
The Quantification of Spike Proteins in the Inactivated SARS-CoV-2 Vaccines of the Prototype, Delta, and Omicron Variants by LC–MS
 , ,
, ,
Abstract
:1. Introduction
2. Methods
2.1. Selection and Synthesis of Native and Isotopically Labeled Strain Specific SARS-CoV-2 Peptides
2.2. Samples
2.3. Preparation of Standard Solutions and the Samples
2.4. LC/MS/MS Instrumentation Parameters
2.5. Data Analysis
3. Results
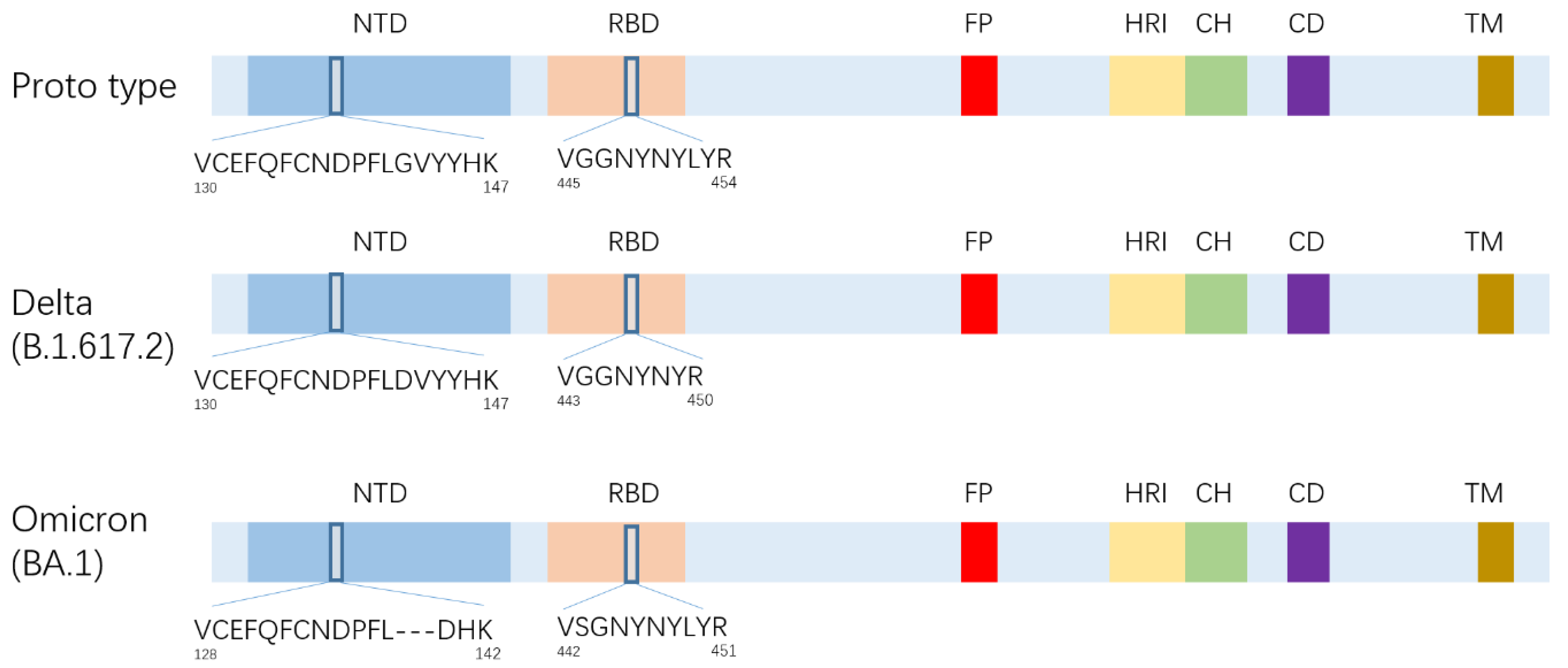
3.1. Select of Strain Specific SARS-CoV-2 Peptides
3.2. Method Validation
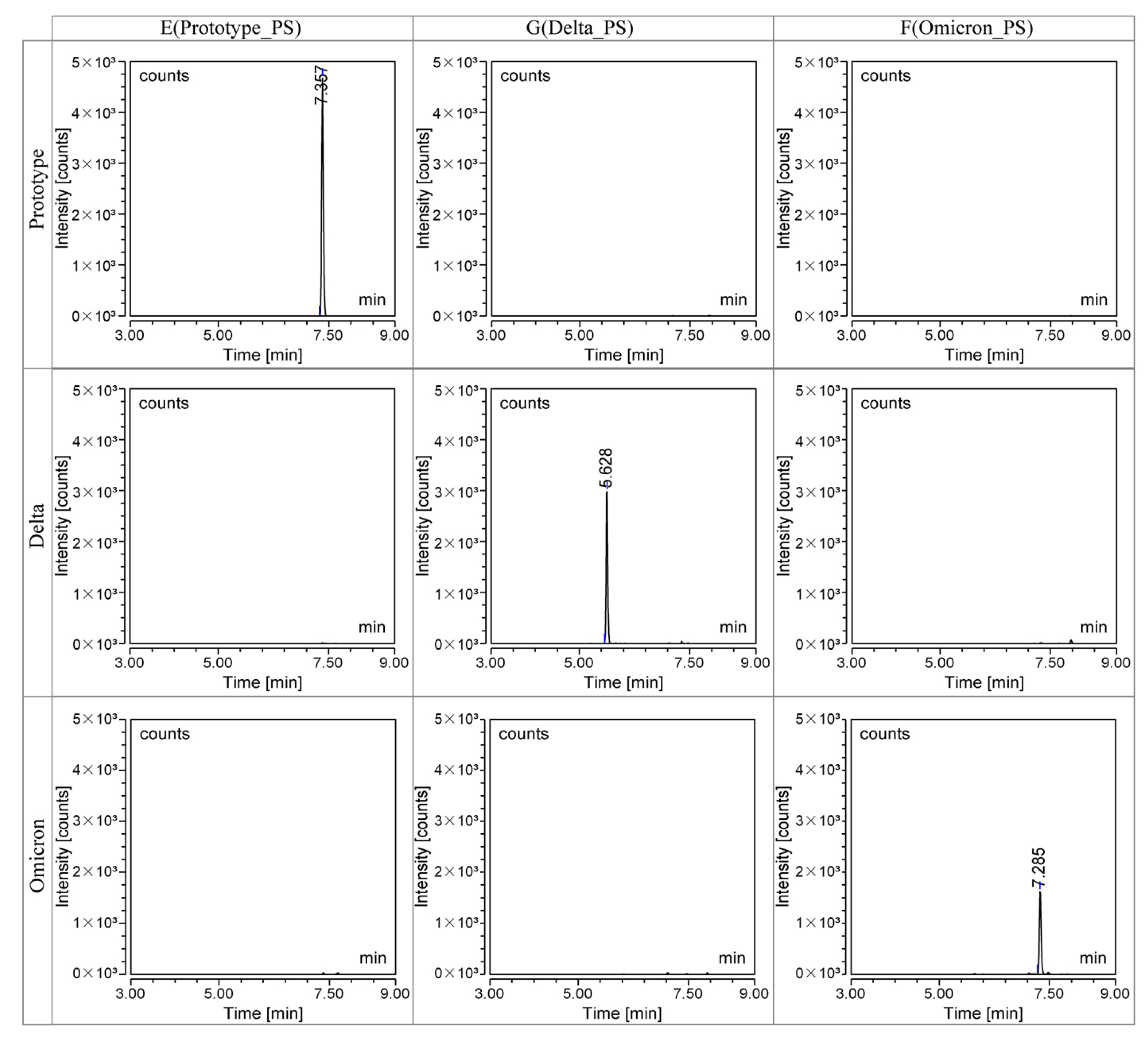
3.2.1. Specificity
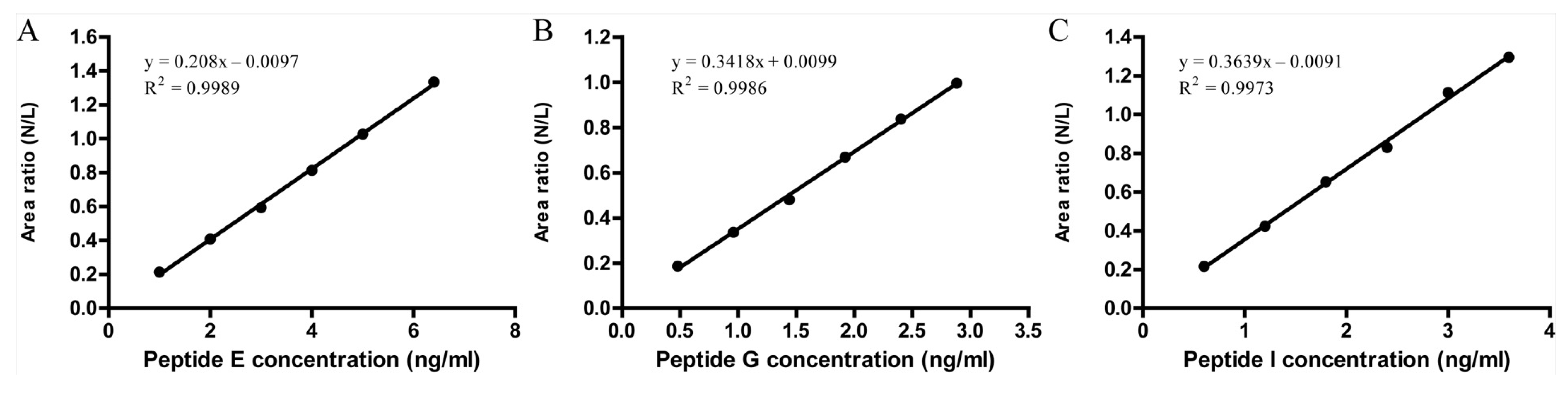
3.2.2. Linearity
3.2.3. Recovery
3.2.4. Precision
3.3. Quantification of S Protein in Inactivated Vaccine Bulks and Trivalent Vaccines
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Weisblum, Y.; Schmidt, F.; Zhang, F.; DaSilva, J.; Poston, D.; Lorenzi, J.C.; Muecksch, F.; Rutkowska, M.; Hoffmann, H.H.; Michailidis, E.; et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. Elife 2020, 9, e61312. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 2020, 183, 739–751.e8. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Coronaviridae Study Group of the International Committee on Taxonomy of V. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef]
- Santacroce, L.; Charitos, I.A.; Carretta, D.M.; De Nitto, E.; Lovero, R. The human coronaviruses (HCoVs) and the molecular mechanisms of SARS-CoV-2 infection. J. Mol. Med. 2021, 99, 93–106. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Souza, P.F.N.; Mesquita, F.P.; Amaral, J.L.; Landim, P.G.C.; Lima, K.R.P.; Costa, M.B.; Farias, I.R.; Lima, L.B.; Montenegro, R.C. The human pandemic coronaviruses on the show: The spike glycoprotein as the main actor in the coronaviruses play. Int. J. Biol. Macromol. 2021, 179, 1–19. [Google Scholar] [CrossRef]
- Frampton, D.; Rampling, T.; Cross, A.; Bailey, H.; Heaney, J.; Byott, M.; Scott, R.; Sconza, R.; Price, J.; Margaritis, M.; et al. Genomic characteristics and clinical effect of the emergent SARS-CoV-2 B.1.1.7 lineage in London, UK: A whole-genome sequencing and hospital-based cohort study. Lancet Infect. Dis. 2021, 21, 1246–1256. [Google Scholar] [CrossRef]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef]
- Araf, Y.; Akter, F.; Tang, Y.D.; Fatemi, R.; Parvez, M.S.A.; Zheng, C.; Hossain, M.G. Omicron variant of SARS-CoV-2: Genomics, transmissibility, and responses to current COVID-19 vaccines. J. Med. Virol. 2022, 94, 1825–1832. [Google Scholar] [CrossRef]
- Tian, D.; Sun, Y.; Xu, H.; Ye, Q. The emergence and epidemic characteristics of the highly mutated SARS-CoV-2 Omicron variant. J. Med. Virol. 2022, 94, 2376–2383. [Google Scholar] [CrossRef]
- Yin, W.; Xu, Y.; Xu, P.; Cao, X.; Wu, C.; Gu, C.; He, X.; Wang, X.; Huang, S.; Yuan, Q.; et al. Structures of the Omicron spike trimer with ACE2 and an anti-Omicron antibody. Science 2022, 375, 1048–1053. [Google Scholar] [CrossRef]
- Cele, S.; Jackson, L.; Khoury, D.S.; Khan, K.; Moyo-Gwete, T.; Tegally, H.; San, J.E.; Cromer, D.; Scheepers, C.; Amoako, D.G.; et al. Omicron extensively but incompletely escapes Pfizer BNT162b2 neutralization. Nature 2022, 602, 654–656. [Google Scholar] [CrossRef]
- Wilhelm, A.; Widera, M.; Grikscheit, K.; Toptan, T.; Schenk, B.; Pallas, C.; Metzler, M.; Kohmer, N.; Hoehl, S.; Marschalek, R.; et al. Limited neutralisation of the SARS-CoV-2 Omicron subvariants BA.1 and BA.2 by convalescent and vaccine serum and monoclonal antibodies. EBioMedicine 2022, 82, 104158. [Google Scholar] [CrossRef]
- Yu, X.; Wei, D.; Xu, W.; Li, Y.; Li, X.; Zhang, X.; Qu, J.; Yang, Z.; Chen, E. Reduced sensitivity of SARS-CoV-2 Omicron variant to antibody neutralization elicited by booster vaccination. Cell Discov. 2022, 8, 4. [Google Scholar] [CrossRef]
- Su, S.; Li, W.; Jiang, S. Developing pan-β-coronavirus vaccines against emerging SARS-CoV-2 variants of concern. Trends Immunol. 2022, 43, 170–172. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, Z.; Zhang, G.; Xu, W.; Xing, L.; Lu, L.; Wang, Q.; Jiang, S. Development of variant-proof severe acute respiratory syndrome coronavirus 2, pan-sarbecovirus, and pan-beta-coronavirus vaccines. J. Med. Virol. 2023, 95, e28172. [Google Scholar] [CrossRef]
- Tan, C.W.; Chia, W.N.; Young, B.E.; Zhu, F.; Lim, B.L.; Sia, W.R.; Thein, T.L.; Chen, M.I.C.; Leo, Y.S.; Lye, D.C.; et al. Pan-Sarbecovirus Neutralizing Antibodies in BNT162b2-Immunized SARS-CoV-1 Survivors. N. Engl. J. Med. 2021, 385, 1401–1406. [Google Scholar] [CrossRef] [PubMed]
- Saunders, K.O.; Lee, E.; Parks, R.; Martinez, D.R.; Li, D.; Chen, H.; Edwards, R.J.; Gobeil, S.; Barr, M.; Mansouri, K.; et al. Neutralizing antibody vaccine for pandemic and pre-emergent coronaviruses. Nature 2021, 594, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Joyce, M.G.; Chen, W.H.; Sankhala, R.S.; Hajduczki, A.; Thomas, P.V.; Choe, M.; Martinez, E.J.; Chang, W.C.; Peterson, C.E.; Morrison, E.B.; et al. SARS-CoV-2 ferritin nanoparticle vaccines elicit broad SARS coronavirus immunogenicity. Cell Rep. 2021, 37, 110143. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Peng, W.J.; Kuo, B.S.; Ho, Y.H.; Wang, M.S.; Yang, Y.T.; Chang, P.Y.; Shen, Y.H.; Hwang, K.P. Toward a pan-SARS-CoV-2 vaccine targeting conserved epitopes on spike and non-spike proteins for potent, broad and durable immune responses. PLoS Pathog. 2023, 19, e1010870. [Google Scholar] [CrossRef]
- Huang, R.; Ying, L.; Wang, J.; Xia, J.; Zhang, Y.; Mao, H.; Zhang, R.; Zang, R.; Le, Z.; Shu, Q.; et al. Non-spike and spike-specific memory T cell responses after the third dose of inactivated COVID-19 vaccine. Front. Immunol. 2023, 14, 1139620. [Google Scholar] [CrossRef]
- Chalkias, S.; Eder, F.; Essink, B.; Khetan, S.; Nestorova, B.; Feng, J.; Chen, X.; Chang, Y.; Zhou, H.; Montefiori, D.; et al. Safety, immunogenicity and antibody persistence of a bivalent Beta-containing booster vaccine against COVID-19: A phase 2/3 trial. Nat. Med. 2022, 28, 2388–2397. [Google Scholar] [CrossRef]
- Hause, A.M.; Marquez, P.; Zhang, B.; Myers, T.R.; Gee, J.; Su, J.R.; Blanc, P.G.; Thomas, A.; Thompson, D.; Shimabukuro, T.T.; et al. Safety Monitoring of Bivalent COVID-19 mRNA Vaccine Booster Doses Among Persons Aged ≥12 Years—United States, August 31–October 23, 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 1401–1406. [Google Scholar] [CrossRef]
- Long, Z.; Wei, C.; Dong, X.; Li, X.; Yang, H.; Deng, H.; Ma, X.; Yin, S.; Qi, Y.; Bo, T. Simultaneous quantification of spike and nucleocapsid protein in inactivated COVID-19 vaccine bulk by liquid chromatography-tandem mass spectrometry. J. Chromatogr. B 2021, 1181, 122884. [Google Scholar] [CrossRef]
- Cazares, L.H.; Chaerkady, R.; Samuel Weng, S.H.; Boo, C.C.; Cimbro, R.; Hsu, H.E.; Rajan, S.; Dall’Acqua, W.; Clarke, L.; Ren, K.; et al. Development of a Parallel Reaction Monitoring Mass Spectrometry Assay for the Detection of SARS-CoV-2 Spike Glycoprotein and Nucleoprotein. Anal. Chem. 2020, 92, 13813–13821. [Google Scholar] [CrossRef]
- Rosen, O.; Jayson, A.; Dor, E.; Epstein, E.; Makovitzki, A.; Cherry, L.; Lupu, E.; Monash, A.; Borni, S.; Baruchi, T.; et al. SARS-CoV-2 spike antigen quantification by targeted mass spectrometry of a virus-based vaccine. J. Virol. Methods 2022, 303, 114498. [Google Scholar] [CrossRef]
- Pierce-Ruiz, C.; Santana, W.I.; Sutton, W.J.H.; Fischler, D.A.; Cooper, H.C.; Marc, L.R.; Barr, J.R.; Williams, T.L. Quantification of SARS-CoV-2 spike and nucleocapsid proteins using isotope dilution tandem mass spectrometry. Vaccine 2021, 39, 5106–5115. [Google Scholar] [CrossRef]
- Hoofnagle, A.N.; Whiteaker, J.R.; Carr, S.A.; Kuhn, E.; Liu, T.; Massoni, S.A.; Thomas, S.N.; Townsend, R.R.; Zimmerman, L.J.; Boja, E.; et al. Recommendations for the Generation, Quantification, Storage, and Handling of Peptides Used for Mass Spectrometry-Based Assays. Clin. Chem. 2016, 62, 48–69. [Google Scholar] [CrossRef]
- Lau, S.Y.; Wang, P.; Mok, B.W.; Zhang, A.J.; Chu, H.; Lee, A.C.; Deng, S.; Chen, P.; Chan, K.H.; Song, W.; et al. Attenuated SARS-CoV-2 variants with deletions at the S1/S2 junction. Emerg. Microbes Infect. 2020, 9, 837–842. [Google Scholar] [CrossRef]
- To, K.K.; Li, X.; Lung, D.C.; Ip, J.D.; Chan, W.M.; Chu, A.W.; Yip, C.C.Y.; Chen, J.H.; Poon, R.W.S.; Tsoi, H.W.; et al. False Coronavirus Disease 2019 Cases due to Contamination by Inactivated Virus Vaccine. Clin. Infect. Dis. 2022, 74, 1485–1488. [Google Scholar] [CrossRef]
- Dong, Y.; Zhao, Y.X.; Zhuge, X.L.; Yang, Z.N.; Li, J.; Li, A.L. Analysis of S protein cross-interference in three different corona virus strains. Chin. J. New Drugs 2022, 31, 2136–2143. [Google Scholar]
- Xu, K.; Li, C.; Gravel, C.; Jiang, Z.; Jaentschke, B.; Van Domselaar, G.; Li, X.; Wang, J. Universal type/subtype-specific antibodies for quantitative analyses of neuraminidase in trivalent influenza vaccines. Sci. Rep. 2018, 8, 1067. [Google Scholar] [CrossRef]
- Izquierdo-Lara, R.; Elsinga, G.; Heijnen, L.; Munnink, B.B.O.; Schapendonk, C.M.E.; Nieuwenhuijse, D.; Kon, M.; Lu, L.; Aarestrup, F.M.; Lycett, S.; et al. Monitoring SARS-CoV-2 Circulation and Diversity through Community Wastewater Sequencing, the Netherlands and Belgium. Emerg. Infect. Dis. 2021, 27, 1405–1415. [Google Scholar] [CrossRef]
- Nasir, J.A.; Kozak, R.A.; Aftanas, P.; Raphenya, A.R.; Smith, K.M.; Maguire, F.; Maan, H.; Alruwaili, M.; Banerjee, A.; Mbareche, H.; et al. A Comparison of Whole Genome Sequencing of SARS-CoV-2 Using Amplicon-Based Sequencing, Random Hexamers, and Bait Capture. Viruses 2020, 12, 895. [Google Scholar] [CrossRef]
- Banada, P.; Green, R.; Banik, S.; Chopoorian, A.; Streck, D.; Jones, R.; Chakravorty, S.; Alland, D. A Simple Reverse Transcriptase PCR Melting-Temperature Assay To Rapidly Screen for Widely Circulating SARS-CoV-2 Variants. J. Clin. Microbiol. 2021, 59, e0084521. [Google Scholar] [CrossRef]
- Dikdan, R.J.; Marras, S.A.E.; Field, A.P.; Brownlee, A.; Cironi, A.; Hill, D.A.; Tyagi, S. Multiplex PCR Assays for Identifying all Major Severe Acute Respiratory Syndrome Coronavirus 2 Variants. J. Mol. Diagn. 2022, 24, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Loos, G.; Van Schepdael, A.; Cabooter, D. Quantitative mass spectrometry methods for pharmaceutical analysis. Philos. Trans. A Math. Phys. Eng. Sci. 2016, 374, 20150366. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.K.; Sharma, I.; Glick, J. The expanding role of mass spectrometry in the field of vaccine development. Mass Spectrom. Rev. 2020, 39, 83–104. [Google Scholar] [CrossRef] [PubMed]
- Brun, V.; Masselon, C.; Garin, J.; Dupuis, A. Isotope dilution strategies for absolute quantitative proteomics. J. Proteom. 2009, 72, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.L.; Pirkle, J.L.; Barr, J.R. Simultaneous quantification of hemagglutinin and neuraminidase of influenza virus using isotope dilution mass spectrometry. Vaccine 2012, 30, 2475–2482. [Google Scholar] [CrossRef]
- Verch, T.; Trausch, J.J.; Shank-Retzlaff, M. Principles of vaccine potency assays. Bioanalysis 2018, 10, 163–180. [Google Scholar] [CrossRef]
- Kouiavskaia, D.; Puligedda, R.D.; Dessain, S.K.; Chumakov, K. Universal ELISA for quantification of D-antigen in inactivated poliovirus vaccines. J. Virol. Methods 2020, 276, 113785. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Variant Strain | Peptide Name | Peptide Sequence | Precursor Ion m/z | Target Ion m/z | Collision Energy (%) |
|---|---|---|---|---|---|
| Prototype strain | E | VGGNYNYLYR | 610.08 (+2) | 1119.41 | 23 |
| F | VGGNYNYLYR | 613.08 (+2) | 1119.58 | 23 | |
| Delta (B.1.617.2) | G | VGGNYNYR | 471.98 (+2) | 843.33 | 16 |
| H | VGGNYNYR | 474.56 (+2) | 843.25 | 18 | |
| Omicron (BA.1) | I | VSGNYNYLYR | 625.05 (+2) | 728.58 | 20 |
| J | VSGNYNYLYR | 628.03 (+2) | 728.5 | 21 |
| Standard Polypeptides | Concentration | (ng/mL) | ||||
|---|---|---|---|---|---|---|
| Working Solution | 1 | 2 | 3 | 4 | 5 | 6 |
| polypeptide E | 1.0 | 2.0 | 3.0 | 4.0 | 5.0 | 6.4 |
| isotope-labeled peptide F | 3.6 | 3.6 | 3.6 | 3.6 | 3.6 | 3.6 |
| polypeptide G | 0.48 | 0.96 | 1.44 | 1.92 | 2.4 | 2.88 |
| isotope-labeled peptide H | 3.0 | 3.0 | 3.0 | 3.0 | 3.0 | 3.0 |
| polypeptide I | 0.6 | 1.2 | 1.8 | 2.4 | 3 | 3.6 |
| isotope-labeled peptide J | 3.6 | 3.6 | 3.6 | 3.6 | 3.6 | 3.6 |
| Samples | Lot | Spike Protein Concentration (pmol/mL) | ||
|---|---|---|---|---|
| Prototype Type | Delta | Omicron | ||
| Vaccine bulks of the prototype strain | 1 | 68.5 | / | / |
| 2 | 68.7 | / | / | |
| 3 | 67.7 | / | / | |
| 4 | 68.3 | / | / | |
| 5 | 71.3 | / | / | |
| 6 | 72.3 | / | / | |
| Vaccine bulks of the Delta strain | 1 | / | 7.6 | / |
| 2 | / | 7.6 | / | |
| 3 | / | 5.6 | / | |
| 4 | / | 5.8 | / | |
| 5 | / | 7.9 | / | |
| 6 | / | 5.1 | / | |
| Vaccine bulks of the Omicron strain | 1 | / | / | 10.3 |
| 2 | / | / | 11.7 | |
| 3 | / | / | 10.3 | |
| 4 | / | / | 9.2 | |
| 5 | / | / | 9 | |
| 6 | / | / | 10.3 | |
| trivalent inactivated vaccine | 1 | 2.6 | 2.5 | 2.4 |
| 2 | 2.6 | 2.4 | 2.6 | |
| 3 | 2.7 | 2.6 | 2.3 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, K.; Sun, H.; Wang, K.; Quan, Y.; Qiao, Z.; Hu, Y.; Li, C. The Quantification of Spike Proteins in the Inactivated SARS-CoV-2 Vaccines of the Prototype, Delta, and Omicron Variants by LC–MS. Vaccines 2023, 11, 1002. https://doi.org/10.3390/vaccines11051002
Xu K, Sun H, Wang K, Quan Y, Qiao Z, Hu Y, Li C. The Quantification of Spike Proteins in the Inactivated SARS-CoV-2 Vaccines of the Prototype, Delta, and Omicron Variants by LC–MS. Vaccines. 2023; 11(5):1002. https://doi.org/10.3390/vaccines11051002
Chicago/Turabian StyleXu, Kangwei, Huang Sun, Kaiqin Wang, Yaru Quan, Zhizhong Qiao, Yaling Hu, and Changgui Li. 2023. "The Quantification of Spike Proteins in the Inactivated SARS-CoV-2 Vaccines of the Prototype, Delta, and Omicron Variants by LC–MS" Vaccines 11, no. 5: 1002. https://doi.org/10.3390/vaccines11051002