
Vaccinia Virus Strain MVA Expressing a Prefusion-Stabilized SARS-CoV-2 Spike Glycoprotein Induces Robust Protection and Prevents Brain Infection in Mouse and Hamster Models
 , , , , , , , , , and add
Show full author list
, , , , , , , , , and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Cells and Viruses
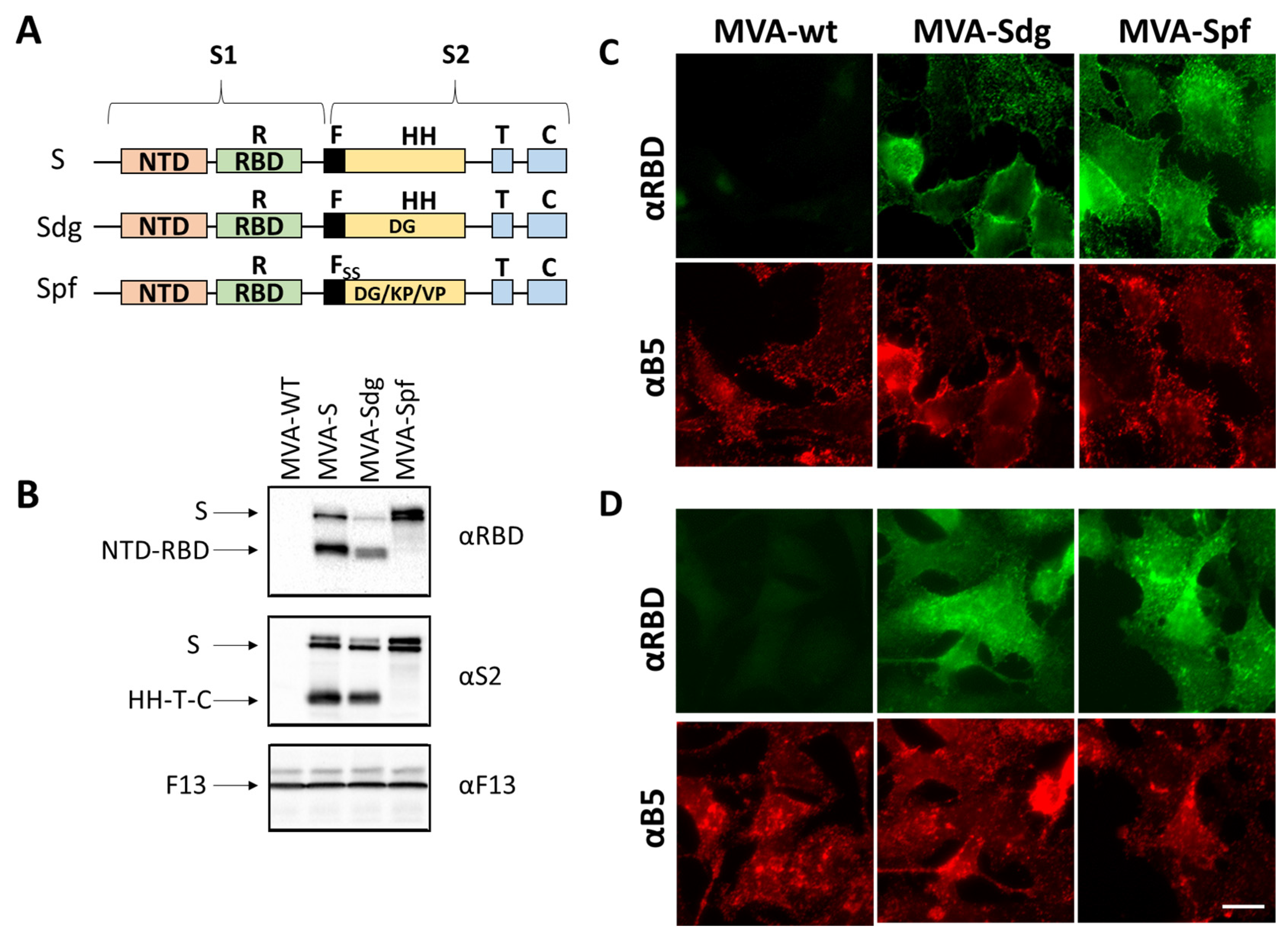
2.3. Design and Generation of MVAs
2.4. Western Blot
2.5. Immunofluorescence
2.6. Protein Expression and Purification
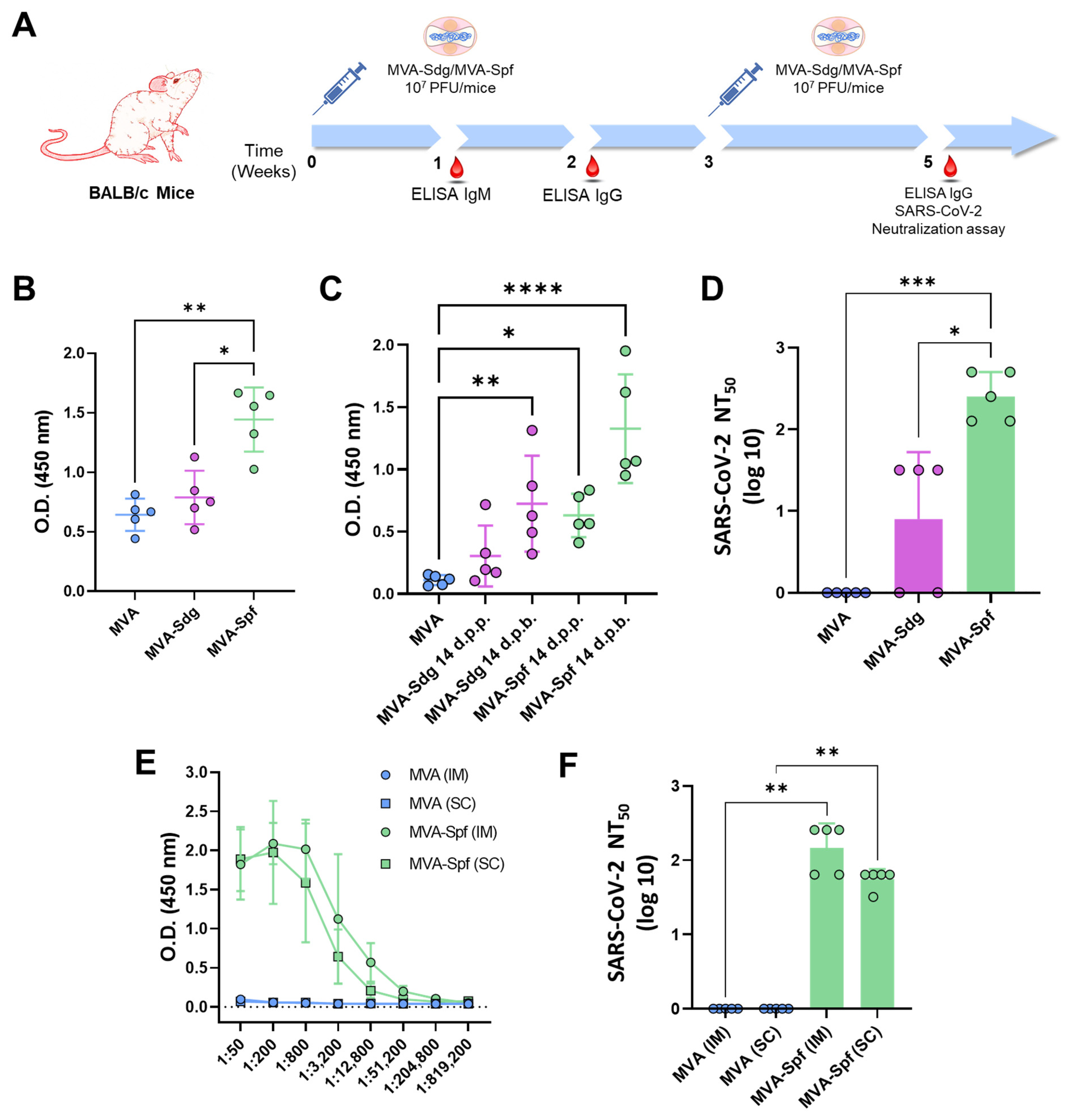
2.7. Mice Immunization and SARS-CoV-2 Challenge
2.8. Enzyme-Linked Immunosorbent Assay (ELISA) to SARS-CoV-2 S
2.9. Ex Vivo Flow Cytometric Analysis
2.10. Plaque Reduction Neutralization (PRNT) Assay
2.11. Bioluminescence Imaging (BLI) of SARS-CoV-2 Infection
2.12. Measurement of Viral Burden and Analysis of Signature Inflammatory Cytokines mRNA
2.13. Experiments in Golden Syrian Hamsters
2.14. Statistical Analysis
3. Results
3.1. Isolation of MVA Recombinants Expressing SARS-CoV-2 S
3.2. S Protein Expression by MVA Recombinants
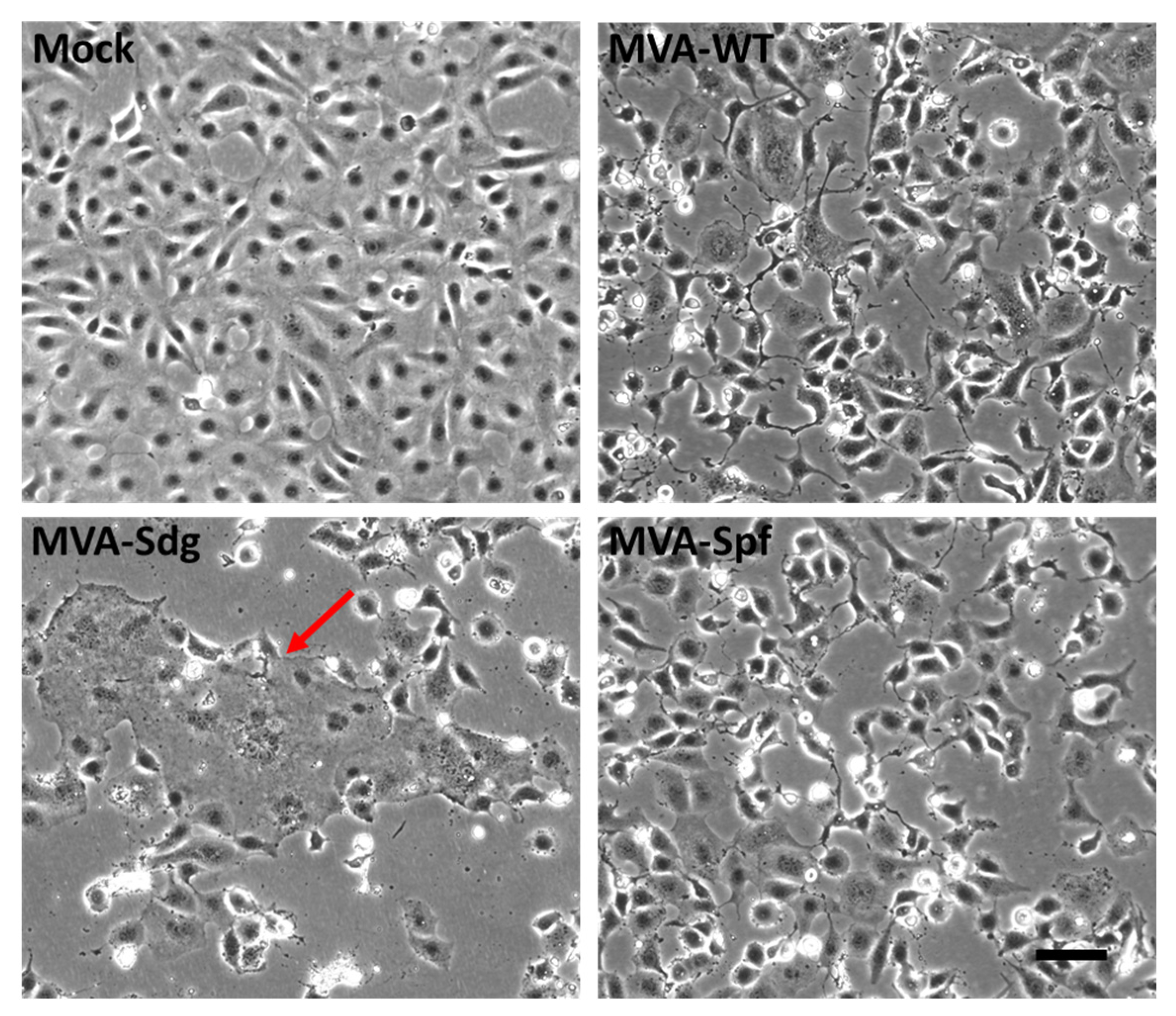
3.3. Fusion Phenotype in Cells Infected by MVA Recombinants
3.4. Induction of Antibody Responses in BALB/c Mice
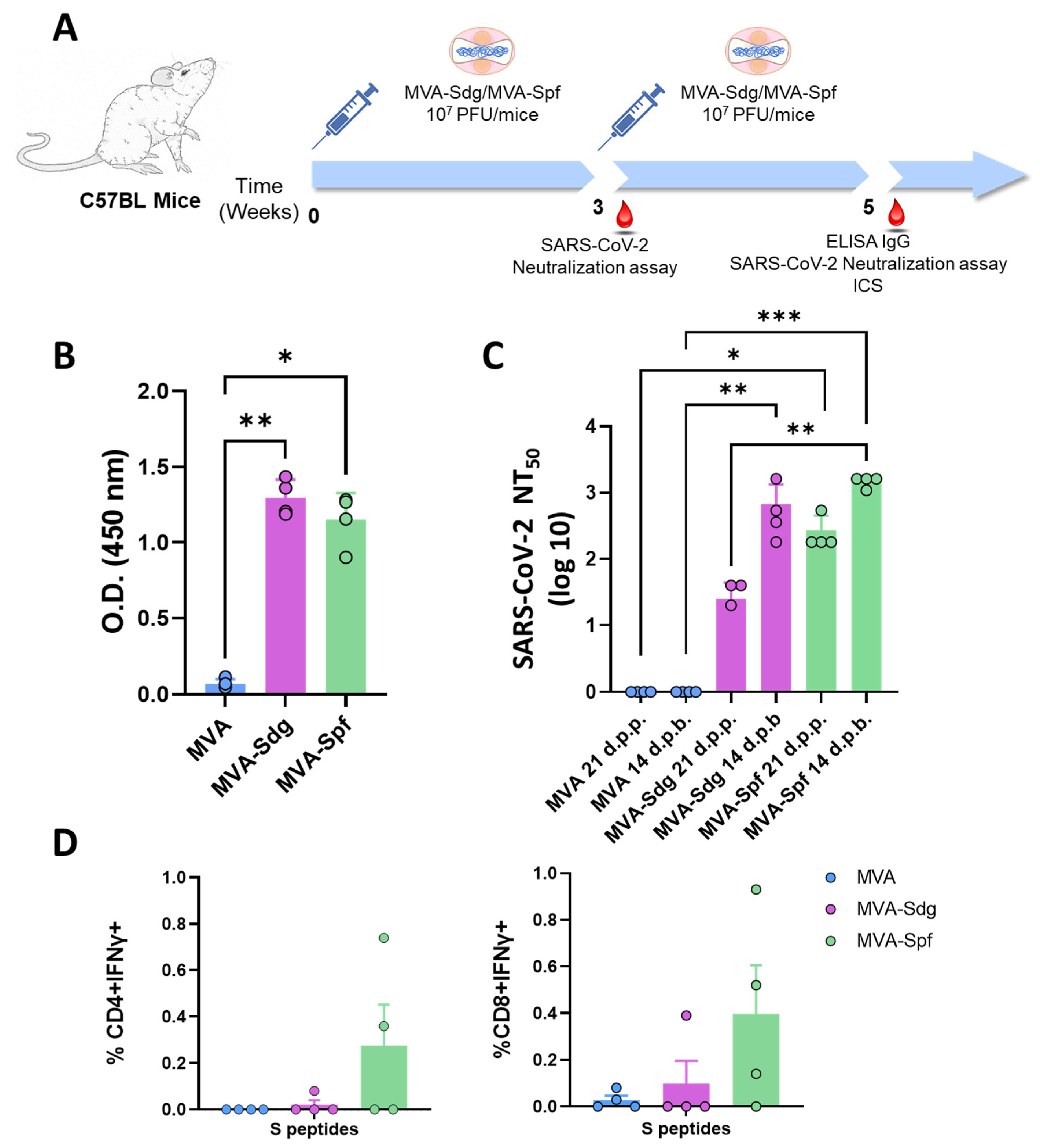
3.5. Induction of Humoral and Cellular Responses in C57BL6 (B6) Mice
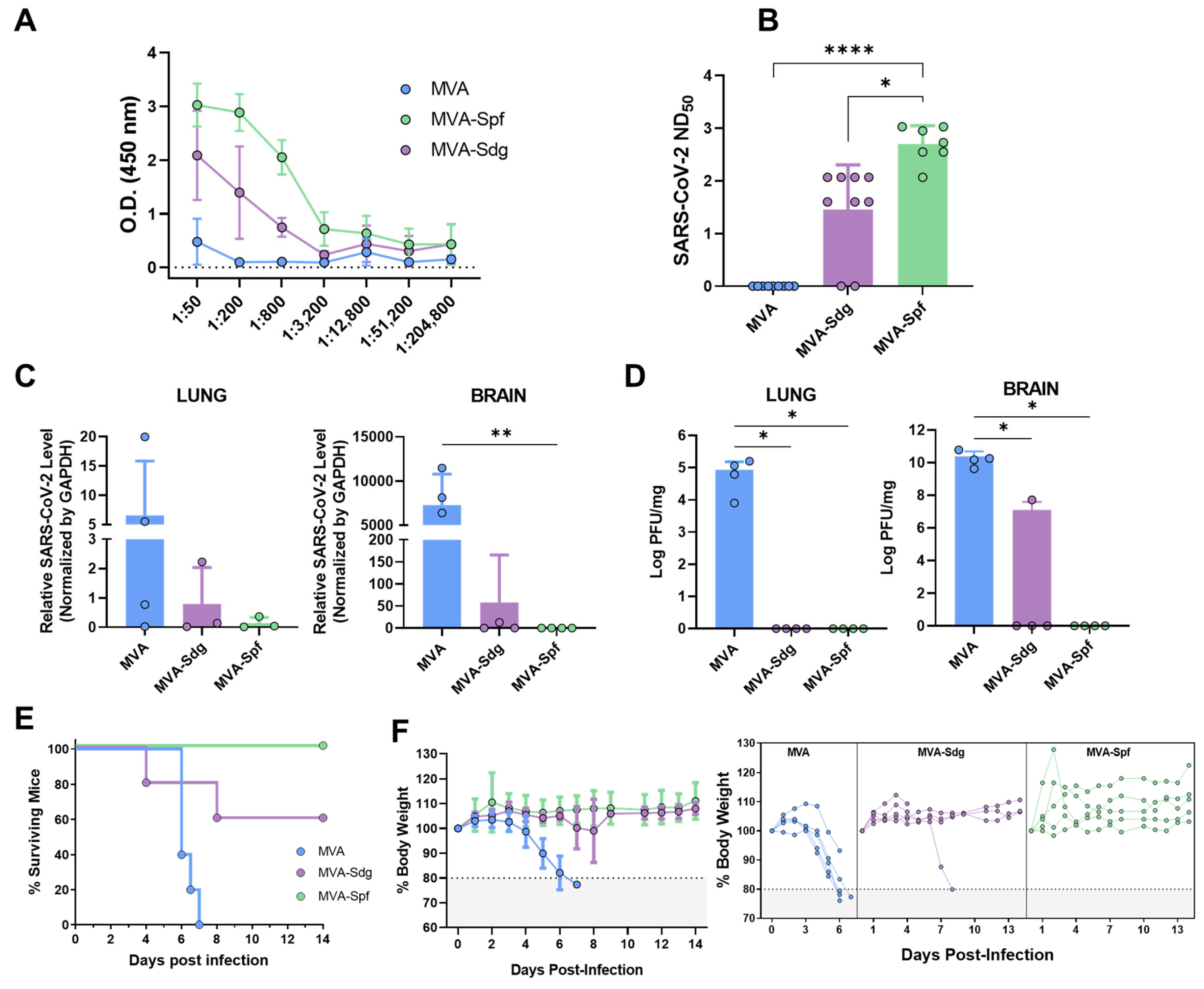
3.6. Immunogenicity and Protection Efficacy of Recombinant MVAs in K18-hACE2 Mice
3.6.1. Humoral Response in K18-hACE2 Mice
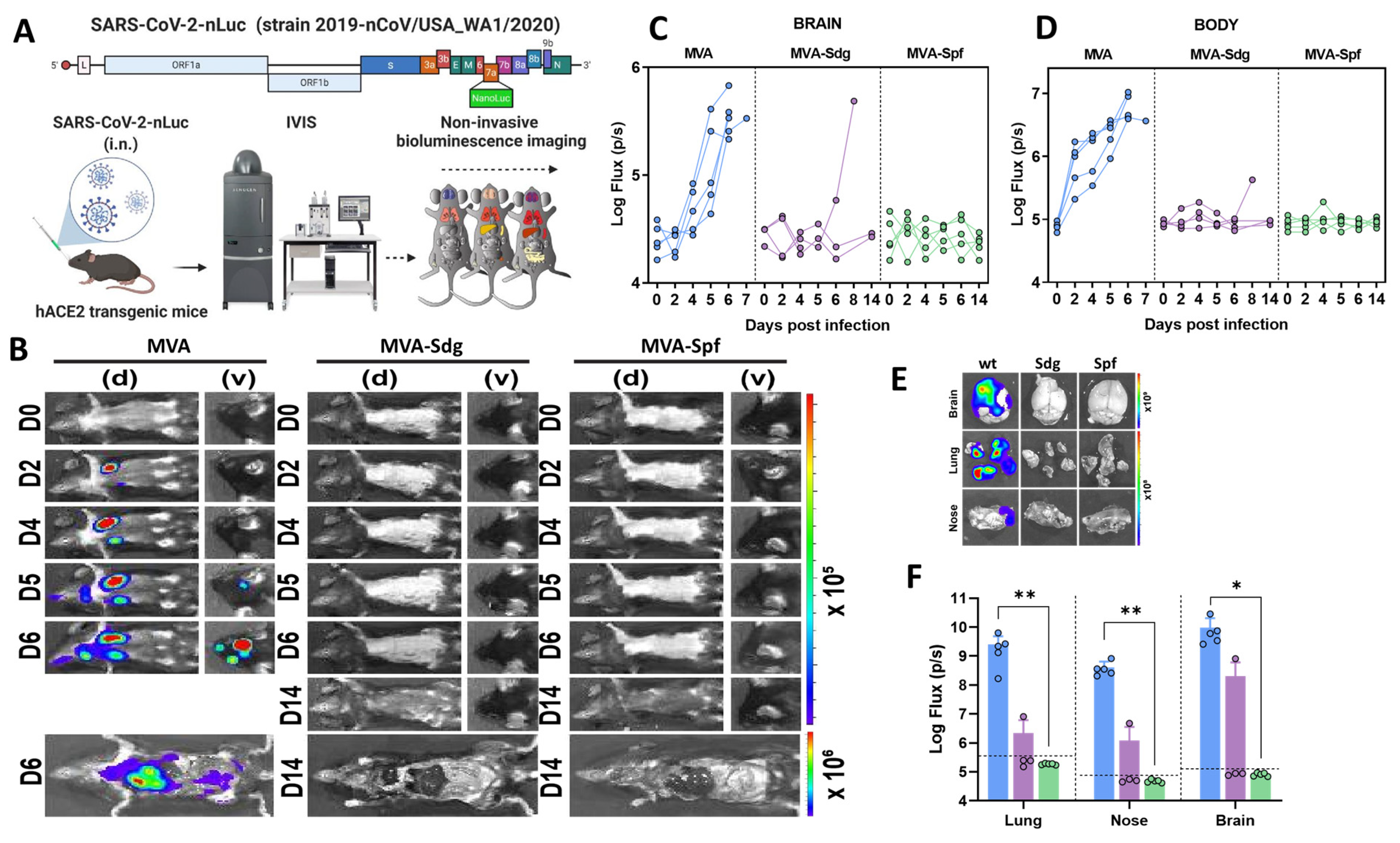
3.6.2. Protection in K18-hACE2 Mice
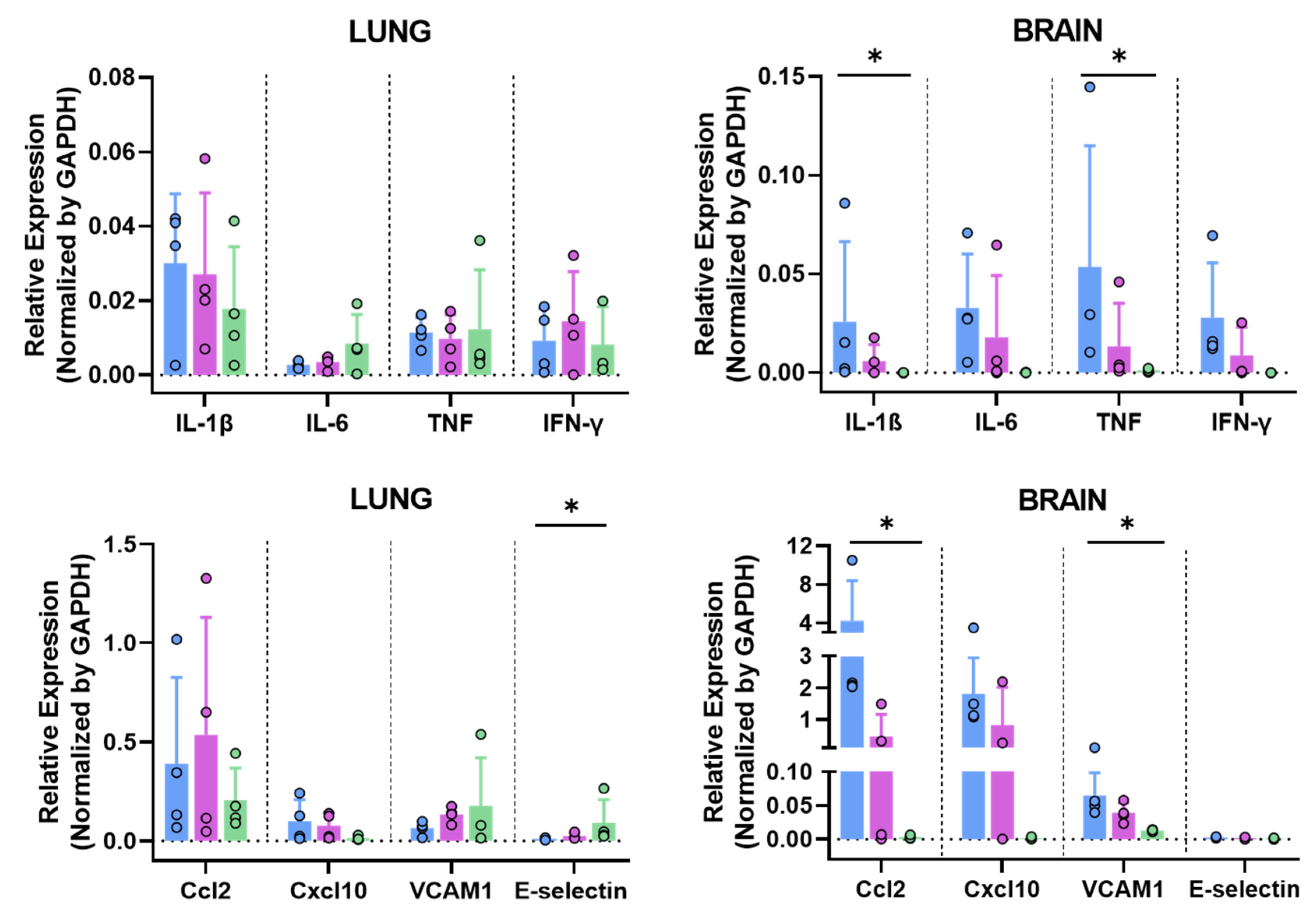
3.6.3. MVA-Spf Controls Cytokine Response in the Brain of SARS-CoV-2 Infected K18-hACE2 Mice
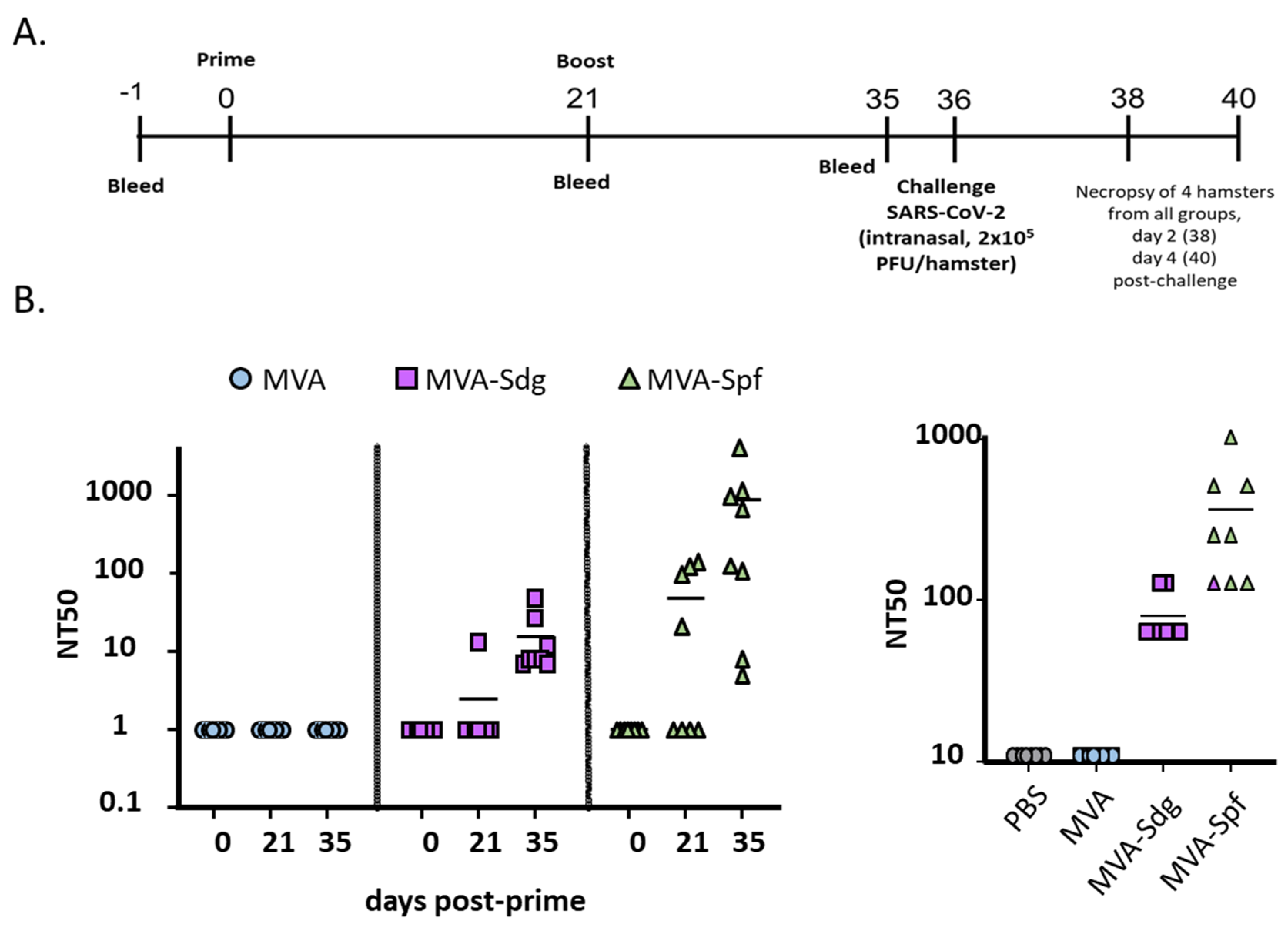
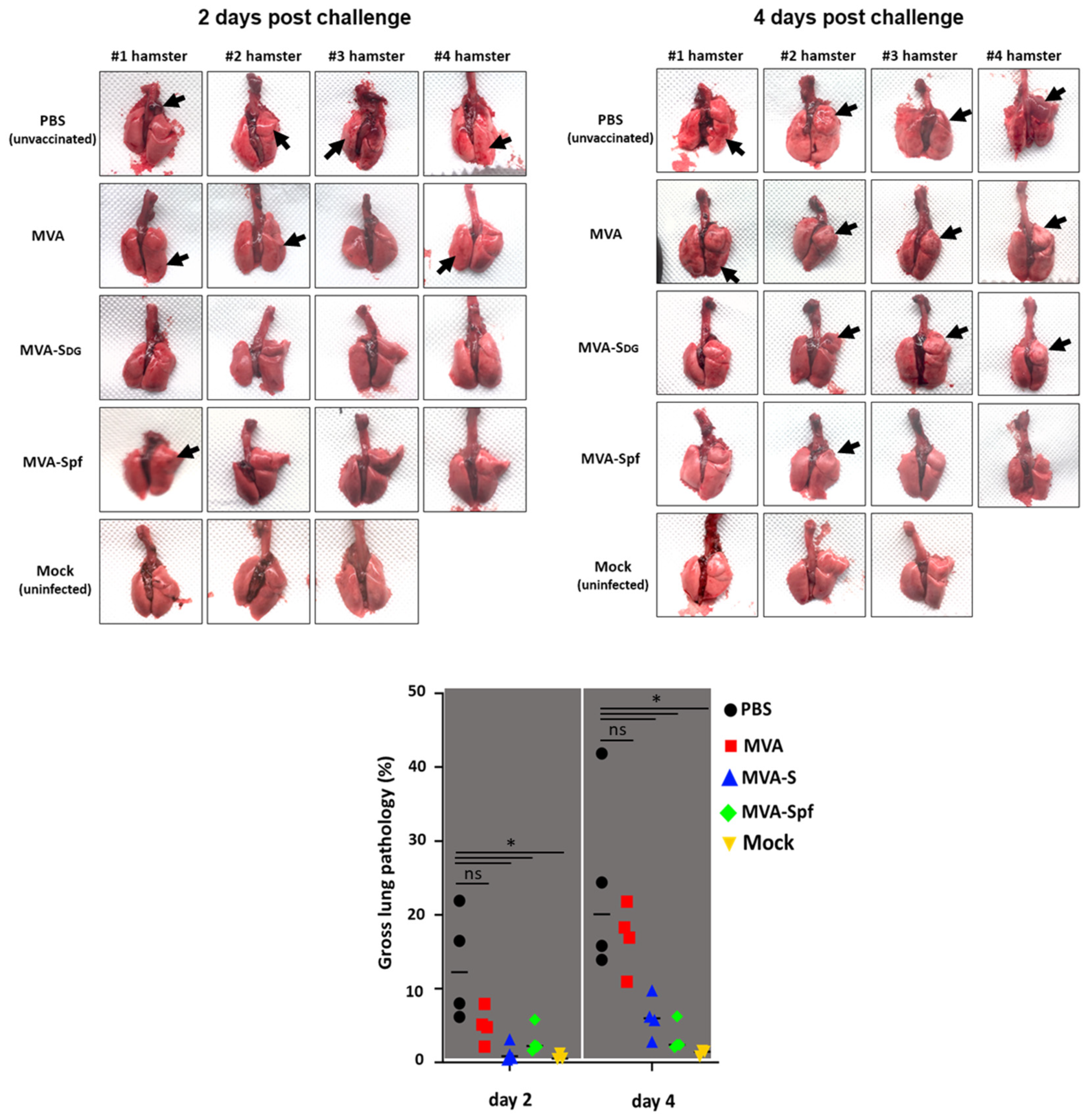
3.7. Protective Capacity of MVA Vaccines in Golden Syrian Hamsters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kyriakidis, N.C.; López-Cortés, A.; González, E.V.; Grimaldos, A.B.; Prado, E.O. SARS-CoV-2 vaccines strategies: A comprehensive review of phase 3 candidates. NPJ Vaccines 2021, 6, 28. [Google Scholar] [CrossRef]
- Du, L.; He, Y.; Zhou, Y.; Liu, S.; Zheng, B.-J.; Jiang, S. The spike protein of SARS-CoV—A target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009, 7, 226–236. [Google Scholar] [CrossRef]
- Jiang, S.; Hillyer, C.; Du, L. Neutralizing Antibodies against SARS-CoV-2 and Other Human Coronaviruses. Trends Immunol. 2020, 41, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Abdulla, Z.A.; Al-Bashir, S.M.; Alzoubi, H.; Al-Salih, N.S.; Aldamen, A.A.; Abdulazeez, A.Z. The Role of Immunity in the Pathogenesis of SARS-CoV-2 Infection and in the Protection Generated by COVID-19 Vaccines in Different Age Groups. Pathogens 2023, 12, 329. [Google Scholar] [CrossRef]
- Cheetham, N.J.; Kibble, M.; Wong, A.; Silverwood, R.J.; Knuppel, A.; Williams, D.M.; Hamilton, O.K.; Lee, P.H.; Staatz, C.B.; Di Gessa, G.; et al. Antibody levels following vaccination against SARS-CoV-2: Associations with post-vaccination infection and risk factors in two UK longitudinal studies. Elife 2023, 12, 80428. [Google Scholar] [CrossRef]
- Samrat, S.K.; Tharappel, A.M.; Li, Z.; Li, H. Prospect of SARS-CoV-2 spike protein: Potential role in vaccine and therapeutic development. Virus Res. 2020, 288, 198141. [Google Scholar] [CrossRef]
- Cattel, L.; Giordano, S.; Traina, S.; Lupia, T.; Corcione, S.; Angelone, L.; La Valle, G.; De Rosa, F.G.; Cattel, F. Vaccine development and technology for SARS-CoV-2: Current insight. J. Med. Virol. 2021, 94, 878–896. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xie, Y.; Li, C. Understanding the mechanisms for COVID-19 vaccine’s protection against infection and severe disease. Expert Rev. Vaccines 2023, 22, 186–192. [Google Scholar] [CrossRef]
- Wilder-Smith, A. What is the vaccine effect on reducing transmission in the context of the SARS-CoV-2 delta variant? Lancet Infect. Dis. 2022, 22, 152–153. [Google Scholar] [CrossRef] [PubMed]
- Feikin, D.R.; Higdon, M.M.; Abu-Raddad, L.J.; Andrews, N.; Araos, R.; Goldberg, Y.; Groome, M.J.; Huppert, A.; O’Brien, K.L.; Smith, P.G.; et al. Duration of effectiveness of vaccines against SARS-CoV-2 infection and COVID-19 disease: Results of a systematic review and meta-regression. Lancet 2022, 399, 924–944. [Google Scholar] [CrossRef]
- Volz, A.; Sutter, G. Modified Vaccinia Virus Ankara: History, Value in Basic Research, and Current Perspectives for Vaccine Development. Adv. Virus Res. 2017, 97, 187–243. [Google Scholar] [CrossRef]
- Sutter, G.; Staib, C. Vaccinia vectors as candidate vaccines: The development of modified vaccinia virus Ankara for antigen delivery. Curr. Drug Targets Infect. Disord. 2003, 3, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Volkmann, A.; Williamson, A.-L.; Weidenthaler, H.; Meyer, T.P.; Robertson, J.S.; Excler, J.-L.; Condit, R.C.; Evans, E.; Smith, E.R.; Kim, D.; et al. The Brighton Collaboration standardized template for collection of key information for risk/benefit assessment of a Modified Vaccinia Ankara (MVA) vaccine platform. Vaccine 2020, 39, 3067–3080. [Google Scholar] [CrossRef] [PubMed]
- Stittelaar, K.J.; van Amerongen, G.; Kondova, I.; Kuiken, T.; van Lavieren, R.F.; Pistoor, F.H.M.; Niesters, H.G.M.; van Doornum, G.; van der Zeijst, B.A.M.; Mateo, L.; et al. Modified Vaccinia Virus Ankara Protects Macaques against Respiratory Challenge with Monkeypox Virus. J. Virol. 2005, 79, 7845–7851. [Google Scholar] [CrossRef]
- Zaeck, L.M.; Lamers, M.M.; Verstrepen, B.E.; Bestebroer, T.M.; van Royen, M.E.; Götz, H.; Shamier, M.C.; van Leeuwen, L.P.M.; Schmitz, K.S.; Alblas, K.; et al. Low levels of monkeypox virus-neutralizing antibodies after MVA-BN vaccination in healthy individuals. Nat. Med. 2022, 29, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Sutter, G.; Wyatt, L.S.; Foley, P.L.; Bennink, J.R.; Moss, B. A recombinant vector derived from the host range-restricted and highly attenuated MVA strain of vaccinia virus stimulates protective immunity in mice to influenza virus. Vaccine 1994, 12, 1032–1040. [Google Scholar] [CrossRef] [PubMed]
- Altenburg, A.F.; Kreijtz, J.H.C.M.; De Vries, R.D.; Song, F.; Fux, R.; Rimmelzwaan, G.F.; Sutter, G.; Volz, A. Modified Vaccinia Virus Ankara (MVA) as Production Platform for Vaccines against Influenza and Other Viral Respiratory Diseases. Viruses 2014, 6, 2735–2761. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; Amara, R.R. DNA/MVA Vaccines for HIV/AIDS. Vaccines 2014, 2, 160–178. [Google Scholar] [CrossRef]
- Ondondo, B.O.; Yang, H.; Dong, T.; di Gleria, K.; Suttill, A.; Conlon, C.; Brown, D.; Williams, P.; Rowland-Jones, S.L.; Hanke, T.; et al. Immunisation with recombinant modified vaccinia virus Ankara expressing HIV-1 gag in HIV-1-infected subjects stimulates broad functional CD4+ T cell responses. Eur. J. Immunol. 2006, 36, 2585–2594. [Google Scholar] [CrossRef]
- McShane, H.; Behboudi, S.; Goonetilleke, N.; Brookes, R.; Hill, A.V.S. Protective Immunity against Mycobacterium tuberculosis Induced by Dendritic Cells Pulsed with both CD8+- and CD4+-T-Cell Epitopes from Antigen 85A. Infect. Immun. 2002, 70, 1623–1626. [Google Scholar] [CrossRef]
- Schneider, R.; Gilbert, S.C.; Blanchard, T.J.; Hanke, T.; Robson, K.J.; Hannan, C.M.; Becker, M.; Sinden, R.; Smith, G.L.; Hill, A.V.S. Enhanced immunogenicity for CD8+ T cell induction and complete protective efficacy of malaria DNA vaccination by boosting with modified vaccinia virus Ankara. Nat. Med. 1998, 4, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Volz, A.; Lim, S.; Kaserer, M.; Lülf, A.; Marr, L.; Jany, S.; Deeg, C.A.; Pijlman, G.P.; Koraka, P.; Osterhaus, A.D.; et al. Immunogenicity and protective efficacy of recombinant Modified Vaccinia virus Ankara candidate vaccines delivering West Nile virus envelope antigens. Vaccine 2016, 34, 1915–1926. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, T. Third-generation smallpox vaccine strain-based recombinant vaccines for viral hemorrhagic fevers. Vaccine 2021, 39, 6174–6181. [Google Scholar] [CrossRef] [PubMed]
- Tomori, O.; Kolawole, M.O. Ebola virus disease: Current vaccine solutions. Curr. Opin. Immunol. 2021, 71, 27–33. [Google Scholar] [CrossRef]
- Milligan, I.D.; Gibani, M.M.; Sewell, R.; Clutterbuck, E.A.; Campbell, D.; Plested, E.; Nuthall, E.; Voysey, M.; Silva-Reyes, L.; McElrath, M.J.; et al. Safety and immunogenicity of novel adenovirus type 26-and modified vaccinia Ankara-vectored Ebola vaccines: A randomized clinical trial. J. Am. Med. Assoc. 2016, 315, 1610–1623. [Google Scholar] [CrossRef] [PubMed]
- Ewer, K.; Rampling, T.; Venkatraman, N.; Bowyer, G.; Wright, D.; Lambe, T.; Imoukhuede, E.B.; Payne, R.; Fehling, S.K.; Strecker, T.; et al. A Monovalent Chimpanzee Adenovirus Ebola Vaccine Boosted with MVA. N. Engl. J. Med. 2016, 374, 1635–1646. [Google Scholar] [CrossRef]
- Tapia, M.D.; O Sow, S.; E Lyke, K.; Haidara, F.C.; Diallo, F.; Doumbia, M.; Traore, A.; Coulibaly, F.; Kodio, M.; Onwuchekwa, U.; et al. Use of ChAd3-EBO-Z Ebola virus vaccine in Malian and US adults, and boosting of Malian adults with MVA-BN-Filo: A phase 1, single-blind, randomised trial, a phase 1b, open-label and double-blind, dose-escalation trial, and a nested, randomised, double-blind, placebo-controlled trial. Lancet Infect. Dis. 2015, 16, 31–42. [Google Scholar] [CrossRef]
- Nova, N. Cross-Species Transmission of Coronaviruses in Humans and Domestic Mammals, What Are the Ecological Mechanisms Driving Transmission, Spillover, and Disease Emergence? Front. Public Health 2021, 9, 717941. [Google Scholar] [CrossRef]
- He, Y.; Zhou, Y.; Wu, H.; Luo, B.; Chen, J.; Li, W.; Jiang, S. Identification of Immunodominant Sites on the Spike Protein of Severe Acute Respiratory Syndrome (SARS) Coronavirus: Implication for Developing SARS Diagnostics and Vaccines. J. Immunol. 2004, 173, 4050–4057. [Google Scholar] [CrossRef]
- Sui, J.; Li, W.; Murakami, A.; Tamin, A.; Matthews, L.J.; Wong, S.K.; Moore, M.J.; Tallarico, A.S.C.; Olurinde, M.; Choe, H.; et al. Potent neutralization of severe acute respiratory syndrome (SARS) coronavirus by a human mAb to S1 protein that blocks receptor association. Proc. Natl. Acad. Sci. USA 2004, 101, 2536–2541. [Google Scholar] [CrossRef]
- Bisht, H.; Roberts, A.; Vogel, L.; Bukreyev, A.; Collins, P.L.; Murphy, B.R.; Subbarao, K.; Moss, B. Severe acute respiratory syndrome coronavirus spike protein expressed by attenuated vaccinia virus protectively immunizes mice. Proc. Natl. Acad. Sci. USA 2004, 101, 6641–6646. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, L.; Qin, C.; Ba, L.; Yi, C.E.; Zhang, F.; Wei, Q.; He, T.; Yu, W.; Yu, J.; et al. Recombinant Modified Vaccinia Virus Ankara Expressing the Spike Glycoprotein of Severe Acute Respiratory Syndrome Coronavirus Induces Protective Neutralizing Antibodies Primarily Targeting the Receptor Binding Region. J. Virol. 2005, 79, 2678–2688. [Google Scholar] [CrossRef]
- Alharbi, N.K.; Aljamaan, F.; Aljami, H.A.; Alenazi, M.W.; Albalawi, H.; Almasoud, A.; Alharthi, F.J.; Azhar, E.I.; Barhoumi, T.; Bosaeed, M.; et al. Immunogenicity of High-Dose MVA-Based MERS Vaccine Candidate in Mice and Camels. Vaccines 2022, 10, 1330. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, N.K.; Padron-Regalado, E.; Thompson, C.P.; Kupke, A.; Wells, D.; Sloan, M.; Grehan, K.; Temperton, N.; Lambe, T.; Warimwe, G.; et al. ChAdOx1 and MVA based vaccine candidates against MERS-CoV elicit neutralising antibodies and cellular immune responses in mice. Vaccine 2017, 35, 3780–3788. [Google Scholar] [CrossRef]
- Haagmans, B.L.; Brand, J.M.A.V.D.; Raj, V.S.; Volz, A.; Wohlsein, P.; Smits, S.L.; Schipper, D.; Bestebroer, T.M.; Okba, N.; Fux, R.; et al. An orthopoxvirus-based vaccine reduces virus excretion after MERS-CoV infection in dromedary camels. Science 2016, 351, 77–81. [Google Scholar] [CrossRef]
- Koch, T.; Dahlke, C.; Fathi, A.; Kupke, A.; Krähling, V.; A Okba, N.M.; Halwe, S.; Rohde, C.; Eickmann, M.; Volz, A.; et al. Safety and immunogenicity of a modified vaccinia virus Ankara vector vaccine candidate for Middle East respiratory syndrome: An open-label, phase 1 trial. Lancet Infect. Dis. 2020, 20, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Fux, R.; Provacia, L.B.; Volz, A.; Eickmann, M.; Becker, S.; Osterhaus, A.D.; Haagmans, B.L.; Sutter, G. Middle East Respiratory Syndrome Coronavirus Spike Protein Delivered by Modified Vaccinia Virus Ankara Efficiently Induces Virus-Neutralizing Antibodies. J. Virol. 2013, 87, 11950–11954. [Google Scholar] [CrossRef]
- Volz, A.; Kupke, A.; Song, F.; Jany, S.; Fux, R.; Shams-Eldin, H.; Schmidt, J.; Becker, C.; Eickmann, M.; Becker, S.; et al. Protective Efficacy of Recombinant Modified Vaccinia Virus Ankara Delivering Middle East Respiratory Syndrome Coronavirus Spike Glycoprotein. J. Virol. 2015, 89, 8651–8656. [Google Scholar] [CrossRef] [PubMed]
- García-Arriaza, J.; Garaigorta, U.; Pérez, P.; Lázaro-Frías, A.; Zamora, C.; Gastaminza, P.; del Fresno, C.; Casasnovas, J.M.; Sorzano, C.S.; Sancho, D.; et al. COVID-19 Vaccine Candidates Based on Modified Vaccinia Virus Ankara Expressing the SARS-CoV-2 Spike Protein Induce Robust T- and B-Cell Immune Responses and Full Efficacy in Mice. J. Virol. 2021, 95, e02260-20. [Google Scholar] [CrossRef]
- Tscherne, A.; Schwarz, J.H.; Rohde, C.; Kupke, A.; Kalodimou, G.; Limpinsel, L.; Okba, N.M.A.; Bošnjak, B.; Sandrock, I.; Odak, I.; et al. Immunogenicity and efficacy of the COVID-19 candidate vector vaccine MVA-SARS-2-S in preclinical vaccination. Proc. Natl. Acad. Sci. USA 2021, 118, e2026207118. [Google Scholar] [CrossRef]
- Lázaro-Frías, A.; Pérez, P.; Zamora, C.; Sánchez-Cordón, P.J.; Guzmán, M.; Luczkowiak, J.; Delgado, R.; Casasnovas, J.M.; Esteban, M.; García-Arriaza, J. Full efficacy and long-term immunogenicity induced by the SARS-CoV-2 vaccine candidate MVA-CoV2-S in mice. NPJ Vaccines 2022, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Mooij, P.; García-Arriaza, J.; Pérez, P.; Lázaro-Frías, A.; Verstrepen, B.E.; Böszörményi, K.P.; Mortier, D.; Fagrouch, Z.; Kiemenyi-Kayere, G.; Niphuis, H.; et al. Poxvirus MVA Expressing SARS-CoV-2 S Protein Induces Robust Immunity and Protects Rhesus Macaques from SARS-CoV-2. Front. Immunol. 2022, 13, 845887. [Google Scholar] [CrossRef] [PubMed]
- Pérez, P.; Lázaro-Frías, A.; Zamora, C.; Sánchez-Cordón, P.J.; Astorgano, D.; Luczkowiak, J.; Delgado, R.; Casasnovas, J.M.; Esteban, M.; García-Arriaza, J. A Single Dose of an MVA Vaccine Expressing a Prefusion-Stabilized SARS-CoV-2 Spike Protein Neutralizes Variants of Concern and Protects Mice from a Lethal SARS-CoV-2 Infection. Front. Immunol. 2022, 12, 824728. [Google Scholar] [CrossRef]
- Kulkarni, R.; Chen, W.-C.; Lee, Y.; Kao, C.-F.; Hu, S.-L.; Ma, H.-H.; Jan, J.-T.; Liao, C.-C.; Liang, J.-J.; Ko, H.-Y.; et al. Vaccinia virus-based vaccines confer protective immunity against SARS-CoV-2 virus in Syrian hamsters. PLoS ONE 2021, 16, e0257191. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Americo, J.L.; Cotter, C.A.; Earl, P.L.; Erez, N.; Peng, C.; Moss, B. One or two injections of MVA-vectored vaccine shields hACE2 transgenic mice from SARS-CoV-2 upper and lower respiratory tract infection. Proc. Natl. Acad. Sci. USA 2021, 118, e2026785118. [Google Scholar] [CrossRef]
- Bošnjak, B.; Odak, I.; Barros-Martins, J.; Sandrock, I.; Hammerschmidt, S.I.; Permanyer, M.; Patzer, G.E.; Greorgiev, H.; Jauregui, R.G.; Tscherne, A.; et al. Intranasal Delivery of MVA Vector Vaccine Induces Effective Pulmonary Immunity against SARS-CoV-2 in Rodents. Front. Immunol. 2021, 12, 772240. [Google Scholar] [CrossRef]
- zu Natrup, C.M.; Tscherne, A.; Dahlke, C.; Ciurkiewicz, M.; Shin, D.-L.; Fathi, A.; Rohde, C.; Kalodimou, G.; Halwe, S.; Limpinsel, L.; et al. Stabilized recombinant SARS-CoV-2 spike antigen enhances vaccine immunogenicity and protective capacity. J. Clin. Investig. 2022, 132, e159895. [Google Scholar] [CrossRef]
- Routhu, N.K.; Gangadhara, S.; Lai, L.; Davis-Gardner, M.E.; Floyd, K.; Shiferaw, A.; Bartsch, Y.C.; Fischinger, S.; Khoury, G.; Rahman, S.A.; et al. A modified vaccinia Ankara vaccine expressing spike and nucleocapsid protects rhesus macaques against SARS-CoV-2 Delta infection. Sci. Immunol. 2022, 7, eabo0226. [Google Scholar] [CrossRef]
- Americo, J.L.; Cotter, C.A.; Earl, P.L.; Liu, R.; Moss, B. Intranasal inoculation of an MVA-based vaccine induces IgA and protects the respiratory tract of hACE2 mice from SARS-CoV-2 infection. Proc. Natl. Acad. Sci. USA 2022, 119, e2202069119. [Google Scholar] [CrossRef]
- Pérez, P.; Astorgano, D.; Albericio, G.; Flores, S.; Sánchez-Cordón, P.J.; Luczkowiak, J.; Delgado, R.; Casasnovas, J.M.; Esteban, M.; García-Arriaza, J. Intranasal administration of a single dose of MVA-based vaccine candidates against COVID-19 induced local and systemic immune responses and protects mice from a lethal SARS-CoV-2 infection. Front. Immunol. 2022, 13, 995235. [Google Scholar] [CrossRef]
- Villadiego, J.; García-Arriaza, J.; Ramírez-Lorca, R.; García-Swinburn, R.; Cabello-Rivera, D.; Rosales-Nieves, A.E.; Álvarez-Vergara, M.I.; Cala-Fernández, F.; García-Roldán, E.; López-Ogáyar, J.L.; et al. Full protection from SARS-CoV-2 brain infection and damage in susceptible transgenic mice conferred by MVA-CoV2-S vaccine candidate. Nat. Neurosci. 2023, 26, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Puig, J.M.; Blasco, R. Isolation of vaccinia MVA recombinants using the viral F13L gene as the selective marker. Biotechniques 2005, 39, 665–666, 668, 670. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Chiem, K.; Park, J.-G.; Oladunni, F.; Platt, R.N.; Anderson, T.; Almazan, F.; de la Torre, J.C.; Martinez-Sobrido, L. Rescue of SARS-CoV-2 from a Single Bacterial Artificial Chromosome. mBio 2020, 11, e02168-20. [Google Scholar] [CrossRef]
- Sánchez-Puig, J.M.; Lorenzo, M.M.; Blasco, R. Isolation of Recombinant MVA Using F13L Selection. Methods Mol. Biol. 2012, 890, 93–111. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, L.S.; Earl, P.L.; Moss, B. Generation of Recombinant Vaccinia Viruses. Curr. Protoc. Mol. Biol. 2017, 117, 16.17.1–16.17.18. [Google Scholar] [CrossRef] [PubMed]
- Stadlbauer, D.; Amanat, F.; Chromikova, V.; Jiang, K.; Strohmeier, S.; Arunkumar, G.A.; Tan, J.; Bhavsar, D.; Capuano, C.; Kirkpatrick, E.; et al. SARS-CoV-2 Seroconversion in Humans: A Detailed Protocol for a Serological Assay, Antigen Production, and Test Setup. Curr. Protoc. Microbiol. 2020, 57, e100. [Google Scholar] [CrossRef]
- Amanat, F.; Stadlbauer, D.; Strohmeier, S.; Nguyen, T.H.O.; Chromikova, V.; McMahon, M.; Jiang, K.; Arunkumar, G.A.; Jurczyszak, D.; Polanco, J.; et al. A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat. Med. 2020, 26, 1033–1036. [Google Scholar] [CrossRef]
- Schmidt, F.; Weisblum, Y.; Muecksch, F.; Hoffmann, H.-H.; Michailidis, E.; Lorenzi, J.C.; Mendoza, P.; Rutkowska, M.; Bednarski, E.; Gaebler, C.; et al. Measuring SARS-CoV-2 neutralizing antibody activity using pseudotyped and chimeric viruses. J. Exp. Med. 2020, 217, e20201181. [Google Scholar] [CrossRef]
- Park, J.-G.; Oladunni, F.S.; Rohaim, M.A.; Whittingham-Dowd, J.; Tollitt, J.; Hodges, M.D.; Fathallah, N.; Assas, M.B.; Alhazmi, W.; Almilaibary, A.; et al. Immunogenicity and protective efficacy of an intranasal live-attenuated vaccine against SARS-CoV-2. iScience 2021, 24, 102941. [Google Scholar] [CrossRef]
- Piepenbrink, M.S.; Park, J.-G.; Oladunni, F.S.; Deshpande, A.; Basu, M.; Sarkar, S.; Loos, A.; Woo, J.; Lovalenti, P.; Sloan, D.; et al. Therapeutic activity of an inhaled potent SARS-CoV-2 neutralizing human monoclonal antibody in hamsters. Cell Rep. Med. 2021, 2, 100218. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Pallesen, J.; Wang, N.; Corbett, K.S.; Wrapp, D.; Kirchdoerfer, R.N.; Turner, H.L.; Cottrell, C.A.; Becker, M.M.; Wang, L.; Shi, W.; et al. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc. Natl. Acad. Sci. USA 2017, 114, E7348–E7357. [Google Scholar] [CrossRef]
- Wrobel, A.G.; Benton, D.J.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat. Struct. Mol. Biol. 2020, 27, 763–767. [Google Scholar] [CrossRef]
- Ullah, I.; Prévost, J.; Ladinsky, M.S.; Stone, H.; Lu, M.; Anand, S.P.; Beaudoin-Bussières, G.; Symmes, K.; Benlarbi, M.; Ding, S.; et al. Live imaging of SARS-CoV-2 infection in mice reveals that neutralizing antibodies require Fc function for optimal efficacy. Immunity 2021, 54, 2143–2158.e15. [Google Scholar] [CrossRef]
- Howe, C.L.; LaFrance-Corey, R.G.; Goddery, E.N.; Johnson, R.K.; Mirchia, K. Neuronal CCL2 expression drives inflammatory monocyte infiltration into the brain during acute virus infection. J. Neuroinflamm. 2017, 14, 238. [Google Scholar] [CrossRef]
- Boudewijns, R.; Pérez, P.; Lázaro-Frías, A.; Van Looveren, D.; Vercruysse, T.; Thibaut, H.J.; Weynand, B.; Coelmont, L.; Neyts, J.; Astorgano, D.; et al. MVA-CoV2-S Vaccine Candidate Neutralizes Distinct Variants of Concern and Protects against SARS-CoV-2 Infection in Hamsters. Front. Immunol. 2022, 13, 845969. [Google Scholar] [CrossRef] [PubMed]
- Dangi, T.; Class, J.; Palacio, N.; Richner, J.M.; MacMaster, P.P. Combining spike- and nucleocapsid-based vaccines improves distal control of SARS-CoV-2. Cell Rep. 2021, 36, 109664. [Google Scholar] [CrossRef] [PubMed]
- Ku, M.; Authié, P.; Bourgine, M.; Anna, F.; Noirat, A.; Moncoq, F.; Vesin, B.; Nevo, F.; Lopez, J.; Souque, P.; et al. Brain cross-protection against SARS-CoV-2 variants by a lentiviral vaccine in new transgenic mice. EMBO Mol. Med. 2021, 13, e14459. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorenzo, M.M.; Marín-López, A.; Chiem, K.; Jimenez-Cabello, L.; Ullah, I.; Utrilla-Trigo, S.; Calvo-Pinilla, E.; Lorenzo, G.; Moreno, S.; Ye, C.; et al. Vaccinia Virus Strain MVA Expressing a Prefusion-Stabilized SARS-CoV-2 Spike Glycoprotein Induces Robust Protection and Prevents Brain Infection in Mouse and Hamster Models. Vaccines 2023, 11, 1006. https://doi.org/10.3390/vaccines11051006
Lorenzo MM, Marín-López A, Chiem K, Jimenez-Cabello L, Ullah I, Utrilla-Trigo S, Calvo-Pinilla E, Lorenzo G, Moreno S, Ye C, et al. Vaccinia Virus Strain MVA Expressing a Prefusion-Stabilized SARS-CoV-2 Spike Glycoprotein Induces Robust Protection and Prevents Brain Infection in Mouse and Hamster Models. Vaccines. 2023; 11(5):1006. https://doi.org/10.3390/vaccines11051006
Chicago/Turabian StyleLorenzo, María M., Alejandro Marín-López, Kevin Chiem, Luis Jimenez-Cabello, Irfan Ullah, Sergio Utrilla-Trigo, Eva Calvo-Pinilla, Gema Lorenzo, Sandra Moreno, Chengjin Ye, and et al. 2023. "Vaccinia Virus Strain MVA Expressing a Prefusion-Stabilized SARS-CoV-2 Spike Glycoprotein Induces Robust Protection and Prevents Brain Infection in Mouse and Hamster Models" Vaccines 11, no. 5: 1006. https://doi.org/10.3390/vaccines11051006