curvature. J. Cell Biol. 2011, 193, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Pichler, H.; Gaigg, B.; Hrastnik, C.; Achleitner, G.; Kohlwein, S.D.; Zellnig, G.; Perktold, A.; Daum, G. A subfraction of the yeast endoplasmic reticulum associates with the plasma membrane and has a high capacity to synthesize lipids. Eur. J. Biochem. 2001, 268, 2351–2361. [Google Scholar] [CrossRef]
- Quon, E.; Sere, Y.Y.; Chauhan, N.; Johansen, J.; Sullivan, D.P.; Dittman, J.S.; Rice, W.J.; Chan, R.B.; Paolo, G.D.; Beh, C.T.; et al. Endoplasmic reticulum-plasma membrane contact sites integrate sterol and phospholipid regulation. PLoS Biol. 2018, 16, e2003864. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, T.; Gecht, M.; Covino, R.; Hummer, G.; Surma, M.A.; Klose, C.; Arai, H.; Kono, N.; Stefan, C.J. Osh proteins control nanoscale lipid organization necessary for PI (4, 5) P2 synthesis. Mol. Cell 2019, 75, 1043–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefan, C.J.; Manford, A.G.; Baird, D.; Yamada-Hanff, J.; Mao, Y.; Emr, S.D. Osh proteins regulate phosphoinositide metabolism at ER-plasma membrane contact sites. Cell 2011, 144, 389–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, J.; Torta, F.; Masai, K.; Lucast, L.; Czapla, H.; Tanner, L.B.; Narayanaswamy, P.; Wenk, M.R.; Nakatsu, F.; De Camilli, P. PI4P/phosphatidylserine countertransport at ORP5-and ORP8-mediated ER–plasma membrane contacts. Science 2015, 349, 428–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zewe, J.P.; Wills, R.C.; Sangappa, S.; Goulden, B.D.; Hammond, G.R. SAC1 degrades its lipid substrate PtdIns4P in the endoplasmic reticulum to maintain a steep chemical gradient with donor membranes. eLife 2018, 7, e35588. [Google Scholar] [CrossRef]
- de Saint-Jean, M.; Delfosse, V.; Douguet, D.; Chicanne, G.; Payrastre, B.; Bourguet, W.; Antonny, B.; Drin, G. Osh4p exchanges sterols for phosphatidylinositol 4-phosphate between lipid bilayers. J. Cell Biol. 2011, 195, 965–978. [Google Scholar] [CrossRef] [Green Version]
- Von Filseck, J.M.; Vanni, S.; Mesmin, B.; Antonny, B.; Drin, G. A phosphatidylinositol-4-phosphate powered exchange mechanism to create a lipid gradient between membranes. Nat. Commun. 2015, 6, 6671. [Google Scholar] [CrossRef] [Green Version]
- Omnus, D.J.; Manford, A.G.; Bader, J.M.; Emr, S.D.; Stefan, C.J. Phosphoinositide kinase signaling controls ER-PM cross-talk. Mol. Biol. Cell 2016, 27, 1170–1180. [Google Scholar] [CrossRef]
- Schmidt, O.; Weyer, Y.; Sprenger, S.; Widerin, M.A.; Eising, S.; Baumann, V.; Angelova, M.; Loewith, R.; Stefan, C.J.; Hess, M.W.; et al. TOR complex 2 (TORC2) signaling and the ESCRT machinery cooperate in the protection of plasma membrane integrity in yeast. J. Biol. Chem. 2020, 295, 12028–12044. [Google Scholar] [CrossRef]
- Rispal, D.; Eltschinger, S.; Stahl, M.; Vaga, S.; Bodenmiller, B.; Abraham, Y.; Filipuzzi, I.; Movva, N.R.; Aebersold, R.; Helliwell, S.B.; et al. Target of rapamycin complex 2 regulates actin polarization and endocytosis via multiple pathways. J. Biol. Chem. 2015, 290, 14963–14978. [Google Scholar] [CrossRef] [Green Version]
- Riggi, M.; Bourgoint, C.; Macchione, M.; Matile, S.; Loewith, R.; Roux, A. TORC2 controls endocytosis through plasma membrane tension. J. Cell Biol. 2019, 218, 2265–2276. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.S.; Zhang, Z.; Zhao, W.D.; Wang, D.; Luo, F.; Wu, L.G. Calcineurin is universally involved in vesicle endocytosis at neuronal and nonneuronal secretory cells. Cell Rep. 2014, 7, 982–988. [Google Scholar] [CrossRef] [Green Version]
- Stradalova, V.; Blazikova, M.; Grossmann, G.; Opekarová, M.; Tanner, W.; Malinsky, J. Distribution of cortical endoplasmic reticulum determines positioning of endocytic events in yeast plasma membrane. PLoS ONE 2012, 7, e35132. [Google Scholar] [CrossRef] [PubMed]
- Qian, T.; Li, C.; He, R.; Wan, C.; Liu, Y.; Yu, H. Calcium-dependent and-independent lipid transfer mediated by tricalbins in yeast. J. Biol. Chem. 2021, 296, 100729. [Google Scholar] [CrossRef] [PubMed]
- Thomas, F.B.; Omnus, D.J.; Bader, J.M.; Chung, G.H.; Kono, N.; Stefan, C.J. Tricalbin proteins regulate plasma membrane phospholipid homeostasis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Jorgensen, J.R.; Tei, R.; Baskin, J.M.; Michel, A.H.; Kornmann, B.; Emr, S.D. ESCRT-III and ER–PM contacts maintain lipid homeostasis. Mol. Biol. Cell 2020, 31, 1302–1313. [Google Scholar] [CrossRef]
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The ESCRT pathway. Dev. Cell 2011, 21, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Vietri, M.; Radulovic, M.; Stenmark, H. The many functions of ESCRTs. Nat. Rev. Mol. Cell. Biol. 2020, 21, 25–42. [Google Scholar] [CrossRef]
- Mioka, T.; Guo, T.; Wang, S.; Tsuji, T.; Kishimoto, T.; Fujimoto, T.; Tanaka, K. Characterization of micron-scale protein-depleted plasma membrane domains in phosphatidylserine-deficient yeast cells. J. Cell Sci. 2021, 135, jcs256529. [Google Scholar] [CrossRef]
- Cordeiro, R.M. Molecular structure and permeability at the interface between phase-separated membrane domains. J. Phys. Chem. B 2018, 122, 6954–6965. [Google Scholar] [CrossRef]
- Liu, L.K.; Choudhary, V.; Toulmay, A.; Prinz, W.A. An inducible ER–Golgi tether facilitates ceramide transport to alleviate lipotoxicity. J. Cell Biol. 2017, 216, 131–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toulmay, A.; Prinz, W.A. A conserved membrane-binding domain targets proteins to organelle contact sites. J. Cell Sci. 2012, 125, 49–58. [Google Scholar] [CrossRef] [Green Version]
- Kama, R.; Kanneganti, V.; Ungermann, C.; Gerst, J.E. The yeast Batten disease orthologue Btn1 controls endosome–Golgi retrograde transport via SNARE assembly. J. Cell Biol. 2011, 195, 203–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henne, W.M.; Zhu, L.; Balogi, Z.; Stefan, C.; Pleiss, J.A.; Emr, S.D. Mdm1/Snx13 is a novel ER–endolysosomal interorganelle tethering protein. J. Cell Biol. 2015, 210, 541–551. [Google Scholar] [CrossRef]
- Hariri, H.; Speer, N.; Bowerman, J.; Rogers, S.; Fu, G.; Reetz, E.; Datta, S.; Feathers, J.R.; Ugrankar, R.; Nicastro, D.; et al. Mdm1 maintains endoplasmic reticulum homeostasis by spatially regulating lipid droplet biogenesis. J. Cell Biol. 2019, 218, 1319–1334. [Google Scholar] [CrossRef] [Green Version]
- Hanada, K.; Kumagai, K.; Yasuda, S.; Miura, Y.; Kawano, M.; Fukasawa, M.; Nishijima, M. Molecular machinery for non-vesicular trafficking of ceramide. Nature 2003, 426, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, K.; Hanada, K. Structure, functions and regulation of CERT, a lipid-transfer protein for the delivery of ceramide at the ER-Golgi membrane contact sites. FEBS Lett. 2019, 593, 2366–2377. [Google Scholar] [CrossRef] [Green Version]
- Kihara, A.; Igarashi, Y. Identification and characterization of a Saccharomyces cerevisiae gene, RSB1, involved in sphingoid long-chain base release. J. Biol. Chem. 2002, 277, 30048–30054. [Google Scholar] [CrossRef] [Green Version]
- Heidler, S.A.; Radding, J.A. The AUR1 gene in Saccharomyces cerevisiae encodes dominant resistance to the antifungal agent aureobasidin A (LY295337). Antimicrob. Agents Chemother. 1995, 39, 2765–2769. [Google Scholar] [CrossRef] [Green Version]
- Hashida-Okado, T.; Ogawa, A.; Endo, M.; Yasumoto, R.; Takesako, K.; Kato, I. AUR1, a novel gene conferring aureobasidin resistance on Saccharomyces cerevisiae: A study of defective morphologies in Aur1p-depleted cells. Mol. Gen. Genet. 1996, 251, 236–244. [Google Scholar]
- Schorling, S.; Vallée, B.; Barz, W.P.; Riezman, H.; Oesterhelt, D. Lag1p and Lac1p are essential for the Acyl-CoA–dependent ceramide synthase reaction in Saccharomyces cerevisae. Mol. Biol. Cell 2001, 12, 3417–3427. [Google Scholar] [CrossRef] [PubMed]
- Kajiwara, K.; Muneoka, T.; Watanabe, Y.; Karashima, T.; Kitagaki, H.; Funato, K. Perturbation of sphingolipid metabolism induces endoplasmic reticulum stress-mediated mitochondrial apoptosis in budding yeast. Mol. Microbiol. 2012, 86, 1246–1261. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, T.; Büttner, S. Lipids and cell death in yeast. FEMS Yeast Res. 2014, 14, 179–197. [Google Scholar] [CrossRef] [Green Version]
- Rego, A.; Duarte, A.M.; Azevedo, F.; Sousa, M.J.; Côrte-Real, M.; Chaves, S.R. Cell wall dynamics modulate acetic acid-induced apoptotic cell death of Saccharomyces cerevisiae. Microb. Cell 2014, 1, 303. [Google Scholar] [CrossRef] [Green Version]
- Voynova, N.S.; Roubaty, C.; Vazquez, H.M.; Mallela, S.K.; Ejsing, C.S.; Conzelmann, A. Saccharomyces cerevisiae is dependent on vesicular traffic between the Golgi apparatus and the vacuole when inositolphosphorylceramide synthase Aur1 is inactivated. Eukaryot. Cell 2015, 14, 1203–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerantola, V.; Guillas, I.; Roubaty, C.; Vionnet, C.; Uldry, D.; Knudsen, J.; Conzelmann, A. Aureobasidin A arrests growth of yeast cells through both ceramide intoxication and deprivation of essential inositolphosphorylceramides. Mol. Microbiol. 2009, 71, 1523–1537. [Google Scholar] [CrossRef] [Green Version]
- Epstein, S.; Castillon, G.A.; Qin, Y.; Riezman, H. An essential function of sphingolipids in yeast cell division. Mol. Microbiol. 2012, 84, 1018–1032. [Google Scholar] [CrossRef]
- Su, W.C.; Lin, Y.H.; Pagac, M.; Wang, C.W. Seipin negatively regulates sphingolipid production at the ER–LD contact site. J. Cell Biol. 2019, 218, 3663–3680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, C.; Xu, R.; Bielawska, A.; Szulc, Z.M.; Obeid, L.M. Cloning and characterization of a Saccharomyces cerevisiae alkaline ceramidase with specificity for dihydroceramide. J. Biol. Chem. 2000, 275, 31369–31378. [Google Scholar] [CrossRef] [Green Version]
- Murakami, S.; Shimamoto, T.; Nagano, H.; Tsuruno, M.; Okuhara, H.; Hatanaka, H.; Tojo, H.; Kodama, Y.; Funato, K. Producing human ceramide-NS by metabolic engineering using yeast Saccharomyces cerevisiae. Sci. Rep. 2015, 5, 16319. [Google Scholar] [CrossRef]
- Siskind, L.J.; Kolesnick, R.N.; Colombini, M. Ceramide forms channels in mitochondrial outer membranes at physiologically relevant concentrations. Mitochondrion 2006, 6, 118–125. [Google Scholar] [CrossRef] [Green Version]
- Siskind, L.J.; Feinstein, L.; Yu, T.; Davis, J.S.; Jones, D.; Choi, J.; Zuckerman, J.E.; Tan, W.; Hill, R.B.; Hardwick, J.M.; et al. Anti-apoptotic Bcl-2 family proteins disassemble ceramide channels. J. Biol. Chem. 2008, 283, 6622–6630. [Google Scholar] [CrossRef] [Green Version]
- Colombini, M. Ceramide channels and mitochondrial outer membrane permeability. J. Bioenerg. Biomembr. 2017, 49, 57–64. [Google Scholar] [CrossRef]
- Rego, A.; Costa, M.; Chaves, S.R.; Matmati, N.; Pereira, H.; Sousa, M.J.; Moradas-Ferreira, P.; Hannun, Y.A.; Costa, V.; Côrte-Real, M. Modulation of mitochondrial outer membrane permeabilization and apoptosis by ceramide metabolism. PLoS ONE 2012, 7, e48571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wissing, S.; Ludovico, P.; Herker, E.; Büttner, S.; Engelhardt, S.M.; Decker, T.; Link, A.; Proksch, A.; Rodrigues, F.; Corte-Real, M.; et al. An AIF orthologue regulates apoptosis in yeast. J. Cell Biol. 2004, 166, 969–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Büttner, S.; Eisenberg, T.; Carmona-Gutierrez, D.; Ruli, D.; Knauer, H.; Ruckenstuhl, C.; Sigrist, C.; Wissing, S.; Kollroser, M.; Fröhlich, S.; et al. Endonuclease G regulates budding yeast life and death. Mol. Cell 2007, 25, 233–246. [Google Scholar] [CrossRef]
- Martins, V.M.; Fernandes, T.R.; Lopes, D.; Afonso, C.B.; Domingues, M.R.; Côrte-Real, M.; Sousa, M.J. Contacts in death: The role of the ER–Mitochondria axis in acetic acid-induced apoptosis in yeast. J. Mol. Biol. 2019, 431, 273–288. [Google Scholar] [CrossRef]
- Wang, H.; Sreenivasan, U.; Hu, H.; Saladino, A.; Polster, B.M.; Lund, L.M.; Gong, D.W.; Stanley, W.C.; Sztalryd, C. Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. J. Lipid Res. 2011, 52, 2159–2168. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.B.; Olzmann, J.A. Lipid droplets and lipotoxicity during autophagy. Autophagy 2017, 13, 2002–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benador, I.Y.; Veliova, M.; Liesa, M.; Shirihai, O.S. Mitochondria bound to lipid droplets: Where mitochondrial dynamics regulate lipid storage and utilization. Cell Metab. 2019, 29, 827–835. [Google Scholar] [CrossRef] [Green Version]
- Faergeman, N.J.; Feddersen, S.; Christiansen, J.K.; Larsen, M.K.; Schneiter, R.; Ungermann, C.; Mutenda, K.; Roepstorff, P.; Knudsen, J. Acyl-CoA-binding protein, Acb1p, is required for normal vacuole function and ceramide synthesis in Saccharomyces cerevisiae. Biochem. J. 2004, 380, 907–918. [Google Scholar] [CrossRef] [Green Version]
- Elbaz-Alon, Y.; Rosenfeld-Gur, E.; Shinder, V.; Futerman, A.H.; Geiger, T.; Schuldiner, M. A dynamic interface between vacuoles and mitochondria in yeast. Dev. Cell 2014, 30, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Ungermann, C. vCLAMPs—An intimate link between vacuoles and mitochondria. Curr. Opin. Cell Biol. 2015, 35, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Hariri, H.; Rogers, S.; Ugrankar, R.; Liu, Y.L.; Feathers, J.R.; Henne, W.M. Lipid droplet biogenesis is spatially coordinated at ER–vacuole contacts under nutritional stress. EMBO Rep. 2018, 19, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Roberts, P.; Chen, Y.; Kvam, E.; Shulga, N.; Huang, K.; Lemmon, S.; Goldfarb, D.S. Nucleus–vacuole junctions in Saccharomyces cerevisiae are formed through the direct interaction of Vac8p with Nvj1p. Mol. Biol. Cell 2000, 11, 2445–2457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otsu, M.; Toume, M.; Yamaguchi, Y.; Tani, M. Proper regulation of inositolphosphorylceramide levels is required for acquirement of low pH resistance in budding yeast. Sci. Rep. 2020, 10, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Matos, G.S.; Madeira, J.B.; Fernandes, C.M.; Dasilva, D.; Masuda, C.A.; Del Poeta, M.; Montero-Lomelí, M. Regulation of sphingolipid synthesis by the G1/S transcription factor Swi4. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2021, 1866, 158983. [Google Scholar] [CrossRef]
- Wells, G.B.; Dickson, R.C.; Lester, R.L. Heat-induced elevation of ceramide in Saccharomyces cerevisiae via de novo synthesis. J. Biol. Chem. 1998, 273, 7235–7243. [Google Scholar] [CrossRef] [Green Version]
- Piña, F.; Yagisawa, F.; Obara, K.; Gregerson, J.D.; Kihara, A.; Niwa, M. Sphingolipids activate the endoplasmic reticulum stress surveillance pathway. J. Cell Biol. 2018, 217, 495–505. [Google Scholar] [CrossRef]
- Fabri, J.H.T.M.; de Sá, N.P.; Malavazi, I.; Del Poeta, M. The dynamics and role of sphingolipids in eukaryotic organisms upon thermal adaptation. Prog. Lipid Res. 2020, 80, 101063. [Google Scholar] [CrossRef]
- Clay, L.; Caudron, F.; Denoth-Lippuner, A.; Boettcher, B.; Frei, S.B.; Snapp, E.L.; Barral, Y. A sphingolipid-dependent diffusion barrier confines ER stress to the yeast mother cell. eLife 2014, 3, e01883. [Google Scholar] [CrossRef]
- Yabuki, Y.; Ikeda, A.; Araki, M.; Kajiwara, K.; Mizuta, K.; Funato, K. Sphingolipid/Pkh1/2-TORC1/Sch9 signaling regulates ribosome biogenesis in tunicamycin-induced stress response in yeast. Genetics 2019, 212, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.L.; Souza, C.M.; Pichler, H.; Dewhurst, G.; Schaad, O.; Kajiwara, K.; Wakabayashi, H.; Ivanova, T.; Castillon, G.A.; Piccolis, M.; et al. Functional interactions between sphingolipids and sterols in biological membranes regulating cell physiology. Mol. Biol. Cell 2009, 20, 2083–2095. [Google Scholar] [CrossRef] [Green Version]
- Breslow, D.K.; Weissman, J.S. Membranes in balance: Mechanisms of sphingolipid homeostasis. Mol. Cell 2010, 40, 267–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, K.; Sampaio, J.L. Membrane organization and lipid rafts. Cold Spring Harb. Perspect. Biol. 2011, 3, a004697. [Google Scholar] [CrossRef] [PubMed]
- Murley, A.; Yamada, J.; Niles, B.J.; Toulmay, A.; Prinz, W.A.; Powers, T.; Nunnari, J. Sterol transporters at membrane contact sites regulate TORC1 and TORC2 signaling. J. Cell Biol. 2017, 216, 2679–2689. [Google Scholar] [CrossRef] [Green Version]
- Gatta, A.T.; Levine, T.P. Piecing Together the Patchwork of Contact Sites. Trends Cell Biol. 2017, 27, 214–229. [Google Scholar] [CrossRef]
- Tsuji, T.; Fujimoto, T. Lipids and lipid domains of the yeast vacuole. Biochem. Soc. Trans. 2018, 46, 1047–1054. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schlarmann, P.; Ikeda, A.; Funato, K. Membrane Contact Sites in Yeast: Control Hubs of Sphingolipid Homeostasis. Membranes 2021, 11, 971. https://doi.org/10.3390/membranes11120971
Schlarmann P, Ikeda A, Funato K. Membrane Contact Sites in Yeast: Control Hubs of Sphingolipid Homeostasis. Membranes. 2021; 11(12):971. https://doi.org/10.3390/membranes11120971
Chicago/Turabian StyleSchlarmann, Philipp, Atsuko Ikeda, and Kouichi Funato. 2021. "Membrane Contact Sites in Yeast: Control Hubs of Sphingolipid Homeostasis" Membranes 11, no. 12: 971. https://doi.org/10.3390/membranes11120971