Insights into the Membranolytic Activity of Antimalarial Drug-Cell Penetrating Peptide Conjugates

 ,
,  , and
, and 
Abstract
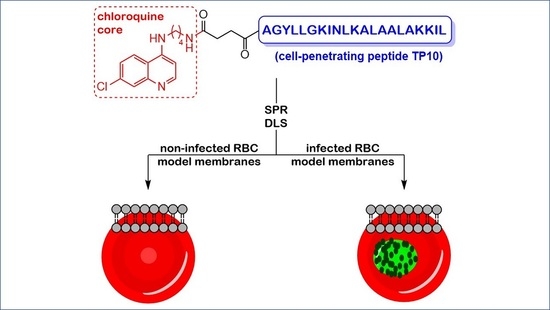
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. SPR Analysis of TP10 and Conjugates Binding to Model Membranes
2.2. Effects of CQ-TP10 Conjugates on the Size of the Lipid Vesicles
2.3. Effect of CQ-TP10 Conjugates on the Fluidity of Model Membranes
3. Discussion
4. Materials and Methods
4.1. Chemical Synthesis
4.2. Preparation of Liposomes
4.3. Determination of the Hydrodynamic Diameter and PDI of the Liposomes
4.4. Determination of Phase Transition Temperature and Cooperativity
4.5. Surface Plasmon Resonance Assays
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. World Malaria Report 2019; WHO: Geneva, Switzerland, 2019. [Google Scholar]
- Tse, E.G.; Korsik, M.; Todd, M.H. The past, present and future of anti-malarial medicines. Malar. J. 2019, 18, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grellier, P.; Rigomier, D.; Clavey, V.; Fruchart, J.C.; Schrevel, J. Lipid traffic between high density lipoproteins and Plasmodium falciparum-infected red blood cells. J. Cell Biol. 1991, 112, 267–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boddey, J.A.; Cowman, A.F. Plasmodium nesting: Remaking the erythrocyte from the inside out. Annu. Rev. Microbiol. 2013, 67, 243–269. [Google Scholar] [CrossRef] [PubMed]
- Ginsburg, H.; Kutner, S.; Krugliak, M.; Cabantchik, Z.I. Characterization of permeation pathways appearing in the host membrane of Plasmodium falciparum infected red blood cells. Mol. Biochem. Parasitol. 1985, 14, 313–322. [Google Scholar] [CrossRef]
- Schwartz, R.S.; Olson, J.A.; Raventos-Suarez, C.; Yee, M.; Health, R.H.; Lubin, B.; Nagel, R.L. Altered plasma membrane phospholipid organization in Plasmodium falciparum-infected human erythrocytes. Blood 1987, 69, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Maguire, P.A.; Sherman, I.W. Phospholipid composition, cholesterol content and cholesterol exchange in Plasmodium falciparum-infected red cells. Mol. Biochem. Parasitol. 1990, 38, 105–112. [Google Scholar] [CrossRef]
- Sherman, I.W. Biochemistry of Plasmodium (malaria parasites). Microbiol. Rev. 1979, 43, 453–495. [Google Scholar] [CrossRef]
- Hsiao, L.L.; Howard, R.J.; Aikawa, M.; Taraschi, T.F. Modification of host cell membrane lipid composition by the intra-erythrocytic human malaria parasite Plasmodium falciparum. Biochem. J. 1991, 274, 121–132. [Google Scholar] [CrossRef] [Green Version]
- Taraschi, T.F.; Parashar, A.; Hooks, M.; Rubin, H. Perturbation of red cell membrane structure during intracellular maturation of Plasmodium falciparum. Science 1986, 232, 102–104. [Google Scholar] [CrossRef]
- Kirk, K.; Lehane, A.M. Membrane transport in the malaria parasite and its host erythrocyte. Biochem. J. 2013, 457, 1–18. [Google Scholar] [CrossRef]
- Smith, J.E. Erythrocyte membrane: Structure, function, and pathophysiology. Vet. Pathol. 1987, 24, 471–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holz, G.G., Jr. Lipids and the malarial parasite. Bull. World Health Organ. 1977, 55, 237–248. [Google Scholar] [PubMed]
- Eda, S.; Sherman, I.W. Cytoadherence of malaria-infected red blood cells involves exposure of phosphatidylserine. Cell. Physiol. Biochem. 2002, 12, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Arrighi, R.B.G.; Ebikeme, C.; Jiang, Y.; Ranford-Cartwright, L.; Barrett, M.P.; Langel, Ü.; Faye, I. Cell-penetrating peptide TP10 shows broad-spectrum activity against both Plasmodium falciparum and Trypanosoma brucei brucei. Antimicrob. Agents Chemother. 2008, 52, 3414–3417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achtman, A.H.; Pilat, S.; Law, C.W.; Lynn, D.J.; Janot, L.; Mayer, M.L.; Ma, S.; Kindrachuk, J.; Finlay, B.B.; Brinkman, F.S.L.; et al. Effective adjunctive therapy by an innate defense regulatory peptide in a preclinical model of severe malaria. Sci. Transl. Med. 2012, 4, 135ra64. [Google Scholar] [CrossRef] [PubMed]
- Guergnon, J.; Dessauge, F.; Dominguez, V.; Viallet, J.; Bonnefoy, S.; Yuste, V.J.; Mercereau-Puijalon, O.; Cayla, X.; Rebollo, A.; Susin, S.A.; et al. Use of penetrating peptides interacting with PP1/PP2A proteins as a general approach for a drug phosphatase technology. Mol. Pharmacol. 2005, 69, 1115–1124. [Google Scholar] [CrossRef] [Green Version]
- Aguiar, L.; Biosca, A.; Lantero, E.; Gut, J.; Vale, N.; Rosenthal, P.J.; Nogueira, F.; Andreu, D.; Fernàndez-Busquets, X.; Gomes, P. Coupling the antimalarial cell penetrating peptide TP10 to classical antimalarial drugs primaquine and chloroquine produces strongly hemolytic conjugates. Molecules 2019, 24, 4559. [Google Scholar] [CrossRef] [Green Version]
- Aguiar, L.; Machado, M.; Sanches-Vaz, M.; Prudêncio, M.; Vale, N.; Gomes, P. Coupling the cell-penetrating peptides transportan and transportan 10 to primaquine enhances its activity against liver-stage malaria parasites. MedChemComm 2018, 10, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Deplazes, E.; Henriques, S.T.; Smith, J.J.; King, G.F.; Craik, D.J.; Mark, A.E.; Schroeder, C.I. Membrane-binding properties of gating modifier and pore-blocking toxins: Membrane interaction is not a prerequisite for modification of channel gating. Biochim. Biophys. Acta (BBA) Biomembr. 2016, 1858, 872–882. [Google Scholar] [CrossRef]
- Ferraz, R.; Pinheiro, M.; Gomes, A.; Teixeira, C.; Prudêncio, C.; Reis, S.; Gomes, P. Effects of novel triple-stage antimalarial ionic liquids on lipid membrane models. Bioorg. Med. Chem. Lett. 2017, 27, 4190–4193. [Google Scholar] [CrossRef]
- Porcar, I.; Codoñer, A.; Gómez, C.M.; Abad, C.; Campos, A. Interaction of quinine with model lipid membranes of different compositions. J. Pharm. Sci. 2003, 92, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.E.; Caseli, L. The interaction of mefloquine hydrochloride with cell membrane models at the air–water interface is modulated by the monolayer lipid composition. J. Colloid Interface Sci. 2014, 431, 24–30. [Google Scholar] [CrossRef] [PubMed]
- IUPAC-IUB Joint Commission on Biochemical Nomenclature. Nomenclature and symbolism for amino acids and peptides. Eur. J. Biochem. 1984, 138, 9–37. [Google Scholar] [CrossRef]
- Mitchell, J.S. Small molecule immunosensing using surface plasmon resonance. Sensors 2010, 10, 7323–7346. [Google Scholar] [CrossRef] [Green Version]
- Kanásová, M.; Nesměrák, K. Systematic review of liposomes’ characterization methods. Mon. Chem. Chem. Mon. 2017, 148, 1581–1593. [Google Scholar] [CrossRef]
- Michel, N.; Fabiano, A.-S.; Polidori, A.; Jack, R.; Pucci, B. Determination of phase transition temperatures of lipids by light scattering. Chem. Phys. Lipids 2006, 139, 11–19. [Google Scholar] [CrossRef]
- Cañadas, O.; Casals, C. Differential scanning calorimetry—Lipid interactions. In Protein-Lipid Interactions; Kleinschmidt, J.H., Ed.; Humana Press: New York, NY, USA, 2013; pp. 55–71. [Google Scholar]
- Nunes, C.; Brezesinski, G.; Pereira-Leite, C.; Lima, J.L.; Reis, S.; Lúcio, M.; Pereira-Leite, C. NSAIDs interactions with membranes: A biophysical approach. Langmuir 2011, 27, 10847–10858. [Google Scholar] [CrossRef]
- Akaki, M.; Nagayasu, E.; Nakano, Y.; Aikawa, M. Surface charge of Plasmodium falciparum merozoites as revealed by atomic force microscopy with surface potential spectroscopy. Parasitol. Res. 2002, 88, 16–20. [Google Scholar] [CrossRef]
- Makler, M.T. P. falciparum invasion of human red cells and cytoadherence to endothelial cells is dependent upon a parasite produced glycosidase. Biochem. Biophys. Res. Commun. 1987, 143, 461–466. [Google Scholar] [CrossRef]
- Florens, L.; Washburn, M.P.; Raine, J.D.; Anthony, R.M.; Grainger, M.; Haynes, J.D.; Moch, J.K.; Muster, N.; Sacci, J.B.; Tabb, D.L.; et al. A proteomic view of the Plasmodium falciparum life cycle. Nature 2002, 419, 520–526. [Google Scholar] [CrossRef]
- Ptaszyńska, N.; Gucwa, K.; Olkiewicz, K.; Heldt, M.; Serocki, M.; Stupak, A.; Martynow, D.; Dębowski, D.; Gitlin-Domagalska, A.; Lica, J.; et al. Conjugates of ciprofloxacin and levofloxacin with cell-penetrating peptide exhibit antifungal activity and mammalian cytotoxicity. Int. J. Mol. Sci. 2020, 21, 4696. [Google Scholar] [CrossRef] [PubMed]
- Pérez, B.C.; Teixeira, C.; Albuquerque, I.S.; Gut, J.; Rosenthal, P.J.; Gomes, J.R.B.; Prudêncio, M.; Gomes, P. N-cinnamoylated chloroquine analogues as dual-stage antimalarial leads. J. Med. Chem. 2013, 56, 556–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinheiro, M.; Arêde, M.; Caio, J.M.; Moiteiro, C.; Lúcio, M.; Reis, S. Drug-membrane interaction studies applied to N’-acetyl-rifabutin. Eur. J. Pharm. Biopharm. 2013, 85, 597–603. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguiar, L.; Pinheiro, M.; Neves, A.R.; Vale, N.; Defaus, S.; Andreu, D.; Reis, S.; Gomes, P. Insights into the Membranolytic Activity of Antimalarial Drug-Cell Penetrating Peptide Conjugates. Membranes 2021, 11, 4. https://doi.org/10.3390/membranes11010004
Aguiar L, Pinheiro M, Neves AR, Vale N, Defaus S, Andreu D, Reis S, Gomes P. Insights into the Membranolytic Activity of Antimalarial Drug-Cell Penetrating Peptide Conjugates. Membranes. 2021; 11(1):4. https://doi.org/10.3390/membranes11010004
Chicago/Turabian StyleAguiar, Luísa, Marina Pinheiro, Ana Rute Neves, Nuno Vale, Sira Defaus, David Andreu, Salette Reis, and Paula Gomes. 2021. "Insights into the Membranolytic Activity of Antimalarial Drug-Cell Penetrating Peptide Conjugates" Membranes 11, no. 1: 4. https://doi.org/10.3390/membranes11010004