
Nanoparticles and Antiviral Vaccines


Abstract
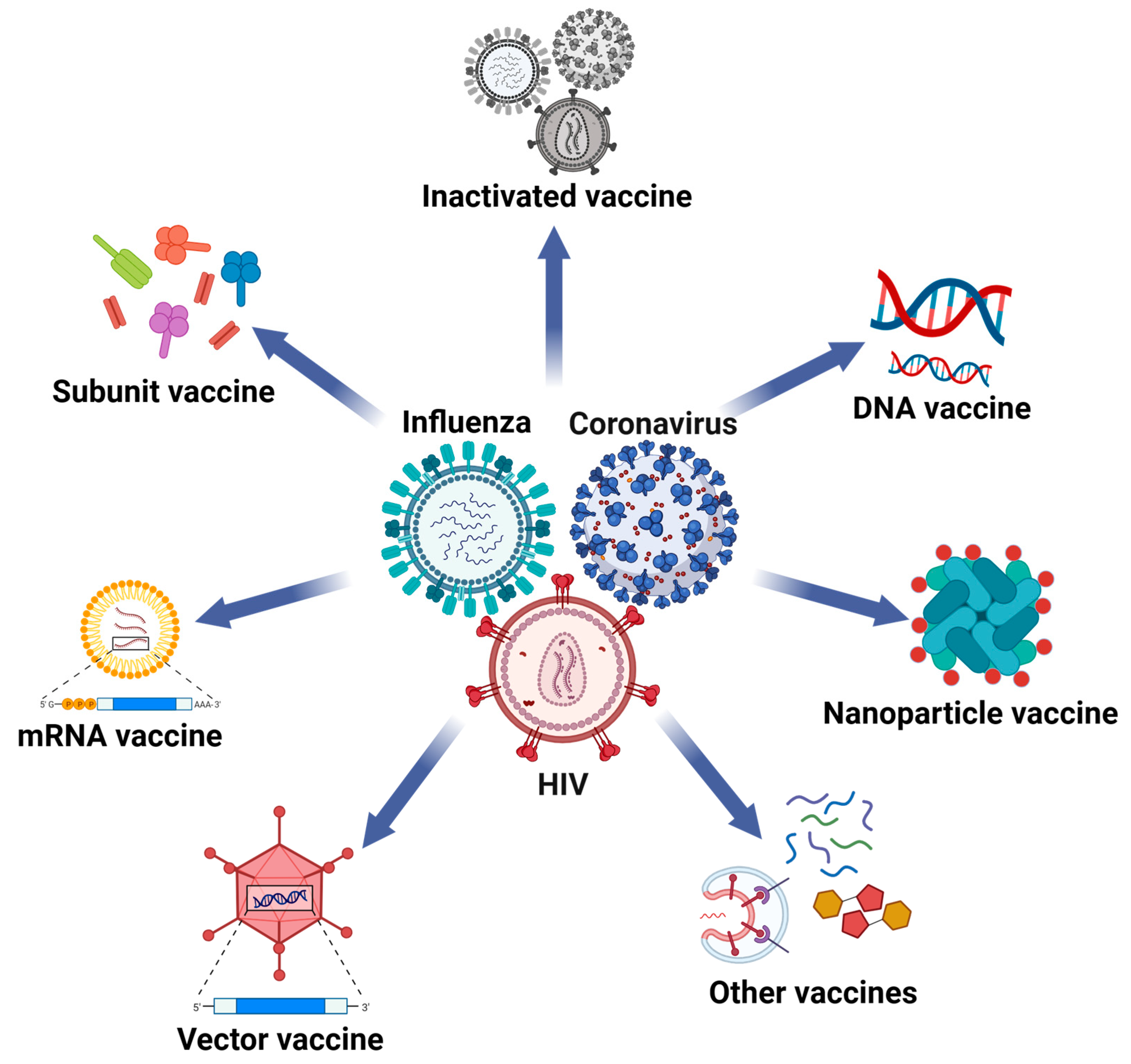
:1. Introduction
1.1. Major Pathogenic Viruses and Their Pandemics
1.2. Antiviral Vaccines and Adjuvants
2. The Applications of Nanoparticles in Influenza Vaccines

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Viruses | Vaccines | Nanoparticles | Sizes | Functions | Status | Ref. |
|---|---|---|---|---|---|---|
| Influenza | CNPs-KAg | Chitosan | 571.7 nm | Induce high levels of antigen-specific IgG antibodies and mucosal IgA antibodies | Pre-clinical | [85] |
| PLGA-KAg | PLGA | 200 nm to 300 nm | Reinforce the expansion of antigen-specific lymphocytes | Pre-clinical | [86] | |
| PNP hydrogel | PEG-PLA/ HPMC-C12 | 31 nm | Enhance the potency, durability, and breadth of humoral immune responses | Pre-clinical | [87] | |
| HA-ferritin | Ferritin | 30 nm | Induce stronger immune responses than licensed inactivated vaccines | Phase I | [88] | |
| H2HA-ferritin | Ferritin | 37.23 nm | Induce high titers of broadly neutralizing antibodies | Phase I | [89] | |
| qsMosaic-I53-dn5/qsCocktail-I53-dn5 | I53-dn5 | 50 nm | Induce broad-spectrum nAbs against both autologous and heterologous viral strains | Phase I | [90] | |
| 3M2e-rHF | Human heavy-chain ferritin | 15.9 nm | Induce M2e-specific IgG humoral immune responses and T cell immune responses, as well as mucosal IgA immune responses | Pre-clinical | [91] | |
| HMNF | Human heavy-chain ferritin | 18 nm | Induce robust and durable titers of antigen-specific antibody responses, along with sustained levels of cellular immune responses | Pre-clinical | [92] | |
| H7N9 VLP | HA-, NA-, and M1-based VLP | 113.9 nm | Elicit potent humoral and cellular immune responses in murine and avian animal models | Pre-clinical | [93,94] | |
| PapMV-CP-M2e | PapMV VLP | 86 nm | Induce stronger humoral immune responses upon co-administration with M2e peptides | Pre-clinical | [95] | |
| HBc VLP | M2e- and NP-containing HBc VLP | 30 nm | Induce stronger humoral immunity and cellular response, which provide protection against the lethal challenge of viruses | Pre-clinical | [96] | |
| AuNPs-HA/FliC | Gold nanoparticles (AuNPs) | 145 nm | Trigger protective immunity by inducing nAbs and cytokine secretion | Pre-clinical | [98] | |
| M2e-AuNPs | AuNPs | 12 nm | Provide strong and long-lasting protection against influenza attacks | Pre-clinical | [99] | |
| Inactivated vaccine with AgNPs | Silver nanoparticles (AgNPs) | 18 nm | Induce stronger IgA production and lower toxicity | Pre-clinical | [102] | |
| Inactivated vaccine with CaPNPs | Calcium phosphate nanoparticles (CaPNPs) | 450 nm to 500 nm | Induce high titers of antiviral antibodies and balanced T cell response | Pre-clinical | [105] | |
| HA-SiNP/c-di-GMP | Silica nanoparticles (SiNPs) | 100 nm to 500 nm | Induce high local IgG, IgA, and T cell responses within BAL | Pre-clinical | [108] | |
| H7-ND | Nanodiamond (ND) | 58 nm to 580 nm | Elicit stronger humoral immune responses than free H7 trimers | Pre-clinical | [109] |
3. The Applications of Nanoparticles in Coronavirus Vaccines
| Viruses | Vaccines | Nanoparticles | Size | Functions | Status | Ref. |
|---|---|---|---|---|---|---|
| Coronavirus | mRNA-1273 | LNP | 80 nm to 150 nm | Elicit potent neutralizing antibodies and mitigate severe illness | Approved (Moderna) | [135] |
| BNT162b2 | LNP | 80 nm to 150 nm | Elicit a high nAbs titer and provide efficient preventive protection | Approved (Pfizer-BioNTech) | [135] | |
| SYS6006 | LNP | 80 nm to 150 nm | Induce nAbs against SARS-CoV-2 VOCs in vivo and SARS-CoV-2-specific memory B and T cell immune responses | Approved (CSPC) | [136] | |
| ARCoV | LNP | 88.85 nm | Induce strong nAbs and T cell immunity with high stability | Approved (Walvax) | [137] | |
| mRNA-S+N | LNP | 80 nm to 150 nm | Elicit enhanced protection against both Delta and Omicron variants | Pre-clinical | [138] | |
| circRNA-RBD | LNP | 100 nm | Generate higher amounts and more persistent antigens and induce elevated levels of nAbs and Th1-biased immune responses | Pre-clinical | [139] | |
| RBD-I53-50 | I53-50 | 37 nm to 41 nm | Induce high nAb titers with low antigen doses | Approved (SK Bioscience) | [150] | |
| RBD/HR-ferritin | Ferritin | 15 nm to 20 nm | Induce strong nAbs with potential cross-neutralizing activity | Pre-clinical | [147] | |
| RBD-mi3 | mi3 | 20.7 nm | Thermostable and induce stronger nAbs responses than those in convalescent sera | Pre-clinical | [145] | |
| RBD-Fc-LS | Lumazine synthase (LS) | 15 nm | Elicit potent and persistent nAbs against SARS-CoV-2 mutants, SARS-CoV, and other bat coronaviruses | Pre-clinical | [55] | |
| DCFHP | Ferritin | 15 nm to 30 nm | Elicit potent, durable, and broad-spectrum neutralizing antibodies against nearly all the VOCs | Pre-clinical | [151] | |
| S-LS | LS | 63.2 nm | Elicit 25-fold higher nAbs than S only | Pre-clinical | [152] | |
| Mosaic RBD-mi3 | mi3 | 33 nm to 48 nm | Exhibit strong cross-reactive binding and neutralizing activities and prevent multiple betacoronaviruses attacks | Pre-clinical | [153,154] | |
| S/S1/S2 VLP | M1-based VLP | 80 nm to 200 nm | S- or S1-containing VLPs induce nAbs | Pre-clinical | [157] | |
| S/M/N/E VLP | VLP | 117 nm to 127 nm | Trigger high titers of IgG, leading to multifunctional Th1-biased T cell responses | Phase II | [158] | |
| CoVLP | Plant-derived VLP | 80 nm to 120 nm | Well tolerated and induces 10-fold higher nAbs than convalescent sera | Phase II/III | [159] | |
| SPG-eVLP | MLV Gag-based VLP | 100 nm to 140 nm | Induce robust and sustained nAbs and protect against authentic viruses | Phase I/II | [160] | |
| S-EABR eVLP | ESCRT-based VLP | 40 nm to 60 nm | Achieve in vivo assembly and provide long-term protection against viral mutants | Pre-clinical | [161] | |
| NVX-CoV2373 | PS80/Matrix-M | 27.2 nm | Elicit multifunctional T cell and B cell immune responses | Approved (Novavax) | [162,163,164,165,166] | |
| rS1-E-PLGA | PLGA | 670 nm | Induce high nAbs titers and enhance cellular immune responses | Pre-clinical | [168] | |
| PAPE-RBD | PAPE | 3551 nm | Induce higher antigen-specific nAb titers and more IFN-γ-secreting T cells than conventional alum-RBD vaccines | Pre-clinical | [169] | |
| RBD-AuNP | AuNP | 50 nm to 60 nm | Induce stronger long-term humoral responses than monomers | Pre-clinical | [170] | |
| S DNA vaccine with AuNS-chitosan | Gold nanostar (AuNS)/Chitosan | 35 nm to 48 nm | Induce both strong mucosal immunity and potent systemic humoral immune responses | Pre-clinical | [172] |
4. The Applications of Nanoparticles in HIV Vaccines
| Viruses | Vaccines | Nanoparticles | Size | Functions | Status | Ref. |
|---|---|---|---|---|---|---|
| HIV | BG505 SOSIP.664 gp140-ferritin | Ferritin | 30 nm to 40 nm | Enhance the immunogenicity of gp140 | Pre-clinical | [183] |
| CH848 10.17DT SOSIP-ferritin | Ferritin | 30 nm to 50 nm | Upregulate GC B cells and Tfh cells | Pre-clinical | [184,186] | |
| MD39 gp140-ferritin | Ferritin | 40 nm | Quickly enter into GCs and be captured by FDCs | Pre-clinical | [185] | |
| eOD-GT6 LS | LS | 32 nm | Activate both germline (GL) and mature B cells and elicit potent bNAbs | Phase I | [149] | |
| MPER-E2p | E2p | 24 nm | Elicit strong MPER-specific antibodies and bNAbs | Pre-clinical | [188] | |
| gp140-E2p | E2p | 37.6 nm | Induce a robust stimulation of cognate bNAb VRC01 receptor-containing B cells | Pre-clinical | [189] | |
| BG505 UFO gp140-E2p | E2p | 41.7 nm | Exhibit long retention in lymph node follicles and a great presentation on FDC dendrites | Pre-clinical | [190] | |
| P24-Nef/FLiC | PLGA | 198 nm | Enhance the immunogenicity of antigens and reduce required antigenic doses | Pre-clinical | [192] | |
| Env/NP | P1M10 | 20 nm | They have a high stability and induce more potent and broader neutralizing antibodies | Pre-clinical | [193] | |
| Env DNA vaccine with AuNR | Gold nanorod (AuNR) | 15 nm | Activate APCs better and enhance cellular and humoral immune responses | Pre-clinical | [196] | |
| Peptide vaccine with AuNP | AuNP | 2.3 nm | Enhance HIV-specific cellular immune responses | Pre-clinical | [197] | |
| Env-CaPNP | CaPNP | 44 nm to 120 nm | Activate Env-specific B cells and induce Env-specific antibody responses | Pre-clinical | [198] | |
| Env DNA vaccine with SCP | Silica-coated CaPNP (SCP) | 158.8 nm | Induce broad humoral and robust cellular immune responses | Pre-clinical | [199] |
5. The Applications of Nanoparticles in Hepatitis Virus Vaccines
| Viruses | Vaccines | Nanoparticles | Size | Functions | Status | Ref. |
|---|---|---|---|---|---|---|
| Hepatitis viruses | HBsAg/HBcAg VLP | VLP | 22 nm to 25 nm | Elicit both humoral and cellular immune responses | Pre-clinical | [205] |
| S/preS1/preS2 VLP | VLP | 22 nm | Achieve high rates of seroprotection by eliciting elevated nAb levels at low antigen doses | Approved (VBI Vaccines) | [208,209] | |
| preS1-ferritin | Ferritin | 15 nm to 30 nm | Activate both T follicular helper cells and B cells and induce high levels of preventive and therapeutic nAbs | Pre-clinical | [210] | |
| HBsAg-MNP | PLGA | 186.6 nm | Elicit persistent humoral immunity and enhance cellular immune response | Pre-clinical | [211] | |
| HBcAg-MPLA | PLGA | 279 nm | Induce strong Th1-biased cellular immune responses with more IFN-γ production | Pre-clinical | [212] | |
| HCV core/E1/E2 VLP | VLP | 40 nm to 60 nm | Induce robust and broad humoral and cellular immune responses in mice, baboons, and chimpanzees | Pre-clinical | [215] | |
| HCV core/E1/E2 VLP targeting genotypes 1a, 1b, 2a, and 3a | VLP | 50 nm | Induce strong antibodies, nAbs, and memory B and T cell responses | Pre-clinical | [216,218] | |
| MLV-Gag-based E1/E2 HCV VLP | VLP | 100 nm to 120 nm | Induce cross-neutralizing antibodies against wide ranges of genotypes | Pre-clinical | [217] | |
| Adenovirus-vectored HCV VLP | VLP | 80 nm to 120 nm | Reinforce immunogenicity by eliciting high-titer, broad-spectrum, and cross-reactive T cell responses | Pre-clinical | [219] | |
| HCV E2E1-I53-50 | I53-50 | 35 nm | Elicit significant nAb responses with cross-reactivity | Pre-clinical | [221] | |
| Optimized HCV E2 NP | Ferritin/E2p | 24.5 nm/34.5 nm | Elicit more effective nAbs to potently neutralize both autologous and heterologous HCV genotypes | Pre-clinical | [222] |
6. The Applications of Nanoparticles in Other Antiviral Vaccines
| Viruses | Vaccines | Nanoparticles | Size | Functions | Status | Ref. |
|---|---|---|---|---|---|---|
| Other viruses | ZIKV E-ferritin | Ferritin | 14 nm to 15 nm | Induce high nAb responses and protect mice from lethal ZIKV challenges | Pre-clinical | [226] |
| Live ZIKV-entrapped hydrogel (Vax) | Chitosan/CaCO3 | 200 nm | Directly activate innate immunity by the activation of pattern recognition receptors (PRRs) and generate local inflammatory niche | Pre-clinical | [227] | |
| DENV-2 E-AuNP | AuNP | 24.8 nm to 83.0 nm | Induce the proliferation of IFN-γ and IL-4-producing T cells and lead to the generation of potent nAbs | Pre-clinical | [232] | |
| Inactivated DENV-2 TMC NP | N, N, and N-trimethyl chitosan (TMC) | 483.5 nm | Induce both Th1 and Th2 types of immune responses and increase activated cytotoxic T cells and potent nAbs | Pre-clinical | [233] | |
| RSV DS-Cav1-I53-50 | I53-50 | 44 nm | Induce more potent nAb responses and cellular responses than trimeric DS-Cav1 | Phase I | [240] | |
| RSV FsIIm VLP (V-306) | VLP | 25 nm to 30 nm | Induce well-tolerated FsIIm-specific antibodies, which persist over 4 months | Phase I | [241] | |
| EBOV GP/VP40/NP VLP | VLP | 70 nm to 150 nm | Induce high titers of EBOV-specific nAbs, which protect mice and cynomolgus macaques against lethal EBOV challenges | Pre-clinical | [244,245] | |
| EBOV/SUDV GP-Gag VLP | VLP | 110 nm to 130 nm | Induce high titers of broad-spectrum nAbs that neutralize all four Ebola viruses | Pre-clinical | [246] | |
| rGP-ICMV VLP | Interbilayer-crosslinked multilamellar vesicle (ICMV) | 117 nm | Efficiently generate GC B cells and polyfunctional T cells and elicit robust nAbs responses | Pre-clinical | [248] | |
| EBOV GP-ferritin/E2p/I3-01 | Ferritin/E2p/I3-01 | 34.6 nm/45.9 nm/49.2 nm | Preserve thermostable native-like GP trimers and elicit cross-ebolavirus nAbs | Pre-clinical | [249] | |
| EBOV GP vaccine with Matrix-M | Matrix-M | 40 nm | Elicit robust and persistent B cell and T cell immune responses, characterized by higher titers of nAbs and IFN-γ production | Phase I | [250,251] |
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Excler, J.L.; Saville, M.; Berkley, S.; Kim, J.H. Vaccine development for emerging infectious diseases. Nat. Med. 2021, 27, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Morens, D.M.; Taubenberger, J.K. Influenza Cataclysm, 1918. N. Engl. J. Med. 2018, 379, 2285–2287. [Google Scholar] [CrossRef] [PubMed]
- Trilla, A.; Trilla, G.; Daer, C. The 1918 “Spanish flu” in Spain. Clin. Infect. Dis. 2008, 47, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Javanian, M.; Barary, M.; Ghebrehewet, S.; Koppolu, V.; Vasigala, V.; Ebrahimpour, S. A brief review of influenza virus infection. J. Med. Virol. 2021, 93, 4638–4646. [Google Scholar] [CrossRef] [PubMed]
- Treanor, J.J.; Talbot, H.K.; Ohmit, S.E.; Coleman, L.A.; Thompson, M.G.; Cheng, P.Y.; Petrie, J.G.; Lofthus, G.; Meece, J.K.; Williams, J.V.; et al. Effectiveness of seasonal influenza vaccines in the United States during a season with circulation of all three vaccine strains. Clin. Infect. Dis. 2012, 55, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Osterholm, M.T.; Kelley, N.S.; Sommer, A.; Belongia, E.A. Efficacy and effectiveness of influenza vaccines: A systematic review and meta-analysis. Lancet Infect. Dis. 2012, 12, 36–44. [Google Scholar] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, C.A.; Ghani, A.C.; Leung, G.M.; Hedley, A.J.; Fraser, C.; Riley, S.; Abu-Raddad, L.J.; Ho, L.M.; Thach, T.Q.; Chau, P.; et al. Epidemiological determinants of spread of causal agent of severe acute respiratory syndrome in Hong Kong. Lancet 2003, 361, 1761–1766. [Google Scholar] [CrossRef]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72,314 Cases From the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef]
- Liu, D.X.; Liang, J.Q.; Fung, T.S. Human Coronavirus-229E, -OC43, -NL63, and -HKU1 (Coronaviridae). In Encyclopedia of Virology, 4th ed.; Bamford, D.H., Zuckerman, M., Eds.; Academic Press: Oxford, UK, 2021; pp. 428–440. [Google Scholar]
- Weaver, S.C.; Charlier, C.; Vasilakis, N.; Lecuit, M. Zika, Chikungunya, and Other Emerging Vector-Borne Viral Diseases. Annu. Rev. Med. 2018, 69, 395–408. [Google Scholar]
- Guzman, M.G.; Harris, E. Dengue. Lancet 2015, 385, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Musso, D.; Gubler, D.J. Zika Virus. Clin. Microbiol. Rev. 2016, 29, 487–524. [Google Scholar] [CrossRef] [PubMed]
- Malvy, D.; McElroy, A.K.; de Clerck, H.; Gunther, S.; van Griensven, J. Ebola virus disease. Lancet 2019, 393, 936–948. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Hahn, B.H. Origins of HIV and the AIDS pandemic. Cold Spring Harb. Perspect. Med. 2011, 1, a006841. [Google Scholar] [CrossRef] [PubMed]
- Collaborators, G.H. Global, regional, and national incidence, prevalence, and mortality of HIV, 1980–2017, and forecasts to 2030, for 195 countries and territories: A systematic analysis for the Global Burden of Diseases, Injuries, and Risk Factors Study 2017. Lancet HIV 2019, 6, e831–e859. [Google Scholar]
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef]
- Chen, D.S. Hepatitis B vaccination: The key towards elimination and eradication of hepatitis B. J. Hepatol. 2009, 50, 805–816. [Google Scholar] [CrossRef]
- Dubuisson, J.; Cosset, F.L. Virology and cell biology of the hepatitis C virus life cycle: An update. J. Hepatol. 2014, 61, S3–S13. [Google Scholar] [CrossRef]
- Martinello, M.; Naggie, S.; Rockstroh, J.K.; Matthews, G.V. Direct-Acting Antiviral Therapy for Treatment of Acute and Recent Hepatitis C Virus Infection: A Narrative Review. Clin. Infect. Dis. 2023, 77, S238–S244. [Google Scholar] [CrossRef]
- Ghany, M.G.; Strader, D.B.; Thomas, D.L.; Seeff, L.B.; American Association for the Study of Liver Diseases. Diagnosis, management, and treatment of hepatitis C: An update. Hepatology 2009, 49, 1335–1374. [Google Scholar] [CrossRef] [PubMed]
- Gebre, M.S.; Brito, L.A.; Tostanoski, L.H.; Edwards, D.K.; Carfi, A.; Barouch, D.H. Novel approaches for vaccine development. Cell 2021, 184, 1589–1603. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Li, S.; Yu, H.; Xia, N.; Modis, Y. Virus-like particle-based human vaccines: Quality assessment based on structural and functional properties. Trends Biotechnol. 2013, 31, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Mascola, J.R.; Fauci, A.S. Novel vaccine technologies for the 21st century. Nat. Rev. Immunol. 2020, 20, 87–88. [Google Scholar] [CrossRef] [PubMed]
- Vogel, F.R.; Sarver, N. Nucleic acid vaccines. Clin. Microbiol. Rev. 1995, 8, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liang, B.; Wang, W.; Li, L.; Feng, N.; Zhao, Y.; Wang, T.; Yan, F.; Yang, S.; Xia, X. Viral vectored vaccines: Design, development, preventive and therapeutic applications in human diseases. Signal Transduct. Target. Ther. 2023, 8, 149. [Google Scholar] [CrossRef] [PubMed]
- Pollet, J.; Chen, W.H.; Strych, U. Recombinant protein vaccines, a proven approach against coronavirus pandemics. Adv. Drug Deliv. Rev. 2021, 170, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Salk, J.; Salk, D. Control of influenza and poliomyelitis with killed virus vaccines. Science 1977, 195, 834–847. [Google Scholar] [CrossRef]
- Wood, J.M.; Robertson, J.S. From lethal virus to life-saving vaccine: Developing inactivated vaccines for pandemic influenza. Nat. Rev. Microbiol. 2004, 2, 842–847. [Google Scholar] [CrossRef]
- McGettigan, J.P. Experimental rabies vaccines for humans. Expert Rev. Vaccines 2010, 9, 1177–1186. [Google Scholar] [CrossRef]
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef] [PubMed]
- Gurunathan, S.; Klinman, D.M.; Seder, R.A. DNA vaccines: Immunology, application, and optimization. Annu. Rev. Immunol. 2000, 18, 927–974. [Google Scholar] [CrossRef] [PubMed]
- Seder, R.A.; Gurunathan, S. DNA vaccines—Designer vaccines for the 21st century. N. Engl. J. Med. 1999, 341, 277–278. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Saade, F.; Petrovsky, N. The future of human DNA vaccines. J. Biotechnol. 2012, 162, 171–182. [Google Scholar] [PubMed]
- Pecetta, S.; Rappuoli, R. mRNA, the beginning of a new influenza vaccine game. Proc. Natl. Acad. Sci. USA 2022, 119, e2217533119. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, Y.; Dong, Y. Lipid Nanoparticle-mRNA Formulations for Therapeutic Applications. Acc. Chem. Res. 2021, 54, 4283–4293. [Google Scholar] [CrossRef]
- Lam, K.; Leung, A.; Martin, A.; Wood, M.; Schreiner, P.; Palmer, L.; Daly, O.; Zhao, W.; McClintock, K.; Heyes, J. Unsaturated, Trialkyl Ionizable Lipids are Versatile Lipid-Nanoparticle Components for Therapeutic and Vaccine Applications. Adv. Mater. 2023, 35, e2209624. [Google Scholar]
- Moreira, E.D., Jr.; Kitchin, N.; Xu, X.; Dychter, S.S.; Lockhart, S.; Gurtman, A.; Perez, J.L.; Zerbini, C.; Dever, M.E.; Jennings, T.W.; et al. Safety and Efficacy of a Third Dose of BNT162b2 COVID-19 Vaccine. N. Engl. J. Med. 2022, 386, 1910–1921. [Google Scholar]
- de Vrieze, J. Pfizer’s vaccine raises allergy concerns. Science 2021, 371, 10–11. [Google Scholar] [CrossRef]
- Chen, J.; Chen, J.; Xu, Q. Current Developments and Challenges of mRNA Vaccines. Annu. Rev. Biomed. Eng. 2022, 24, 85–109. [Google Scholar] [CrossRef]
- Ewer, K.J.; Lambe, T.; Rollier, C.S.; Spencer, A.J.; Hill, A.V.; Dorrell, L. Viral vectors as vaccine platforms: From immunogenicity to impact. Curr. Opin. Immunol. 2016, 41, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Zhuang, C.; Chu, K.; Zhang, L.; Zhao, H.; Huang, S.; Su, Y.; Lin, H.; Yang, C.; Jiang, H.; et al. Safety and immunogenicity of a live-attenuated influenza virus vector-based intranasal SARS-CoV-2 vaccine in adults: Randomised, double-blind, placebo-controlled, phase 1 and 2 trials. Lancet Respir. Med. 2022, 10, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Mackett, M.; Smith, G.L.; Moss, B. Vaccinia virus: A selectable eukaryotic cloning and expression vector. Proc. Natl. Acad. Sci. USA 1982, 79, 7415–7419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Seto, D. Chimpanzee Adenovirus Vector Ebola Vaccine—Preliminary Report. N. Engl. J. Med. 2015, 373, 775–776. [Google Scholar] [PubMed]
- Buchbinder, S.P.; Mehrotra, D.V.; Duerr, A.; Fitzgerald, D.W.; Mogg, R.; Li, D.; Gilbert, P.B.; Lama, J.R.; Marmor, M.; Del Rio, C.; et al. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): A double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet 2008, 372, 1881–1893. [Google Scholar] [CrossRef]
- Draper, S.J.; Heeney, J.L. Viruses as vaccine vectors for infectious diseases and cancer. Nat. Rev. Microbiol. 2010, 8, 62–73. [Google Scholar] [CrossRef]
- Dai, L.; Zheng, T.; Xu, K.; Han, Y.; Xu, L.; Huang, E.; An, Y.; Cheng, Y.; Li, S.; Liu, M.; et al. A Universal Design of Betacoronavirus Vaccines against COVID-19, MERS, and SARS. Cell 2020, 182, 722–733.e711. [Google Scholar] [CrossRef]
- Goepfert, P.A.; Fu, B.; Chabanon, A.L.; Bonaparte, M.I.; Davis, M.G.; Essink, B.J.; Frank, I.; Haney, O.; Janosczyk, H.; Keefer, M.C.; et al. Safety and immunogenicity of SARS-CoV-2 recombinant protein vaccine formulations in healthy adults: Interim results of a randomised, placebo-controlled, phase 1–2, dose-ranging study. Lancet Infect. Dis. 2021, 21, 1257–1270. [Google Scholar] [CrossRef]
- Van Der Meeren, O.; Hatherill, M.; Nduba, V.; Wilkinson, R.J.; Muyoyeta, M.; Van Brakel, E.; Ayles, H.M.; Henostroza, G.; Thienemann, F.; Scriba, T.J.; et al. Phase 2b Controlled Trial of M72/AS01(E) Vaccine to Prevent Tuberculosis. N. Engl. J. Med. 2018, 379, 1621–1634. [Google Scholar] [CrossRef]
- Krammer, F.; Palese, P. Advances in the development of influenza virus vaccines. Nat. Rev. Drug Discov. 2015, 14, 167–182. [Google Scholar] [CrossRef]
- Plotkin, S.A.; Plotkin, S.L. The development of vaccines: How the past led to the future. Nat. Rev. Microbiol. 2011, 9, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.T.; Rehor, A.; Schmoekel, H.G.; Hubbell, J.A.; Swartz, M.A. In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles. J. Control. Release 2006, 112, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhu, M.; Zhao, X.; Nie, G. Nanotechnology-empowered vaccine delivery for enhancing CD8(+) T cells-mediated cellular immunity. Adv. Drug Deliv. Rev. 2021, 176, 113889. [Google Scholar] [CrossRef] [PubMed]
- Geng, Q.; Tai, W.; Baxter, V.K.; Shi, J.; Wan, Y.; Zhang, X.; Montgomery, S.A.; Taft-Benz, S.A.; Anderson, E.J.; Knight, A.C.; et al. Novel virus-like nanoparticle vaccine effectively protects animal model from SARS-CoV-2 infection. PLoS Pathog. 2021, 17, e1009897. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Y.; Zhou, Y.; Li, Y.; Xu, J.; Ai, Y.; Xu, L.; Xiao, X.; Zhang, B.; Jin, J. An RBD virus-like particle vaccine for SARS-CoV-2 induces cross-variant antibody responses in mice and macaques. Signal Transduct. Target. Ther. 2023, 8, 173. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Zhang, J.; Zhai, J.; Hong, J.; Yuan, C.; Liang, M. Ferritin: A Multifunctional Nanoplatform for Biological Detection, Imaging Diagnosis, and Drug Delivery. Acc. Chem. Res. 2021, 54, 3313–3325. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.W.; Thornburg, N.J.; Blum, D.L.; Kuhn, S.J.; Wright, D.W.; Crowe, J.E., Jr. Gold nanorod vaccine for respiratory syncytial virus. Nanotechnology 2013, 24, 295102. [Google Scholar] [CrossRef] [PubMed]
- Bolhassani, A.; Javanzad, S.; Saleh, T.; Hashemi, M.; Aghasadeghi, M.R.; Sadat, S.M. Polymeric nanoparticles: Potent vectors for vaccine delivery targeting cancer and infectious diseases. Hum. Vaccin. Immunother. 2014, 10, 321–332. [Google Scholar] [CrossRef]
- Garg, A.; Dewangan, H.K. Nanoparticles as Adjuvants in Vaccine Delivery. Crit. Rev. Ther. Drug Carrier Syst. 2020, 37, 183–204. [Google Scholar] [CrossRef]
- Wu, J.; Ma, G. Biomimic strategies for modulating the interaction between particle adjuvants and antigen-presenting cells. Biomater. Sci. 2020, 8, 2366–2375. [Google Scholar] [CrossRef]
- Zhao, T.; Cai, Y.; Jiang, Y.; He, X.; Wei, Y.; Yu, Y.; Tian, X. Vaccine adjuvants: Mechanisms and platforms. Signal Transduct. Target. Ther. 2023, 8, 283. [Google Scholar] [CrossRef] [PubMed]
- Lima, K.M.; dos Santos, S.A.; Rodrigues, J.M., Jr.; Silva, C.L. Vaccine adjuvant: It makes the difference. Vaccine 2004, 22, 2374–2379. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; O’Hagan, D. Advances in vaccine adjuvants. Nat. Biotechnol. 1999, 17, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Di Pasquale, A.; Preiss, S.; Tavares Da Silva, F.; Garcon, N. Vaccine Adjuvants: From 1920 to 2015 and Beyond. Vaccines 2015, 3, 320–343. [Google Scholar] [CrossRef] [PubMed]
- Parums, D.V. Editorial: First Approval of the Protein-Based Adjuvanted Nuvaxovid (NVX-CoV2373) Novavax Vaccine for SARS-CoV-2 Could Increase Vaccine Uptake and Provide Immune Protection from Viral Variants. Med. Sci. Monit. 2022, 28, e936523. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D.T.; MacKichan, M.L.; Singh, M. Recent developments in adjuvants for vaccines against infectious diseases. Biomol. Eng. 2001, 18, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, R.P.; Gordon, M.L. An overview of influenza A virus genes, protein functions, and replication cycle highlighting important updates. Virus Genes 2022, 58, 255–269. [Google Scholar] [CrossRef]
- Krammer, F.; Smith, G.J.D.; Fouchier, R.A.M.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis. Primers 2018, 4, 3. [Google Scholar] [CrossRef]
- To, J.; Torres, J. Viroporins in the Influenza Virus. Cells 2019, 8, 654. [Google Scholar] [CrossRef]
- Bergeron, H.C.; Reneer, Z.B.; Arora, A.; Reynolds, S.; Nagy, T.; Tripp, R.A. Influenza B Virus (IBV) Immune-Mediated Disease in C57BL/6 Mice. Vaccines 2022, 10, 1440. [Google Scholar] [CrossRef]
- Horthongkham, N.; Athipanyasilp, N.; Pattama, A.; Kaewnapan, B.; Sornprasert, S.; Srisurapanont, S.; Kantakamalakul, W.; Amaranond, P.; Sutthent, R. Epidemiological, Clinical and Virological Characteristics of Influenza B Virus from Patients at the Hospital Tertiary Care Units in Bangkok during 2011–2014. PLoS ONE 2016, 11, e0158244. [Google Scholar] [CrossRef] [PubMed]
- Suntronwong, N.; Klinfueng, S.; Korkong, S.; Vichaiwattana, P.; Thongmee, T.; Vongpunsawad, S.; Poovorawan, Y. Characterizing genetic and antigenic divergence from vaccine strain of influenza A and B viruses circulating in Thailand, 2017–2020. Sci. Rep. 2021, 11, 735. [Google Scholar] [CrossRef] [PubMed]
- Morens, D.M.; Fauci, A.S. The 1918 influenza pandemic: Insights for the 21st century. J. Infect. Dis. 2007, 195, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Vijaykrishna, D.; Bahl, J.; Zhu, H.; Wang, J.; Smith, G.J. The emergence of pandemic influenza viruses. Protein Cell 2010, 1, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Jester, B.J.; Uyeki, T.M.; Jernigan, D.B. Fifty Years of Influenza A(H3N2) Following the Pandemic of 1968. Am. J. Public Health 2020, 110, 669–676. [Google Scholar] [CrossRef]
- Sullivan, S.J.; Jacobson, R.M.; Dowdle, W.R.; Poland, G.A. 2009 H1N1 influenza. Mayo. Clin. Proc. 2010, 85, 64–76. [Google Scholar] [CrossRef]
- Farrukee, R.; Leang, S.K.; Butler, J.; Lee, R.T.; Maurer-Stroh, S.; Tilmanis, D.; Sullivan, S.; Mosse, J.; Barr, I.G.; Hurt, A.C. Influenza viruses with B/Yamagata- and B/Victoria-like neuraminidases are differentially affected by mutations that alter antiviral susceptibility. J. Antimicrob. Chemother. 2015, 70, 2004–2012. [Google Scholar] [CrossRef]
- Killingley, B.; Nguyen-Van-Tam, J. Routes of influenza transmission. Influenza Other Respir. Viruses 2013, 7, 42–51. [Google Scholar] [CrossRef]
- Quantius, J.; Schmoldt, C.; Vazquez-Armendariz, A.I.; Becker, C.; El Agha, E.; Wilhelm, J.; Morty, R.E.; Vadasz, I.; Mayer, K.; Gattenloehner, S.; et al. Influenza Virus Infects Epithelial Stem/Progenitor Cells of the Distal Lung: Impact on Fgfr2b-Driven Epithelial Repair. PLoS Pathog. 2016, 12, e1005544. [Google Scholar] [CrossRef]
- Kalil, A.C.; Thomas, P.G. Influenza virus-related critical illness: Pathophysiology and epidemiology. Crit. Care 2019, 23, 258. [Google Scholar] [CrossRef]
- Pati, R.; Shevtsov, M.; Sonawane, A. Nanoparticle Vaccines Against Infectious Diseases. Front. Immunol. 2018, 9, 2224. [Google Scholar] [CrossRef] [PubMed]
- Bezbaruah, R.; Chavda, V.P.; Nongrang, L.; Alom, S.; Deka, K.; Kalita, T.; Ali, F.; Bhattacharjee, B.; Vora, L. Nanoparticle-Based Delivery Systems for Vaccines. Vaccines 2022, 10, 1946. [Google Scholar] [CrossRef] [PubMed]
- Ways, T.M.M.; Lau, W.M.; Khutoryanskiy, V.V. Chitosan and Its Derivatives for Application in Mucoadhesive Drug Delivery Systems. Polymers 2018, 10, 267. [Google Scholar] [CrossRef] [PubMed]
- Dhakal, S.; Renu, S.; Ghimire, S.; Shaan Lakshmanappa, Y.; Hogshead, B.T.; Feliciano-Ruiz, N.; Lu, F.; HogenEsch, H.; Krakowka, S.; Lee, C.W.; et al. Mucosal Immunity and Protective Efficacy of Intranasal Inactivated Influenza Vaccine Is Improved by Chitosan Nanoparticle Delivery in Pigs. Front. Immunol. 2018, 9, 934. [Google Scholar] [CrossRef] [PubMed]
- Dhakal, S.; Hiremath, J.; Bondra, K.; Lakshmanappa, Y.S.; Shyu, D.L.; Ouyang, K.; Kang, K.I.; Binjawadagi, B.; Goodman, J.; Tabynov, K.; et al. Biodegradable nanoparticle delivery of inactivated swine influenza virus vaccine provides heterologous cell-mediated immune response in pigs. J. Control. Release 2017, 247, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.A.; Saouaf, O.M.; Smith, A.A.A.; Gale, E.C.; Hernández, M.A.; Idoyaga, J.; Appel, E.A. Prolonged Codelivery of Hemagglutinin and a TLR7/8 Agonist in a Supramolecular Polymer–Nanoparticle Hydrogel Enhances Potency and Breadth of Influenza Vaccination. ACS Biomater. Sci. Eng. 2021, 7, 1889–1899. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, M.; Wei, C.J.; Yassine, H.M.; McTamney, P.M.; Boyington, J.C.; Whittle, J.R.; Rao, S.S.; Kong, W.P.; Wang, L.; Nabel, G.J. Self-assembling influenza nanoparticle vaccines elicit broadly neutralizing H1N1 antibodies. Nature 2013, 499, 102–106. [Google Scholar] [CrossRef]
- Houser, K.V.; Chen, G.L.; Carter, C.; Crank, M.C.; Nguyen, T.A.; Burgos Florez, M.C.; Berkowitz, N.M.; Mendoza, F.; Hendel, C.S.; Gordon, I.J.; et al. Safety and immunogenicity of a ferritin nanoparticle H2 influenza vaccine in healthy adults: A phase 1 trial. Nat. Med. 2022, 28, 383–391. [Google Scholar] [CrossRef]
- Boyoglu-Barnum, S.; Ellis, D.; Gillespie, R.A.; Hutchinson, G.B.; Park, Y.-J.; Moin, S.M.; Acton, O.J.; Ravichandran, R.; Murphy, M.; Pettie, D.; et al. Quadrivalent influenza nanoparticle vaccines induce broad protection. Nature 2021, 592, 623–628. [Google Scholar] [CrossRef]
- Qi, M.; Zhang, X.E.; Sun, X.; Zhang, X.; Yao, Y.; Liu, S.; Chen, Z.; Li, W.; Zhang, Z.; Chen, J.; et al. Intranasal Nanovaccine Confers Homo- and Hetero-Subtypic Influenza Protection. Small 2018, 14, e1703207. [Google Scholar] [CrossRef]
- Pan, J.; Wang, Q.; Qi, M.; Chen, J.; Wu, X.; Zhang, X.; Li, W.; Zhang, X.E.; Cui, Z. An Intranasal Multivalent Epitope-Based Nanoparticle Vaccine Confers Broad Protection against Divergent Influenza Viruses. ACS Nano 2023, 17, 13474–13487. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.-M.J.; Chien, C.-Y.; Liu, M.-T.; Fang, Z.-S.; Chang, S.-Y.; Juang, R.-H.; Chang, S.-C.; Chen, H.-W. Multi-antigen avian influenza a (H7N9) virus-like particles: Particulate characterizations and immunogenicity evaluation in murine and avian models. BMC Biotechnol. 2017, 17, 2. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Zhao, Y.; Liu, J.; Ji, X.; Meng, L.; Wang, T.; Sun, W.; Zhang, K.; Sang, X.; Yu, Z.; et al. Intramuscular and intranasal immunization with an H7N9 influenza virus-like particle vaccine protects mice against lethal influenza virus challenge. Int. Immunopharmacol. 2018, 58, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Carignan, D.; Thérien, A.; Rioux, G.; Paquet, G.; Gagné, M.-È.L.; Bolduc, M.; Savard, P.; Leclerc, D. Engineering of the PapMV vaccine platform with a shortened M2e peptide leads to an effective one dose influenza vaccine. Vaccine 2015, 33, 7245–7253. [Google Scholar] [CrossRef]
- Wei, J.; Li, Z.; Yang, Y.; Ma, X.; An, W.; Ma, G.; Su, Z.; Zhang, S. A biomimetic VLP influenza vaccine with interior NP/exterior M2e antigens constructed through a temperature shift-based encapsulation strategy. Vaccine 2020, 38, 5987–5996. [Google Scholar] [CrossRef] [PubMed]
- Dykman, L.A. Gold nanoparticles for preparation of antibodies and vaccines against infectious diseases. Expert. Rev. Vaccines 2020, 19, 465–477. [Google Scholar] [CrossRef]
- Wang, C.; Zhu, W.; Wang, B.Z. Dual-linker gold nanoparticles as adjuvanting carriers for multivalent display of recombinant influenza hemagglutinin trimers and flagellin improve the immunological responses in vivo and in vitro. Int. J. Nanomed. 2017, 12, 4747–4762. [Google Scholar] [CrossRef]
- Tao, W.; Gill, H.S. M2e-immobilized gold nanoparticles as influenza A vaccine: Role of soluble M2e and longevity of protection. Vaccine 2015, 33, 2307–2315. [Google Scholar] [CrossRef]
- Yang, E.J.; Kim, S.; Kim, J.S.; Choi, I.H. Inflammasome formation and IL-1beta release by human blood monocytes in response to silver nanoparticles. Biomaterials 2012, 33, 6858–6867. [Google Scholar] [CrossRef]
- Haberl, N.; Hirn, S.; Wenk, A.; Diendorf, J.; Epple, M.; Johnston, B.D.; Krombach, F.; Kreyling, W.G.; Schleh, C. Cytotoxic and proinflammatory effects of PVP-coated silver nanoparticles after intratracheal instillation in rats. Beilstein J. Nanotechnol. 2013, 4, 933–940. [Google Scholar] [CrossRef]
- Sanchez-Guzman, D.; Le Guen, P.; Villeret, B.; Sola, N.; Le Borgne, R.; Guyard, A.; Kemmel, A.; Crestani, B.; Sallenave, J.M.; Garcia-Verdugo, I. Silver nanoparticle-adjuvanted vaccine protects against lethal influenza infection through inducing BALT and IgA-mediated mucosal immunity. Biomaterials 2019, 217, 119308. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wang, X.; Huang, X.; Zhang, J.; Xia, N.; Zhao, Q. Calcium phosphate nanoparticles as a new generation vaccine adjuvant. Expert Rev. Vaccines 2017, 16, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Musetti, S.; Fu, L.H.; Zhu, Y.J.; Huang, L. Biomolecule-assisted green synthesis of nanostructured calcium phosphates and their biomedical applications. Chem. Soc. Rev. 2019, 48, 2698–2737. [Google Scholar] [CrossRef] [PubMed]
- Morcöl, T.; Hurst, B.L.; Tarbet, E.B. Calcium phosphate nanoparticle (CaPNP) for dose-sparing of inactivated whole virus pandemic influenza A (H1N1) 2009 vaccine in mice. Vaccine 2017, 35, 4569–4577. [Google Scholar] [CrossRef]
- Mercuri, L.P.; Carvalho, L.V.; Lima, F.A.; Quayle, C.; Fantini, M.C.; Tanaka, G.S.; Cabrera, W.H.; Furtado, M.F.; Tambourgi, D.V.; Matos Jdo, R.; et al. Ordered mesoporous silica SBA-15: A new effective adjuvant to induce antibody response. Small 2006, 2, 254–256. [Google Scholar] [CrossRef]
- Navarro-Tovar, G.; Palestino, G.; Rosales-Mendoza, S. An overview on the role of silica-based materials in vaccine development. Expert Rev. Vaccines 2016, 15, 1449–1462. [Google Scholar] [CrossRef]
- Neuhaus, V.; Chichester, J.A.; Ebensen, T.; Schwarz, K.; Hartman, C.E.; Shoji, Y.; Guzman, C.A.; Yusibov, V.; Sewald, K.; Braun, A. A new adjuvanted nanoparticle-based H1N1 influenza vaccine induced antigen-specific local mucosal and systemic immune responses after administration into the lung. Vaccine 2014, 32, 3216–3222. [Google Scholar] [CrossRef]
- Pham, N.B.; Ho, T.T.; Nguyen, G.T.; Le, T.T.; Le, N.T.; Chang, H.C.; Pham, M.D.; Conrad, U.; Chu, H.H. Nanodiamond enhances immune responses in mice against recombinant HA/H7N9 protein. J. Nanobiotechnol. 2017, 15, 69. [Google Scholar] [CrossRef]
- Banerjee, A.; Kulcsar, K.; Misra, V.; Frieman, M.; Mossman, K. Bats and Coronaviruses. Viruses 2019, 11, 41. [Google Scholar] [CrossRef]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904 e899. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Chen, C.; Chen, D.; Zhu, A.; Li, F.; Zhuang, Z.; Mok, C.K.P.; Dai, J.; Li, X.; Jin, Y.; et al. Mouse models susceptible to HCoV-229E and HCoV-NL63 and cross protection from challenge with SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2023, 120, e2202820120. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Liang, H.; Chen, P.; Li, Y.; Li, Z.; Fan, S.; Wu, K.; Li, X.; Chen, W.; Qin, Y.; et al. Viral Vector Vaccine Development and Application during the COVID-19 Pandemic. Microorganisms 2022, 10, 1450. [Google Scholar] [CrossRef] [PubMed]
- Corbett, K.S.; Edwards, D.K.; Leist, S.R.; Abiona, O.M.; Boyoglu-Barnum, S.; Gillespie, R.A.; Himansu, S.; Schäfer, A.; Ziwawo, C.T.; DiPiazza, A.T.; et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 2020, 586, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292 e286. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef]
- Hofmann, H.; Pyrc, K.; van der Hoek, L.; Geier, M.; Berkhout, B.; Pohlmann, S. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc. Natl. Acad. Sci. USA 2005, 102, 7988–7993. [Google Scholar] [CrossRef]
- Wang, N.; Shi, X.; Jiang, L.; Zhang, S.; Wang, D.; Tong, P.; Guo, D.; Fu, L.; Cui, Y.; Liu, X.; et al. Structure of MERS-CoV spike receptor-binding domain complexed with human receptor DPP4. Cell Res. 2013, 23, 986–993. [Google Scholar] [CrossRef]
- Yeager, C.L.; Ashmun, R.A.; Williams, R.K.; Cardellichio, C.B.; Shapiro, L.H.; Look, A.T.; Holmes, K.V. Human aminopeptidase N is a receptor for human coronavirus 229E. Nature 1992, 357, 420–422. [Google Scholar] [CrossRef] [PubMed]
- Hulswit, R.J.G.; Lang, Y.; Bakkers, M.J.G.; Li, W.; Li, Z.; Schouten, A.; Ophorst, B.; van Kuppeveld, F.J.M.; Boons, G.J.; Bosch, B.J.; et al. Human coronaviruses OC43 and HKU1 bind to 9-O-acetylated sialic acids via a conserved receptor-binding site in spike protein domain A. Proc. Natl. Acad. Sci. USA 2019, 116, 2681–2690. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; He, Y.; Zhou, Y.; Liu, S.; Zheng, B.J.; Jiang, S. The spike protein of SARS-CoV--a target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009, 7, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wong, G.; Lu, G.; Yan, J.; Gao, G.F. MERS-CoV spike protein: Targets for vaccines and therapeutics. Antiviral. Res. 2016, 133, 165–177. [Google Scholar] [CrossRef] [PubMed]
- McCallum, M.; De Marco, A.; Lempp, F.A.; Tortorici, M.A.; Pinto, D.; Walls, A.C.; Beltramello, M.; Chen, A.; Liu, Z.; Zatta, F.; et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell 2021, 184, 2332–2347.e2316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xiao, T.; Cai, Y.; Chen, B. Structure of SARS-CoV-2 spike protein. Curr. Opin. Virol. 2021, 50, 173–182. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef]
- Alameh, M.-G.; Tombácz, I.; Bettini, E.; Lederer, K.; Sittplangkoon, C.; Wilmore, J.R.; Gaudette, B.T.; Soliman, O.Y.; Pine, M.; Hicks, P.; et al. Lipid nanoparticles enhance the efficacy of mRNA and protein subunit vaccines by inducing robust T follicular helper cell and humoral responses. Immunity 2022, 55, 1136–1138. [Google Scholar] [CrossRef]
- Maeki, M.; Uno, S.; Niwa, A.; Okada, Y.; Tokeshi, M. Microfluidic technologies and devices for lipid nanoparticle-based RNA delivery. J. Control. Release 2022, 344, 80–96. [Google Scholar] [CrossRef]
- Dilliard, S.A.; Cheng, Q.; Siegwart, D.J. On the mechanism of tissue-specific mRNA delivery by selective organ targeting nanoparticles. Proc. Natl. Acad. Sci. USA 2021, 118, e2109256118. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, N.; Weissman, D.; Whitehead, K.A. mRNA vaccines for infectious diseases: Principles, delivery and clinical translation. Nat. Rev. Drug Discov. 2021, 20, 817–838. [Google Scholar] [CrossRef] [PubMed]
- Dalpke, A.; Helm, M. RNA mediated Toll-like receptor stimulation in health and disease. RNA Biol. 2012, 9, 828–842. [Google Scholar] [CrossRef] [PubMed]
- Teo, S.P. Review of COVID-19 mRNA Vaccines: BNT162b2 and mRNA-1273. J. Pharm. Pract. 2022, 35, 947–951. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Lei, W.; Kang, B.; Yang, H.; Wang, Y.; Lu, Y.; Lv, L.; Sun, Y.; Zhang, J.; Wang, X.; et al. A novel mRNA vaccine, SYS6006, against SARS-CoV-2. Front. Immunol. 2023, 13, 1051576. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.-N.; Li, X.-F.; Deng, Y.-Q.; Zhao, H.; Huang, Y.-J.; Yang, G.; Huang, W.-J.; Gao, P.; Zhou, C.; Zhang, R.-R.; et al. A Thermostable mRNA Vaccine against COVID-19. Cell 2020, 182, 1271–1283.e1216. [Google Scholar] [CrossRef]
- Hajnik, R.L.; Plante, J.A.; Liang, Y.; Alameh, M.-G.; Tang, J.; Bonam, S.R.; Zhong, C.; Adam, A.; Scharton, D.; Rafael, G.H.; et al. Dual spike and nucleocapsid mRNA vaccination confer protection against SARS-CoV-2 Omicron and Delta variants in preclinical models. Sci. Transl. Med. 2022, 14, eabq1945. [Google Scholar] [CrossRef]
- Qu, L.; Yi, Z.; Shen, Y.; Lin, L.; Chen, F.; Xu, Y.; Wu, Z.; Tang, H.; Zhang, X.; Tian, F.; et al. Circular RNA vaccines against SARS-CoV-2 and emerging variants. Cell 2022, 185, 1728–1744.e1716. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2020, 384, 403–416. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Kustin, T.; Harel, N.; Finkel, U.; Perchik, S.; Harari, S.; Tahor, M.; Caspi, I.; Levy, R.; Leshchinsky, M.; Ken Dror, S.; et al. Evidence for increased breakthrough rates of SARS-CoV-2 variants of concern in BNT162b2-mRNA-vaccinated individuals. Nat. Med. 2021, 27, 1379–1384. [Google Scholar] [CrossRef] [PubMed]
- Schoenmaker, L.; Witzigmann, D.; Kulkarni, J.A.; Verbeke, R.; Kersten, G.; Jiskoot, W.; Crommelin, D.J.A. mRNA-lipid nanoparticle COVID-19 vaccines: Structure and stability. Int. J. Pharm. 2021, 601, 120586. [Google Scholar] [CrossRef] [PubMed]
- Reddington, S.C.; Howarth, M. Secrets of a covalent interaction for biomaterials and biotechnology: SpyTag and SpyCatcher. Curr. Opin. Chem. Biol. 2015, 29, 94–99. [Google Scholar] [CrossRef]
- Tan, T.K.; Rijal, P.; Rahikainen, R.; Keeble, A.H.; Schimanski, L.; Hussain, S.; Harvey, R.; Hayes, J.W.P.; Edwards, J.C.; McLean, R.K.; et al. A COVID-19 vaccine candidate using SpyCatcher multimerization of the SARS-CoV-2 spike protein receptor-binding domain induces potent neutralising antibody responses. Nat. Commun. 2021, 12, 542. [Google Scholar] [CrossRef] [PubMed]
- Bruun, T.U.J.; Andersson, A.C.; Draper, S.J.; Howarth, M. Engineering a Rugged Nanoscaffold To Enhance Plug-and-Display Vaccination. ACS Nano 2018, 12, 8855–8866. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Zou, F.; Yu, F.; Li, R.; Yuan, Y.; Zhang, Y.; Zhang, X.; Deng, J.; Chen, T.; Song, Z.; et al. Nanoparticle Vaccines Based on the Receptor Binding Domain (RBD) and Heptad Repeat (HR) of SARS-CoV-2 Elicit Robust Protective Immune Responses. Immunity 2020, 53, 1315–1330.e1319. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.F.; Sun, C.; Sun, J.; Xie, C.; Zhuang, Z.; Xu, H.Q.; Liu, Z.; Liu, Y.H.; Peng, S.; Yuan, R.Y.; et al. Quadrivalent mosaic HexaPro-bearing nanoparticle vaccine protects against infection of SARS-CoV-2 variants. Nat. Commun. 2022, 13, 2674. [Google Scholar] [CrossRef] [PubMed]
- Jardine, J.; Julien, J.P.; Menis, S.; Ota, T.; Kalyuzhniy, O.; McGuire, A.; Sok, D.; Huang, P.S.; MacPherson, S.; Jones, M.; et al. Rational HIV immunogen design to target specific germline B cell receptors. Science 2013, 340, 711–716. [Google Scholar] [CrossRef]
- Walls, A.C.; Fiala, B.; Schafer, A.; Wrenn, S.; Pham, M.N.; Murphy, M.; Tse, L.V.; Shehata, L.; O’Connor, M.A.; Chen, C.; et al. Elicitation of Potent Neutralizing Antibody Responses by Designed Protein Nanoparticle Vaccines for SARS-CoV-2. Cell 2020, 183, 1367–1382.e1317. [Google Scholar] [CrossRef]
- Weidenbacher, P.A.; Sanyal, M.; Friedland, N.; Tang, S.; Arunachalam, P.S.; Hu, M.; Kumru, O.S.; Morris, M.K.; Fontenot, J.; Shirreff, L.; et al. A ferritin-based COVID-19 nanoparticle vaccine that elicits robust, durable, broad-spectrum neutralizing antisera in non-human primates. Nat. Commun. 2023, 14, 2149. [Google Scholar] [CrossRef]
- Zhang, B.; Chao, C.W.; Tsybovsky, Y.; Abiona, O.M.; Hutchinson, G.B.; Moliva, J.I.; Olia, A.S.; Pegu, A.; Phung, E.; Stewart-Jones, G.B.E.; et al. A platform incorporating trimeric antigens into self-assembling nanoparticles reveals SARS-CoV-2-spike nanoparticles to elicit substantially higher neutralizing responses than spike alone. Sci. Rep. 2020, 10, 18149. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.A.; Gnanapragasam, P.N.P.; Lee, Y.E.; Hoffman, P.R.; Ou, S.; Kakutani, L.M.; Keeffe, J.R.; Wu, H.J.; Howarth, M.; West, A.P.; et al. Mosaic nanoparticles elicit cross-reactive immune responses to zoonotic coronaviruses in mice. Science 2021, 371, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.A.; van Doremalen, N.; Greaney, A.J.; Andersen, H.; Sharma, A.; Starr, T.N.; Keeffe, J.R.; Fan, C.; Schulz, J.E.; Gnanapragasam, P.N.P.; et al. Mosaic RBD nanoparticles protect against challenge by diverse sarbecoviruses in animal models. Science 2022, 377, eabq0839. [Google Scholar] [CrossRef] [PubMed]
- Nooraei, S.; Bahrulolum, H.; Hoseini, Z.S.; Katalani, C.; Hajizade, A.; Easton, A.J.; Ahmadian, G. Virus-like particles: Preparation, immunogenicity and their roles as nanovaccines and drug nanocarriers. J. Nanobiotechnol. 2021, 19, 59. [Google Scholar] [CrossRef] [PubMed]
- Zepeda-Cervantes, J.; Ramírez-Jarquín, J.O.; Vaca, L. Interaction Between Virus-Like Particles (VLPs) and Pattern Recognition Receptors (PRRs) From Dendritic Cells (DCs): Toward Better Engineering of VLPs. Front. Immunol. 2020, 11, 1100. [Google Scholar] [CrossRef]
- Chu, K.B.; Kang, H.J.; Yoon, K.W.; Lee, H.A.; Moon, E.K.; Han, B.K.; Quan, F.S. Influenza Virus-like Particle (VLP) Vaccines Expressing the SARS-CoV-2 S Glycoprotein, S1, or S2 Domains. Vaccines 2021, 9, 920. [Google Scholar] [CrossRef]
- Yilmaz, I.C.; Ipekoglu, E.M.; Bulbul, A.; Turay, N.; Yildirim, M.; Evcili, I.; Yilmaz, N.S.; Guvencli, N.; Aydin, Y.; Gungor, B.; et al. Development and preclinical evaluation of virus-like particle vaccine against COVID-19 infection. Allergy 2022, 77, 258–270. [Google Scholar] [CrossRef]
- Ward, B.J.; Gobeil, P.; Seguin, A.; Atkins, J.; Boulay, I.; Charbonneau, P.Y.; Couture, M.; D’Aoust, M.A.; Dhaliwall, J.; Finkle, C.; et al. Phase 1 randomized trial of a plant-derived virus-like particle vaccine for COVID-19. Nat. Med. 2021, 27, 1071–1078. [Google Scholar] [CrossRef]
- Fluckiger, A.C.; Ontsouka, B.; Bozic, J.; Diress, A.; Ahmed, T.; Berthoud, T.; Tran, A.; Duque, D.; Liao, M.; McCluskie, M.; et al. An enveloped virus-like particle vaccine expressing a stabilized prefusion form of the SARS-CoV-2 spike protein elicits highly potent immunity. Vaccine 2021, 39, 4988–5001. [Google Scholar] [CrossRef]
- Hoffmann, M.A.G.; Yang, Z.; Huey-Tubman, K.E.; Cohen, A.A.; Gnanapragasam, P.N.P.; Nakatomi, L.M.; Storm, K.N.; Moon, W.J.; Lin, P.J.C.; West, A.P.; et al. ESCRT recruitment to SARS-CoV-2 spike induces virus-like particles that improve mRNA vaccines. Cell 2023, 186, 2380–2391.e2389. [Google Scholar] [CrossRef]
- Tian, J.-H.; Patel, N.; Haupt, R.; Zhou, H.; Weston, S.; Hammond, H.; Logue, J.; Portnoff, A.D.; Norton, J.; Guebre-Xabier, M.; et al. SARS-CoV-2 spike glycoprotein vaccine candidate NVX-CoV2373 immunogenicity in baboons and protection in mice. Nat. Commun. 2021, 12, 372. [Google Scholar] [CrossRef] [PubMed]
- Bangaru, S.; Ozorowski, G.; Turner, H.L.; Antanasijevic, A.; Huang, D.; Wang, X.; Torres, J.L.; Diedrich, J.K.; Tian, J.-H.; Portnoff, A.D.; et al. Structural analysis of full-length SARS-CoV-2 spike protein from an advanced vaccine candidate. Science 2020, 370, 1089–1094. [Google Scholar] [CrossRef] [PubMed]
- Heath, P.T.; Galiza, E.P.; Baxter, D.N.; Boffito, M.; Browne, D.; Burns, F.; Chadwick, D.R.; Clark, R.; Cosgrove, C.; Galloway, J.; et al. Safety and Efficacy of NVX-CoV2373 COVID-19 Vaccine. N. Engl. J. Med. 2021, 385, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Dunkle, L.M.; Kotloff, K.L.; Gay, C.L.; Áñez, G.; Adelglass, J.M.; Barrat Hernández, A.Q.; Harper, W.L.; Duncanson, D.M.; McArthur, M.A.; Florescu, D.F.; et al. Efficacy and Safety of NVX-CoV2373 in Adults in the United States and Mexico. N. Engl. J. Med. 2021, 386, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Áñez, G.; Dunkle, L.M.; Gay, C.L.; Kotloff, K.L.; Adelglass, J.M.; Essink, B.; Campbell, J.D.; Cloney-Clark, S.; Zhu, M.; Plested, J.S.; et al. Safety, Immunogenicity, and Efficacy of the NVX-CoV2373 COVID-19 Vaccine in Adolescents: A Randomized Clinical Trial. JAMA Netw. Open 2023, 6, e239135. [Google Scholar] [CrossRef] [PubMed]
- Chereddy, K.K.; Vandermeulen, G.; Preat, V. PLGA based drug delivery systems: Promising carriers for wound healing activity. Wound Repair Regen. 2016, 24, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Ding, Y.; Yao, J.; Peng, K.; Deng, K.; Zhang, M.; Zhang, Y.; Zuo, J. The SARS-CoV-2 rS1-E-PLGA nanovaccine and evaluation of its immune effect in BALB/c mice. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 5255–5263. [Google Scholar]
- Peng, S.; Cao, F.; Xia, Y.; Gao, X.-D.; Dai, L.; Yan, J.; Ma, G. Particulate Alum via Pickering Emulsion for an Enhanced COVID-19 Vaccine Adjuvant. Adv. Mater. 2020, 32, 2004210. [Google Scholar] [CrossRef]
- Moshref Javadi, M.; Taghdisi Hosseinzadeh, M.; Soleimani, N.; Rommasi, F. Evaluating the immunogenicity of gold nanoparticles conjugated RBD with Freund’s adjuvant as a potential vaccine against SARS-CoV-2. Microb. Pathog. 2022, 170, 105687. [Google Scholar] [CrossRef]
- Sekimukai, H.; Iwata-Yoshikawa, N.; Fukushi, S.; Tani, H.; Kataoka, M.; Suzuki, T.; Hasegawa, H.; Niikura, K.; Arai, K.; Nagata, N. Gold nanoparticle-adjuvanted S protein induces a strong antigen-specific IgG response against severe acute respiratory syndrome-related coronavirus infection, but fails to induce protective antibodies and limit eosinophilic infiltration in lungs. Microbiol. Immunol. 2020, 64, 33–51. [Google Scholar] [CrossRef]
- Kumar, U.S.; Afjei, R.; Ferrara, K.; Massoud, T.F.; Paulmurugan, R. Gold-Nanostar-Chitosan-Mediated Delivery of SARS-CoV-2 DNA Vaccine for Respiratory Mucosal Immunization: Development and Proof-of-Principle. ACS Nano 2021, 15, 17582–17601. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.-W.; Stuyver, L.; Mizell, S.B.; Ehler, L.A.; Mican, J.A.M.; Baseler, M.; Lloyd, A.L.; Nowak, M.A.; Fauci, A.S. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 1997, 94, 13193–13197. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a Reservoir for HIV-1 in Patients on Highly Active Antiretroviral Therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.K.; Hezareh, M.; Günthard, H.F.; Havlir, D.V.; Ignacio, C.C.; Spina, C.A.; Richman, D.D. Recovery of Replication-Competent HIV Despite Prolonged Suppression of Plasma Viremia. Science 1997, 278, 1291–1295. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Ma, X.; Liu, B.; Chen, C.; Zhang, H. HIV-1 functional cure: Will the dream come true? BMC Med. 2015, 13, 284. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, W.H. Inactivated- or Killed-Virus HIV/AIDS Vaccines. Curr. Drug Targets Infect. Disord. 2005, 5, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Perez-Losada, M.; Jobes, D.V.; Sinangil, F.; Crandall, K.A.; Arenas, M.; Posada, D.; Berman, P.W. Phylodynamics of HIV-1 from a phase III AIDS vaccine trial in Bangkok, Thailand. PLoS ONE 2011, 6, e16902. [Google Scholar] [CrossRef]
- Flynn, N.M.; Forthal, D.N.; Harro, C.D.; Judson, F.N.; Mayer, K.H.; Para, M.F.; rgp120 HIV Vaccine Study Group. Placebo-controlled phase 3 trial of a recombinant glycoprotein 120 vaccine to prevent HIV-1 infection. J. Infect. Dis. 2005, 191, 654–665. [Google Scholar]
- Capon, D.J.; Ward, R.H.R. The CD4-gpl20 Interaction and Aids Pathogenesis. Annu. Rev. Immunol. 1991, 9, 649–678. [Google Scholar] [CrossRef]
- Kowalski, M.; Potz, J.; Basiripour, L.; Dorfman, T.; Goh, W.C.; Terwilliger, E.; Dayton, A.; Rosen, C.; Haseltine, W.; Sodroski, J. Functional regions of the envelope glycoprotein of human immunodeficiency virus type 1. Science 1987, 237, 1351–1355. [Google Scholar] [CrossRef]
- Rerks-Ngarm, S.; Pitisuttithum, P.; Nitayaphan, S.; Kaewkungwal, J.; Chiu, J.; Paris, R.; Premsri, N.; Namwat, C.; de Souza, M.; Adams, E.; et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 2009, 361, 2209–2220. [Google Scholar] [CrossRef] [PubMed]
- Sliepen, K.; Ozorowski, G.; Burger, J.A.; van Montfort, T.; Stunnenberg, M.; LaBranche, C.; Montefiori, D.C.; Moore, J.P.; Ward, A.B.; Sanders, R.W. Presenting native-like HIV-1 envelope trimers on ferritin nanoparticles improves their immunogenicity. Retrovirology 2015, 12, 82. [Google Scholar] [CrossRef] [PubMed]
- Saunders, K.O.; Wiehe, K.; Tian, M.; Acharya, P.; Bradley, T.; Alam, S.M.; Go, E.P.; Scearce, R.; Sutherland, L.; Henderson, R.; et al. Targeted selection of HIV-specific antibody mutations by engineering B cell maturation. Science 2019, 366, eaay7199. [Google Scholar] [CrossRef] [PubMed]
- Tokatlian, T.; Read, B.J.; Jones, C.A.; Kulp, D.W.; Menis, S.; Chang, J.Y.H.; Steichen, J.M.; Kumari, S.; Allen, J.D.; Dane, E.L.; et al. Innate immune recognition of glycans targets HIV nanoparticle immunogens to germinal centers. Science 2019, 363, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Mu, Z.; Wiehe, K.; Saunders, K.O.; Henderson, R.; Cain, D.W.; Parks, R.; Martik, D.; Mansouri, K.; Edwards, R.J.; Newman, A.; et al. mRNA-encoded HIV-1 Env trimer ferritin nanoparticles induce monoclonal antibodies that neutralize heterologous HIV-1 isolates in mice. Cell Rep. 2022, 38, 110514. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Ofek, G.; Laub, L.; Louder, M.K.; Doria-Rose, N.A.; Longo, N.S.; Imamichi, H.; Bailer, R.T.; Chakrabarti, B.; Sharma, S.K.; et al. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature 2012, 491, 406–412. [Google Scholar] [CrossRef]
- Krebs, S.J.; McBurney, S.P.; Kovarik, D.N.; Waddell, C.D.; Jaworski, J.P.; Sutton, W.F.; Gomes, M.M.; Trovato, M.; Waagmeester, G.; Barnett, S.J.; et al. Multimeric Scaffolds Displaying the HIV-1 Envelope MPER Induce MPER-Specific Antibodies and Cross-Neutralizing Antibodies when Co-Immunized with gp160 DNA. PLoS ONE 2014, 9, e113463. [Google Scholar] [CrossRef]
- He, L.; de Val, N.; Morris, C.D.; Vora, N.; Thinnes, T.C.; Kong, L.; Azadnia, P.; Sok, D.; Zhou, B.; Burton, D.R.; et al. Presenting native-like trimeric HIV-1 antigens with self-assembling nanoparticles. Nat. Commun. 2016, 7, 12041. [Google Scholar] [CrossRef]
- Zhang, Y.-N.; Paynter, J.; Antanasijevic, A.; Allen, J.D.; Eldad, M.; Lee, Y.-Z.; Copps, J.; Newby, M.L.; He, L.; Chavez, D.; et al. Single-component multilayered self-assembling protein nanoparticles presenting glycan-trimmed uncleaved prefusion optimized envelope trimers as HIV-1 vaccine candidates. Nat. Commun. 2023, 14, 1985. [Google Scholar] [CrossRef]
- Gutjahr, A.; Phelip, C.; Coolen, A.L.; Monge, C.; Boisgard, A.S.; Paul, S.; Verrier, B. Biodegradable Polymeric Nanoparticles-Based Vaccine Adjuvants for Lymph Nodes Targeting. Vaccines 2016, 4, 34. [Google Scholar] [CrossRef]
- Rostami, H.; Ebtekar, M.; Ardestani, M.S.; Yazdi, M.H.; Mahdavi, M. Co-utilization of a TLR5 agonist and nano-formulation of HIV-1 vaccine candidate leads to increased vaccine immunogenicity and decreased immunogenic dose: A preliminary study. Immunol. Lett. 2017, 187, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Xin, X.; Liu, Y.; Guo, L.; Wang, H.; Lu, D.; Chang, Y.; Wan, M.; Zhang, Y.; Shan, Y.; Zhang, Q.; et al. Improvement of B Cell Responses by an HIV-1 Amphiphilic Polymer Nanovaccine. Nano Lett. 2023, 23, 4090–4094. [Google Scholar] [CrossRef] [PubMed]
- Elechiguerra, J.L.; Burt, J.L.; Morones, J.R.; Camacho-Bragado, A.; Gao, X.; Lara, H.H.; Yacaman, M.J. Interaction of silver nanoparticles with HIV-1. J. Nanobiotechnol. 2005, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Lara, H.H.; Ixtepan-Turrent, L.; Garza Trevino, E.N.; Singh, D.K. Use of silver nanoparticles increased inhibition of cell-associated HIV-1 infection by neutralizing antibodies developed against HIV-1 envelope proteins. J. Nanobiotechnol. 2011, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Liu, Y.; Chen, Z.; Li, W.; Liu, Y.; Wang, L.; Liu, Y.; Wu, X.; Ji, Y.; Zhao, Y.; et al. Surface-engineered gold nanorods: Promising DNA vaccine adjuvant for HIV-1 treatment. Nano Lett. 2012, 12, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- Climent, N.; Garcia, I.; Marradi, M.; Chiodo, F.; Miralles, L.; Maleno, M.J.; Gatell, J.M.; Garcia, F.; Penades, S.; Plana, M. Loading dendritic cells with gold nanoparticles (GNPs) bearing HIV-peptides and mannosides enhance HIV-specific T cell responses. Nanomedicine 2018, 14, 339–351. [Google Scholar] [CrossRef]
- Damm, D.; Kostka, K.; Weingartner, C.; Wagner, J.T.; Rojas-Sanchez, L.; Gensberger-Reigl, S.; Sokolova, V.; Uberla, K.; Epple, M.; Temchura, V. Covalent coupling of HIV-1 glycoprotein trimers to biodegradable calcium phosphate nanoparticles via genetically encoded aldehyde-tags. Acta Biomater. 2022, 140, 586–600. [Google Scholar] [CrossRef]
- Li, S.; Wang, B.; Jiang, S.; Pan, Y.; Shi, Y.; Kong, W.; Shan, Y. Surface-Functionalized Silica-Coated Calcium Phosphate Nanoparticles Efficiently Deliver DNA-Based HIV-1 Trimeric Envelope Vaccines against HIV-1. ACS Appl. Mater. Interfaces 2021, 13, 53630–53645. [Google Scholar] [CrossRef]
- Bailey, J.R.; Barnes, E.; Cox, A.L. Approaches, Progress, and Challenges to Hepatitis C Vaccine Development. Gastroenterology 2019, 156, 418–430. [Google Scholar] [CrossRef]
- Hatzakis, A.; Magiorkinis, E.; Haida, C. HBV virological assessment. J. Hepatol. 2006, 44, S71–S76. [Google Scholar] [CrossRef]
- Maupas, P.; Chiron, J.P.; Barin, F.; Coursaget, P.; Goudeau, A.; Perrin, J.; Denis, F.; Mar, I.D. Efficacy of hepatitis B vaccine in prevention of early HBsAg carrier state in children. Controlled trial in an endemic area (Senegal). Lancet 1981, 1, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Scolnick, E.M.; McLean, A.A.; West, D.J.; McAleer, W.J.; Miller, W.J.; Buynak, E.B. Clinical evaluation in healthy adults of a hepatitis B vaccine made by recombinant DNA. JAMA 1984, 251, 2812–2815. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.M.; Simon, J.K.; Baker, J.R., Jr. Applications of nanotechnology for immunology. Nat. Rev. Immunol. 2013, 13, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Mobini, S.; Chizari, M.; Mafakher, L.; Rismani, E.; Rismani, E. Computational Design of a Novel VLP-Based Vaccine for Hepatitis B Virus. Front. Immunol. 2020, 11, 2074. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhou, X.; Zhou, Y.H. Hepatitis B vaccine development and implementation. Hum. Vaccin Immunother. 2020, 16, 1533–1544. [Google Scholar] [CrossRef] [PubMed]
- Mohsen, M.O.; Zha, L.; Cabral-Miranda, G.; Bachmann, M.F. Major findings and recent advances in virus-like particle (VLP)-based vaccines. Semin Immunol. 2017, 34, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, J.N.; Zuckerman, A.J.; Symington, I.; Du, W.; Williams, A.; Dickson, B.; Young, M.D. Evaluation of a new hepatitis B triple-antigen vaccine in inadequate responders to current vaccines. Hepatology 2001, 34, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Vesikari, T.; Finn, A.; van Damme, P.; Leroux-Roels, I.; Leroux-Roels, G.; Segall, N.; Toma, A.; Vallieres, G.; Aronson, R.; Reich, D.; et al. Immunogenicity and Safety of a 3-Antigen Hepatitis B Vaccine vs a Single-Antigen Hepatitis B Vaccine: A Phase 3 Randomized Clinical Trial. JAMA Netw. Open 2021, 4, e2128652. [Google Scholar] [CrossRef]
- Wang, W.; Zhou, X.; Bian, Y.; Wang, S.; Chai, Q.; Guo, Z.; Wang, Z.; Zhu, P.; Peng, H.; Yan, X.; et al. Dual-targeting nanoparticle vaccine elicits a therapeutic antibody response against chronic hepatitis B. Nat. Nanotechnol. 2020, 15, 406–416. [Google Scholar] [CrossRef]
- Zhu, J.; Qin, F.; Ji, Z.; Fei, W.; Tan, Z.; Hu, Y.; Zheng, C. Mannose-Modified PLGA Nanoparticles for Sustained and Targeted Delivery in Hepatitis B Virus Immunoprophylaxis. AAPS PharmSciTech 2019, 21, 13. [Google Scholar] [CrossRef]
- Chong, C.S.; Cao, M.; Wong, W.W.; Fischer, K.P.; Addison, W.R.; Kwon, G.S.; Tyrrell, D.L.; Samuel, J. Enhancement of T helper type 1 immune responses against hepatitis B virus core antigen by PLGA nanoparticle vaccine delivery. J. Control. Release 2005, 102, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Martinello, M.; Hajarizadeh, B.; Grebely, J.; Dore, G.J.; Matthews, G.V. Management of acute HCV infection in the era of direct-acting antiviral therapy. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Offersgaard, A.; Bukh, J.; Gottwein, J.M. Toward a vaccine against hepatitis C virus. Science 2023, 380, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Elmowalid, G.A.; Qiao, M.; Jeong, S.H.; Borg, B.B.; Baumert, T.F.; Sapp, R.K.; Hu, Z.; Murthy, K.; Liang, T.J. Immunization with hepatitis C virus-like particles results in control of hepatitis C virus infection in chimpanzees. Proc. Natl. Acad. Sci. USA 2007, 104, 8427–8432. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, D.; Earnest-Silveira, L.; Chua, B.; Meuleman, P.; Boo, I.; Grubor-Bauk, B.; Jackson, D.C.; Keck, Z.Y.; Foung, S.K.H.; Drummer, H.E.; et al. Immunological responses following administration of a genotype 1a/1b/2/3a quadrivalent HCV VLP vaccine. Sci. Rep. 2018, 8, 6483. [Google Scholar] [CrossRef]
- Garrone, P.; Fluckiger, A.C.; Mangeot, P.E.; Gauthier, E.; Dupeyrot-Lacas, P.; Mancip, J.; Cangialosi, A.; Du Chene, I.; LeGrand, R.; Mangeot, I.; et al. A prime-boost strategy using virus-like particles pseudotyped for HCV proteins triggers broadly neutralizing antibodies in macaques. Sci. Transl. Med. 2011, 3, 94ra71. [Google Scholar] [CrossRef]
- Earnest-Silveira, L.; Christiansen, D.; Herrmann, S.; Ralph, S.A.; Das, S.; Gowans, E.J.; Torresi, J. Large scale production of a mammalian cell derived quadrivalent hepatitis C virus like particle vaccine. J. Virol. Methods 2016, 236, 87–92. [Google Scholar] [CrossRef]
- von Delft, A.; Donnison, T.A.; Lourenco, J.; Hutchings, C.; Mullarkey, C.E.; Brown, A.; Pybus, O.G.; Klenerman, P.; Chinnakannan, S.; Barnes, E. The generation of a simian adenoviral vectored HCV vaccine encoding genetically conserved gene segments to target multiple HCV genotypes. Vaccine 2018, 36, 313–321. [Google Scholar] [CrossRef]
- Toth, E.A.; Andrianov, A.K.; Fuerst, T.R. Prospects for developing an Hepatitis C virus E1E2-based nanoparticle vaccine. Rev. Med. Virol. 2023, 33, e2474. [Google Scholar] [CrossRef]
- Sliepen, K.; Radic, L.; Capella-Pujol, J.; Watanabe, Y.; Zon, I.; Chumbe, A.; Lee, W.H.; de Gast, M.; Koopsen, J.; Koekkoek, S.; et al. Induction of cross-neutralizing antibodies by a permuted hepatitis C virus glycoprotein nanoparticle vaccine candidate. Nat. Commun. 2022, 13, 7271. [Google Scholar] [CrossRef]
- He, L.; Tzarum, N.; Lin, X.; Shapero, B.; Sou, C.; Mann, C.J.; Stano, A.; Zhang, L.; Nagy, K.; Giang, E.; et al. Proof of concept for rational design of hepatitis C virus E2 core nanoparticle vaccines. Sci. Adv. 2020, 6, eaaz6225. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Xu, K.; Li, J.; Huang, Q.; Song, J.; Han, Y.; Zheng, T.; Gao, P.; Lu, X.; Yang, H.; et al. Protective Zika vaccines engineered to eliminate enhancement of dengue infection via immunodominance switch. Nat. Immunol. 2021, 22, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Zanluca, C.; Dos Santos, C.N. Zika virus—An overview. Microbes Infect. 2016, 18, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Pielnaa, P.; Al-Saadawe, M.; Saro, A.; Dama, M.F.; Zhou, M.; Huang, Y.; Huang, J.; Xia, Z. Zika virus-spread, epidemiology, genome, transmission cycle, clinical manifestation, associated challenges, vaccine and antiviral drug development. Virology 2020, 543, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Pattnaik, A.; Sahoo, B.R.; Struble, L.R.; Borgstahl, G.E.O.; Zhou, Y.; Franco, R.; Barletta, R.G.; Osorio, F.A.; Petro, T.M.; Pattnaik, A.K. A Ferritin Nanoparticle-Based Zika Virus Vaccine Candidate Induces Robust Humoral and Cellular Immune Responses and Protects Mice from Lethal Virus Challenge. Vaccines 2023, 11, 821. [Google Scholar] [CrossRef]
- Hao, H.; Wu, S.; Lin, J.; Zheng, Z.; Zhou, Y.; Zhang, Y.; Guo, Q.; Tian, F.; Zhao, M.; Chen, Y.; et al. Immunization against Zika by entrapping live virus in a subcutaneous self-adjuvanting hydrogel. Nat. Biomed Eng. 2023, 7, 928–942. [Google Scholar] [CrossRef]
- Khetarpal, N.; Khanna, I. Dengue Fever: Causes, Complications, and Vaccine Strategies. J. Immunol. Res. 2016, 2016, 6803098. [Google Scholar] [CrossRef]
- Tully, D.; Griffiths, C.L. Dengvaxia: The world’s first vaccine for prevention of secondary dengue. Ther. Adv. Vaccines Immunother. 2021, 9, 25151355211015839. [Google Scholar] [CrossRef]
- Norshidah, H.; Vignesh, R.; Lai, N.S. Updates on Dengue Vaccine and Antiviral: Where Are We Heading? Molecules 2021, 26, 6768. [Google Scholar] [CrossRef]
- Aguiar, M.; Stollenwerk, N.; Halstead, S.B. The risks behind Dengvaxia recommendation. Lancet Infect. Dis. 2016, 16, 882–883. [Google Scholar] [CrossRef]
- Quach, Q.H.; Ang, S.K.; Chu, J.J.; Kah, J.C.Y. Size-dependent neutralizing activity of gold nanoparticle-based subunit vaccine against dengue virus. Acta Biomater. 2018, 78, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Jearanaiwitayakul, T.; Sunintaboon, P.; Chawengkittikul, R.; Limthongkul, J.; Midoeng, P.; Chaisuwirat, P.; Warit, S.; Ubol, S. Whole inactivated dengue virus-loaded trimethyl chitosan nanoparticle-based vaccine: Immunogenic properties in ex vivo and in vivo models. Hum. Vaccin Immunother. 2021, 17, 2793–2807. [Google Scholar] [CrossRef] [PubMed]
- Madhi, S.A.; Polack, F.P.; Piedra, P.A.; Munoz, F.M.; Trenholme, A.A.; Simoes, E.A.F.; Swamy, G.K.; Agrawal, S.; Ahmed, K.; August, A.; et al. Respiratory Syncytial Virus Vaccination during Pregnancy and Effects in Infants. N. Engl. J. Med. 2020, 383, 426–439. [Google Scholar] [CrossRef]
- Shi, T.; McAllister, D.A.; O’Brien, K.L.; Simoes, E.A.F.; Madhi, S.A.; Gessner, B.D.; Polack, F.P.; Balsells, E.; Acacio, S.; Aguayo, C.; et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: A systematic review and modelling study. Lancet 2017, 390, 946–958. [Google Scholar] [CrossRef] [PubMed]
- Mazur, N.I.; Higgins, D.; Nunes, M.C.; Melero, J.A.; Langedijk, A.C.; Horsley, N.; Buchholz, U.J.; Openshaw, P.J.; McLellan, J.S.; Englund, J.A.; et al. The respiratory syncytial virus vaccine landscape: Lessons from the graveyard and promising candidates. Lancet Infect. Dis. 2018, 18, e295–e311. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.S.; Chen, M.; Joyce, M.G.; Sastry, M.; Stewart-Jones, G.B.E.; Yang, Y.; Zhang, B.; Chen, L.; Srivatsan, S.; Zheng, A.; et al. Structure-Based Design of a Fusion Glycoprotein Vaccine for Respiratory Syncytial Virus. Science 2013, 342, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Ison, M.G.; Langley, J.M.; Lee, D.-G.; Leroux-Roels, I.; Martinon-Torres, F.; Schwarz, T.F.; van Zyl-Smit, R.N.; Campora, L.; Dezutter, N.; et al. Respiratory Syncytial Virus Prefusion F Protein Vaccine in Older Adults. N. Engl. J. Med. 2023, 388, 595–608. [Google Scholar] [CrossRef]
- Walsh, E.E.; Pérez Marc, G.; Zareba, A.M.; Falsey, A.R.; Jiang, Q.; Patton, M.; Polack, F.P.; Llapur, C.; Doreski, P.A.; Ilangovan, K.; et al. Efficacy and Safety of a Bivalent RSV Prefusion F Vaccine in Older Adults. N. Engl. J. Med. 2023, 388, 1465–1477. [Google Scholar] [CrossRef]
- Marcandalli, J.; Fiala, B.; Ols, S.; Perotti, M.; de van der Schueren, W.; Snijder, J.; Hodge, E.; Benhaim, M.; Ravichandran, R.; Carter, L.; et al. Induction of Potent Neutralizing Antibody Responses by a Designed Protein Nanoparticle Vaccine for Respiratory Syncytial Virus. Cell 2019, 176, 1420–1431.e1417. [Google Scholar] [CrossRef]
- Leroux-Roels, I.; Bruhwyler, J.; Stergiou, L.; Sumeray, M.; Joye, J.; Maes, C.; Lambert, P.H.; Leroux-Roels, G. Double-Blind, Placebo-Controlled, Dose-Escalating Study Evaluating the Safety and Immunogenicity of an Epitope-Specific Chemically Defined Nanoparticle RSV Vaccine. Vaccines 2023, 11, 367. [Google Scholar] [CrossRef]
- Leligdowicz, A.; Fischer, W.A.; Uyeki, T.M.; Fletcher, T.E.; Adhikari, N.K.J.; Portella, G.; Lamontagne, F.; Clement, C.; Jacob, S.T.; Rubinson, L.; et al. Ebola virus disease and critical illness. Crit. Care 2016, 20, 217. [Google Scholar] [CrossRef] [PubMed]
- Halperin, S.A.; Arribas, J.R.; Rupp, R.; Andrews, C.P.; Chu, L.; Das, R.; Simon, J.K.; Onorato, M.T.; Liu, K.; Martin, J.; et al. Six-Month Safety Data of Recombinant Vesicular Stomatitis Virus-Zaire Ebola Virus Envelope Glycoprotein Vaccine in a Phase 3 Double-Blind, Placebo-Controlled Randomized Study in Healthy Adults. J. Infect. Dis. 2017, 215, 1789–1798. [Google Scholar] [CrossRef] [PubMed]
- Warfield, K.L.; Bosio, C.M.; Welcher, B.C.; Deal, E.M.; Mohamadzadeh, M.; Schmaljohn, A.; Aman, M.J.; Bavari, S. Ebola virus-like particles protect from lethal Ebola virus infection. Proc. Natl. Acad. Sci. USA 2003, 100, 15889–15894. [Google Scholar] [CrossRef] [PubMed]
- Warfield, K.L.; Swenson, D.L.; Olinger, G.G.; Kalina, W.V.; Aman, M.J.; Bavari, S. Ebola Virus-Like Particle-Based Vaccine Protects Nonhuman Primates against Lethal Ebola Virus Challenge. J. Infect. Dis. 2007, 196, S430–S437. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Marasini, B.; Chen, X.; Ding, L.; Wang, J.-J.; Xiao, P.; Villinger, F.; Spearman, P. A Bivalent, Spherical Virus-Like Particle Vaccine Enhances Breadth of Immune Responses against Pathogenic Ebola Viruses in Rhesus Macaques. J. Virol. 2020, 94, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.J.; Suh, H.; Bershteyn, A.; Stephan, M.T.; Liu, H.; Huang, B.; Sohail, M.; Luo, S.; Ho Um, S.; Khant, H.; et al. Interbilayer-crosslinked multilamellar vesicles as synthetic vaccines for potent humoral and cellular immune responses. Nat. Mater. 2011, 10, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Bazzill, J.D.; Stronsky, S.M.; Kalinyak, L.C.; Ochyl, L.J.; Steffens, J.T.; van Tongeren, S.A.; Cooper, C.L.; Moon, J.J. Vaccine nanoparticles displaying recombinant Ebola virus glycoprotein for induction of potent antibody and polyfunctional T cell responses. Nanomed. Nanotechnol. Biol. Med. 2019, 18, 414–425. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Chaudhary, A.; Lin, X.; Sou, C.; Alkutkar, T.; Kumar, S.; Ngo, T.; Kosviner, E.; Ozorowski, G.; Stanfield, R.L.; et al. Single-component multilayered self-assembling nanoparticles presenting rationally designed glycoprotein trimers as Ebola virus vaccines. Nat. Commun. 2021, 12, 2633. [Google Scholar] [CrossRef]
- Fries, L.; Cho, I.; Krähling, V.; Fehling, S.K.; Strecker, T.; Becker, S.; Hooper, J.W.; Kwilas, S.A.; Agrawal, S.; Wen, J.; et al. Randomized, Blinded, Dose-Ranging Trial of an Ebola Virus Glycoprotein Nanoparticle Vaccine With Matrix-M Adjuvant in Healthy Adults. J. Infect. Dis. 2020, 222, 572–582. [Google Scholar] [CrossRef]
- Bengtsson, K.L.; Song, H.; Stertman, L.; Liu, Y.; Flyer, D.C.; Massare, M.J.; Xu, R.-H.; Zhou, B.; Lu, H.; Kwilas, S.A.; et al. Matrix-M adjuvant enhances antibody, cellular and protective immune responses of a Zaire Ebola/Makona virus glycoprotein (GP) nanoparticle vaccine in mice. Vaccine 2016, 34, 1927–1935. [Google Scholar] [CrossRef]
- Holmgren, J.; Czerkinsky, C. Mucosal immunity and vaccines. Nat. Med. 2005, 11, S45–S53. [Google Scholar] [CrossRef] [PubMed]
- Silva-Sanchez, A.; Randall, T.D. Role of iBALT in Respiratory Immunity. Curr. Top. Microbiol. Immunol. 2020, 426, 21–43. [Google Scholar] [PubMed]
- Negri, D.R.M.; Pinto, D.; Vendetti, S.; Patrizio, M.; Sanchez, M.; Riccomi, A.; Ruggiero, P.; Giudice, G.D.; Magistris, M.T.D. Cholera Toxin and Escherichia coli Heat-Labile Enterotoxin, but Not Their Nontoxic Counterparts, Improve the Antigen-Presenting Cell Function of Human B Lymphocytes. Infect. Immun. 2009, 77, 1924–1935. [Google Scholar] [CrossRef] [PubMed]
- Csaba, N.; Garcia-Fuentes, M.; Alonso, M.J. Nanoparticles for nasal vaccination. Adv. Drug Deliv. Rev. 2009, 61, 140–157. [Google Scholar] [PubMed]
- Zhuo, S.-H.; Wu, J.-J.; Zhao, L.; Li, W.-H.; Zhao, Y.-F.; Li, Y.-M. A chitosan-mediated inhalable nanovaccine against SARS-CoV-2. Nano Res. 2022, 15, 4191–4200. [Google Scholar] [CrossRef] [PubMed]
- Knight, F.C.; Gilchuk, P.; Kumar, A.; Becker, K.W.; Sevimli, S.; Jacobson, M.E.; Suryadevara, N.; Wang-Bishop, L.; Boyd, K.L.; Crowe, J.E., Jr.; et al. Mucosal Immunization with a pH-Responsive Nanoparticle Vaccine Induces Protective CD8+ Lung-Resident Memory T Cells. ACS Nano 2019, 13, 10939–10960. [Google Scholar] [CrossRef]
- Fahy, J.V.; Dickey, B.F. Airway Mucus Function and Dysfunction. N. Engl. J. Med. 2010, 363, 2233–2247. [Google Scholar] [CrossRef]
- Davis, J.D.; Wypych, T.P. Cellular and functional heterogeneity of the airway epithelium. Mucosal Immunol. 2021, 14, 978–990. [Google Scholar] [CrossRef]
- Dillon, A.; Lo, D.D. M Cells: Intelligent Engineering of Mucosal Immune Surveillance. Front. Immunol. 2019, 10, 1499. [Google Scholar] [CrossRef]
- Pedersen, G.; Cox, R. The mucosal vaccine quandary: Intranasal vs. sublingual immunization against influenza. Hum. Vaccines Immunother. 2012, 8, 689–693. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Hu, M.; Liu, X.; Liu, X.; Chen, T.; Zhu, Y.; Liang, T.; Xiao, S.; Li, P.; Ma, X. Nanoparticles and Antiviral Vaccines. Vaccines 2024, 12, 30. https://doi.org/10.3390/vaccines12010030
Liu S, Hu M, Liu X, Liu X, Chen T, Zhu Y, Liang T, Xiao S, Li P, Ma X. Nanoparticles and Antiviral Vaccines. Vaccines. 2024; 12(1):30. https://doi.org/10.3390/vaccines12010030
Chicago/Turabian StyleLiu, Sen, Meilin Hu, Xiaoqing Liu, Xingyu Liu, Tao Chen, Yiqiang Zhu, Taizhen Liang, Shiqi Xiao, Peiwen Li, and Xiancai Ma. 2024. "Nanoparticles and Antiviral Vaccines" Vaccines 12, no. 1: 30. https://doi.org/10.3390/vaccines12010030