
Immunoinformatics Approach to Design a Multi-Epitope Vaccine against Cutaneous Leishmaniasis
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
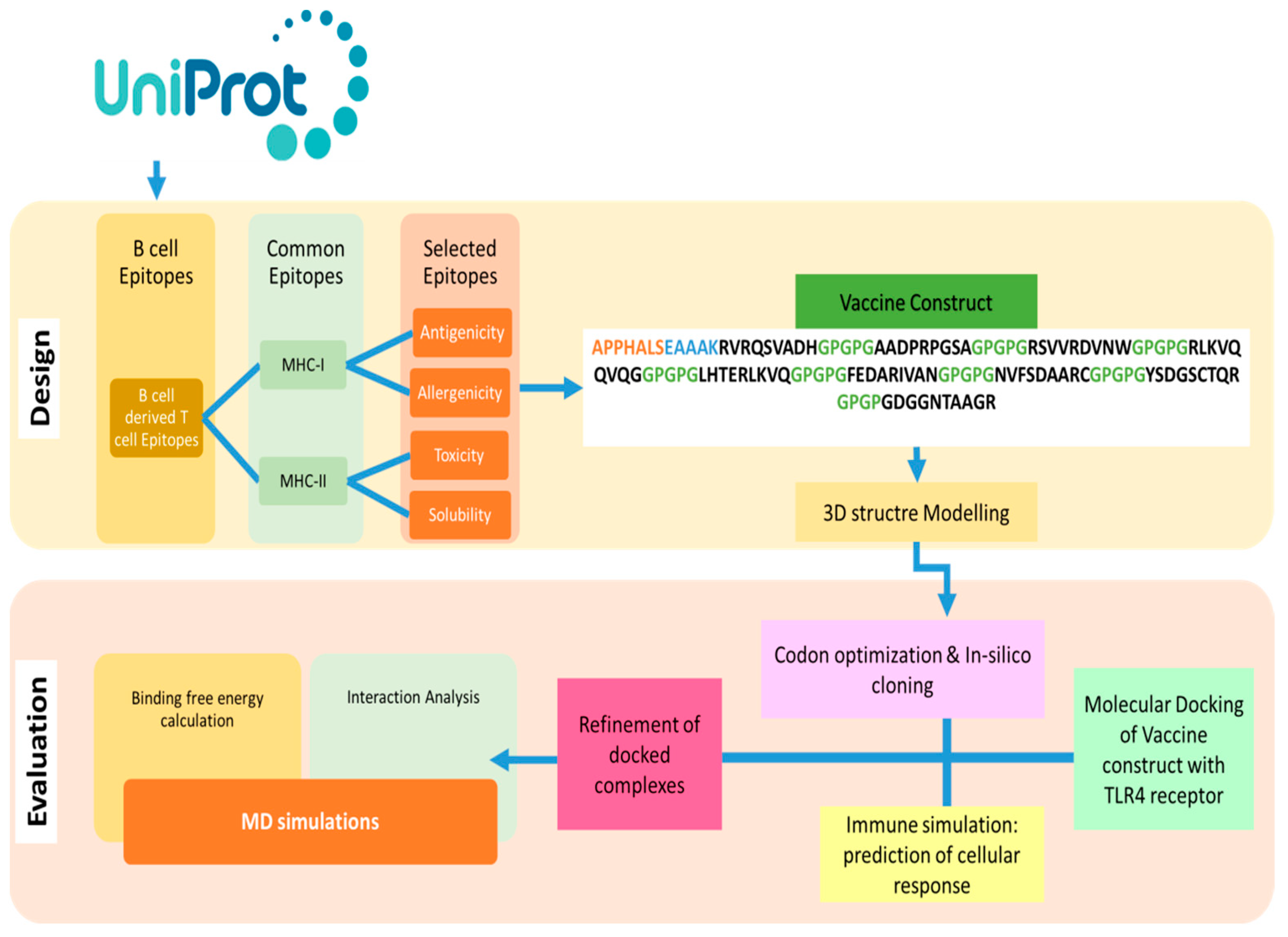
2.1. Study Design
2.2. Sequence Retrieval and Antigenicity Prediction
2.3. Immunoinformatics Analysis
2.3.1. B-Cell Epitope Prediction
2.3.2. MHC-I and MHC-II Epitopes Prediction
2.3.3. Epitopes Mapping
2.3.4. MEVC Designing and Post Analysis
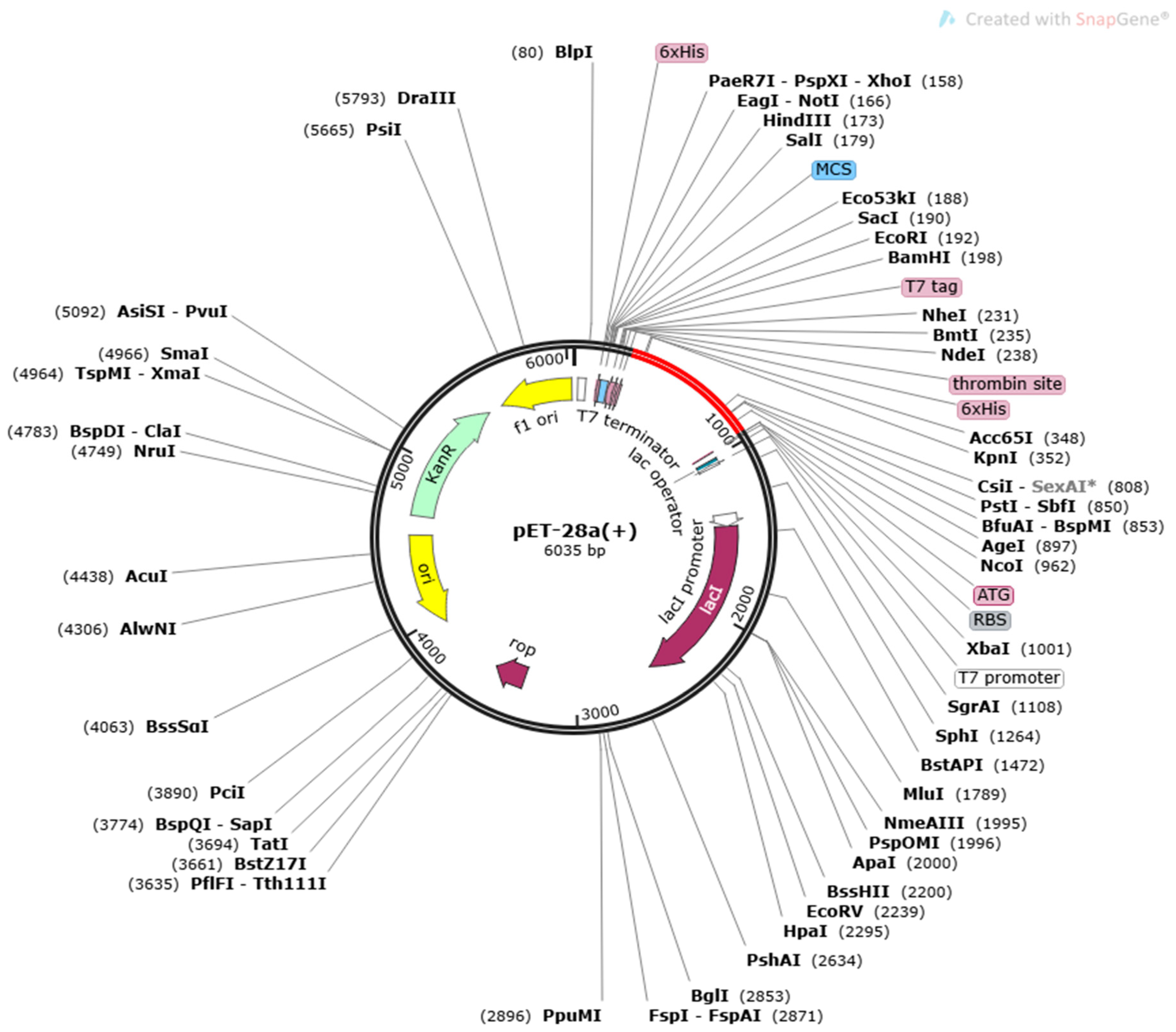
2.3.5. Codon Optimization and In-Silico Cloning
2.4. Molecular Docking of Vaccine with TLR4 Receptor
2.5. Molecular Dynamics Simulation with Vaccine-TLR4 Complex
2.6. Free Energy of Binding and Decomposition
3. Results
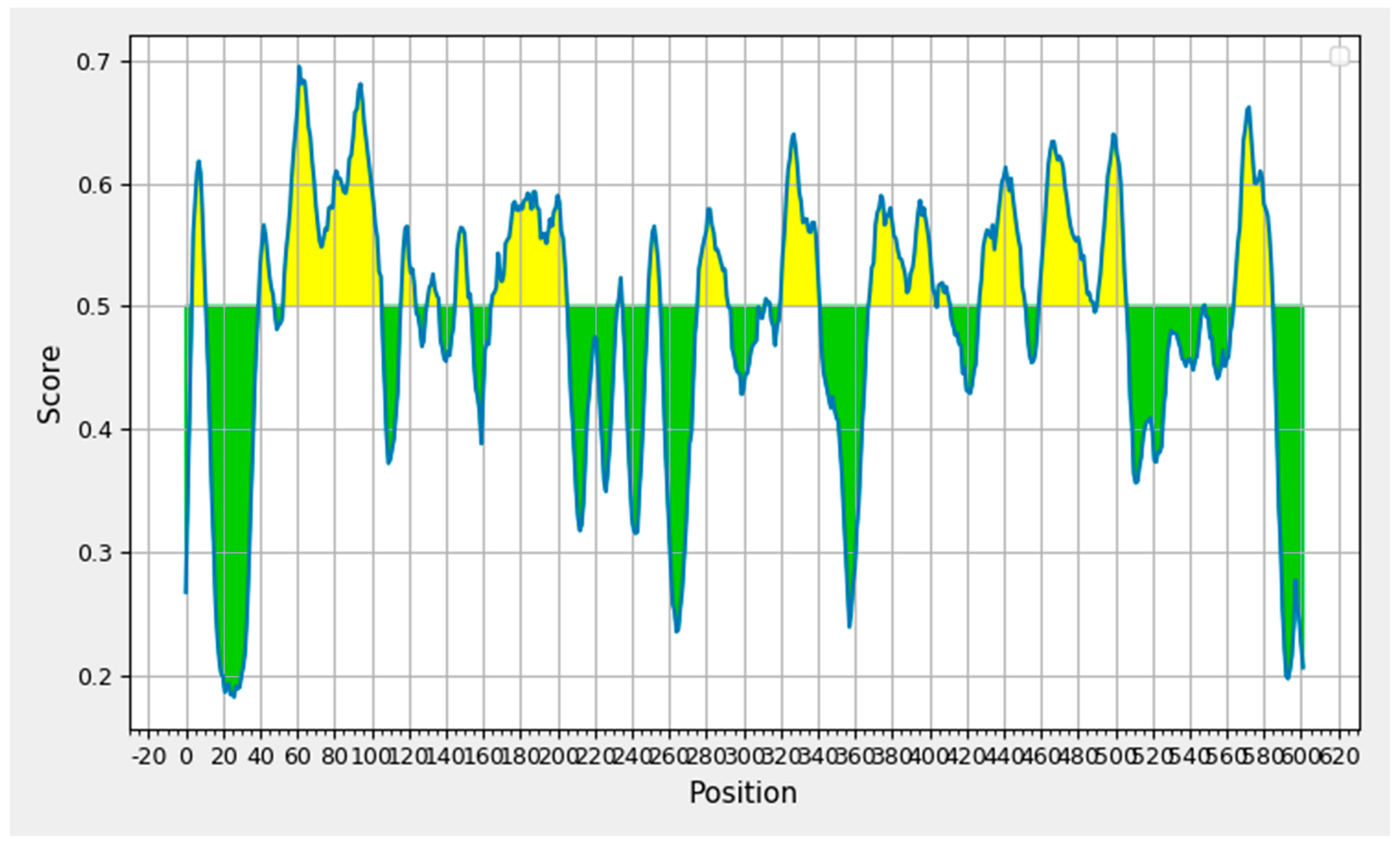
3.1. Protein Antigenicity
3.2. B-Cell Epitope Prediction
3.3. Prediction of MHC-I and MCH-II Binding Epitopes
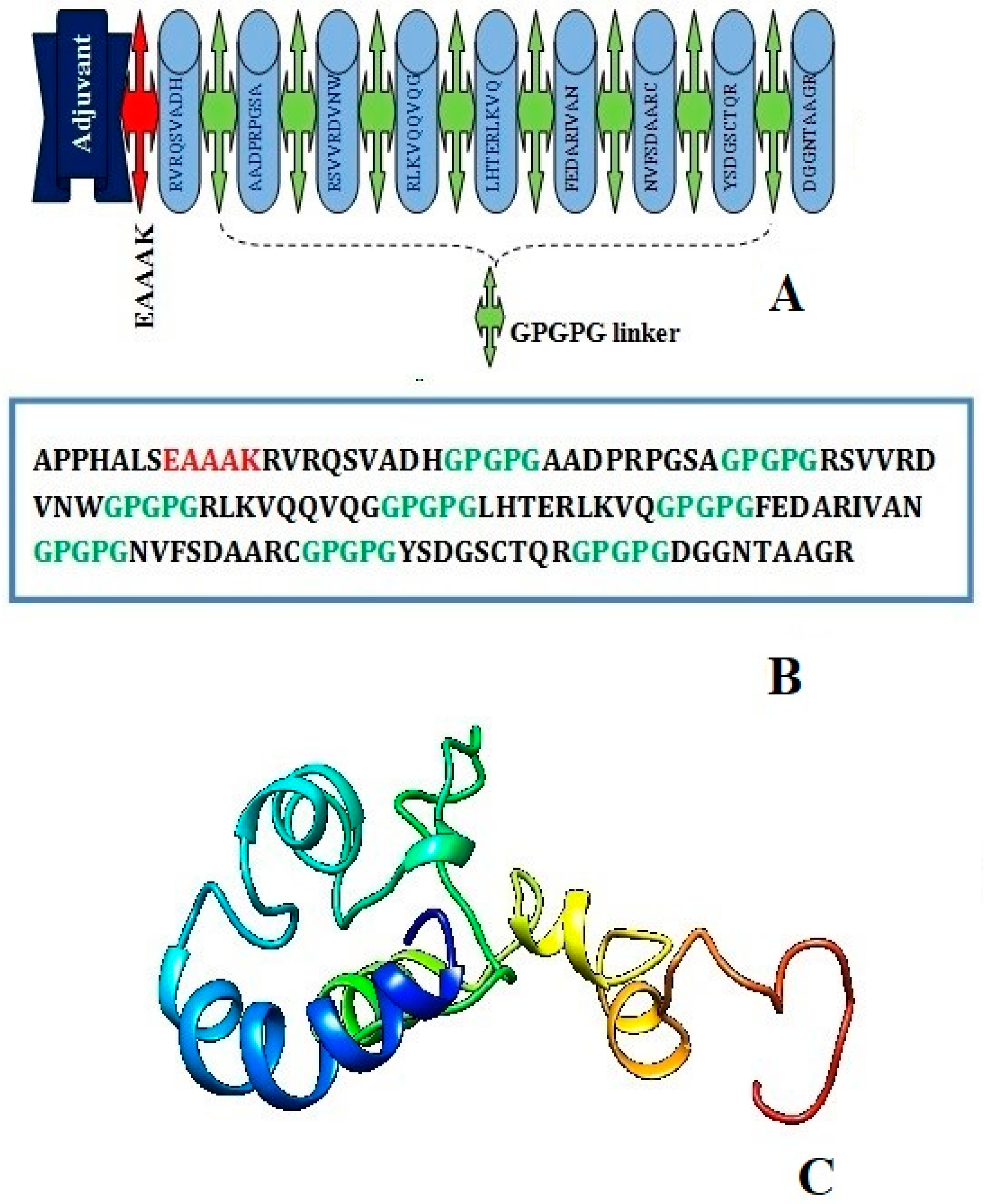
3.4. Construction of Multi-Epitope Peptide Vaccine (MEPVC)
3.5. Antigenic and Non-Allergic Evaluation of MEPVC
3.6. Physiochemical Assessment and Protein Stability
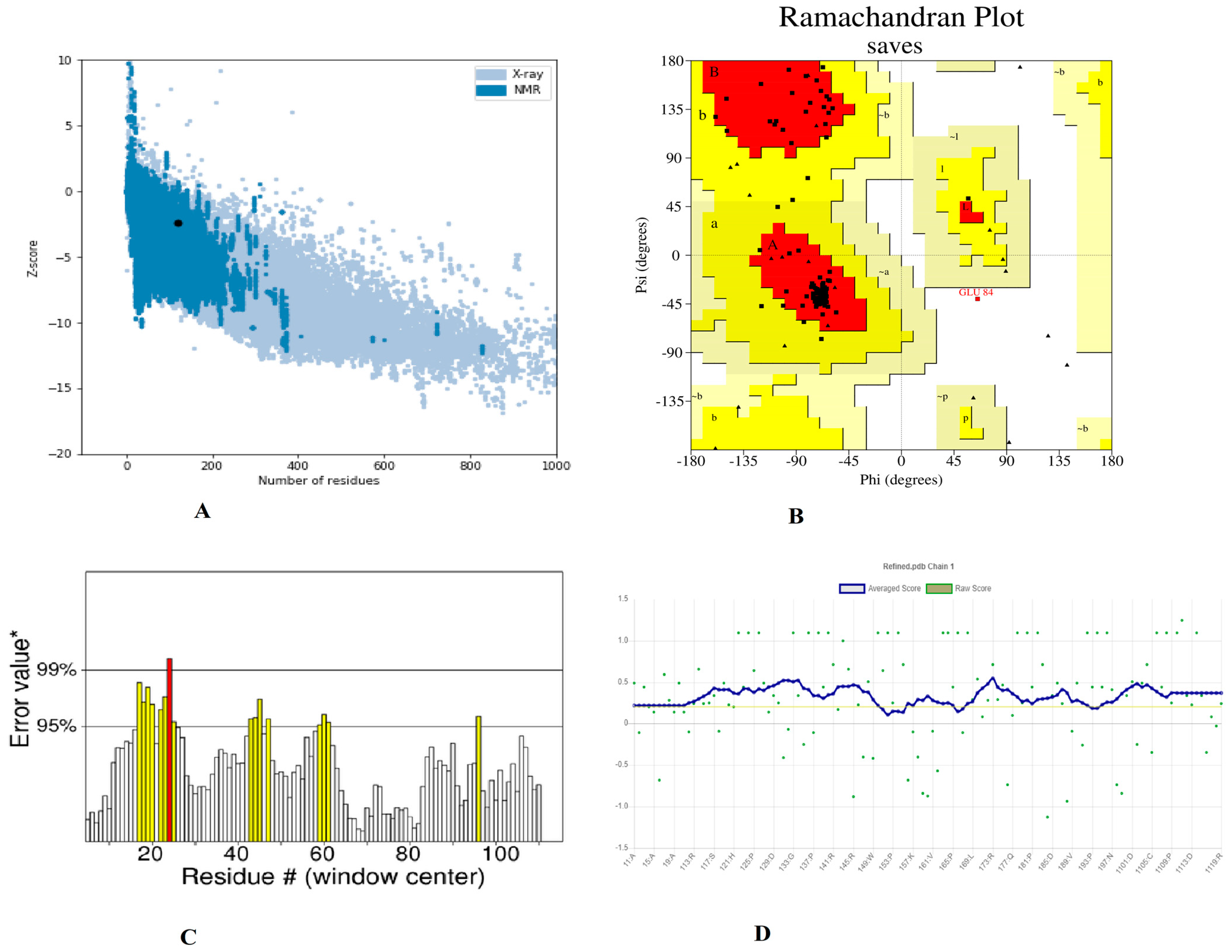
3.7. Prediction of Secondary and Tertiary Structure and Validation
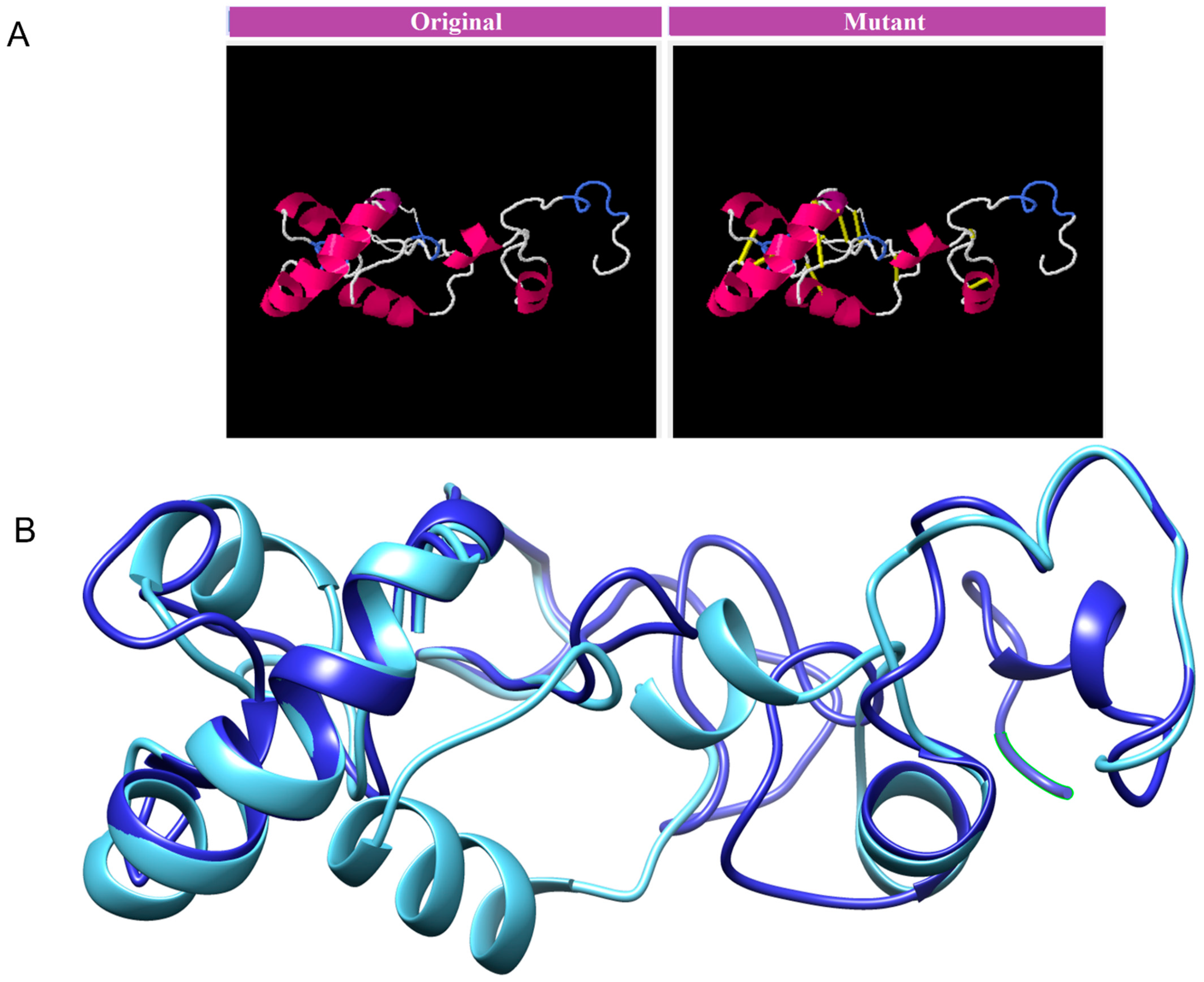
3.8. Disulphide Engineering, Codon Optimization and In Silico Cloning Analysis
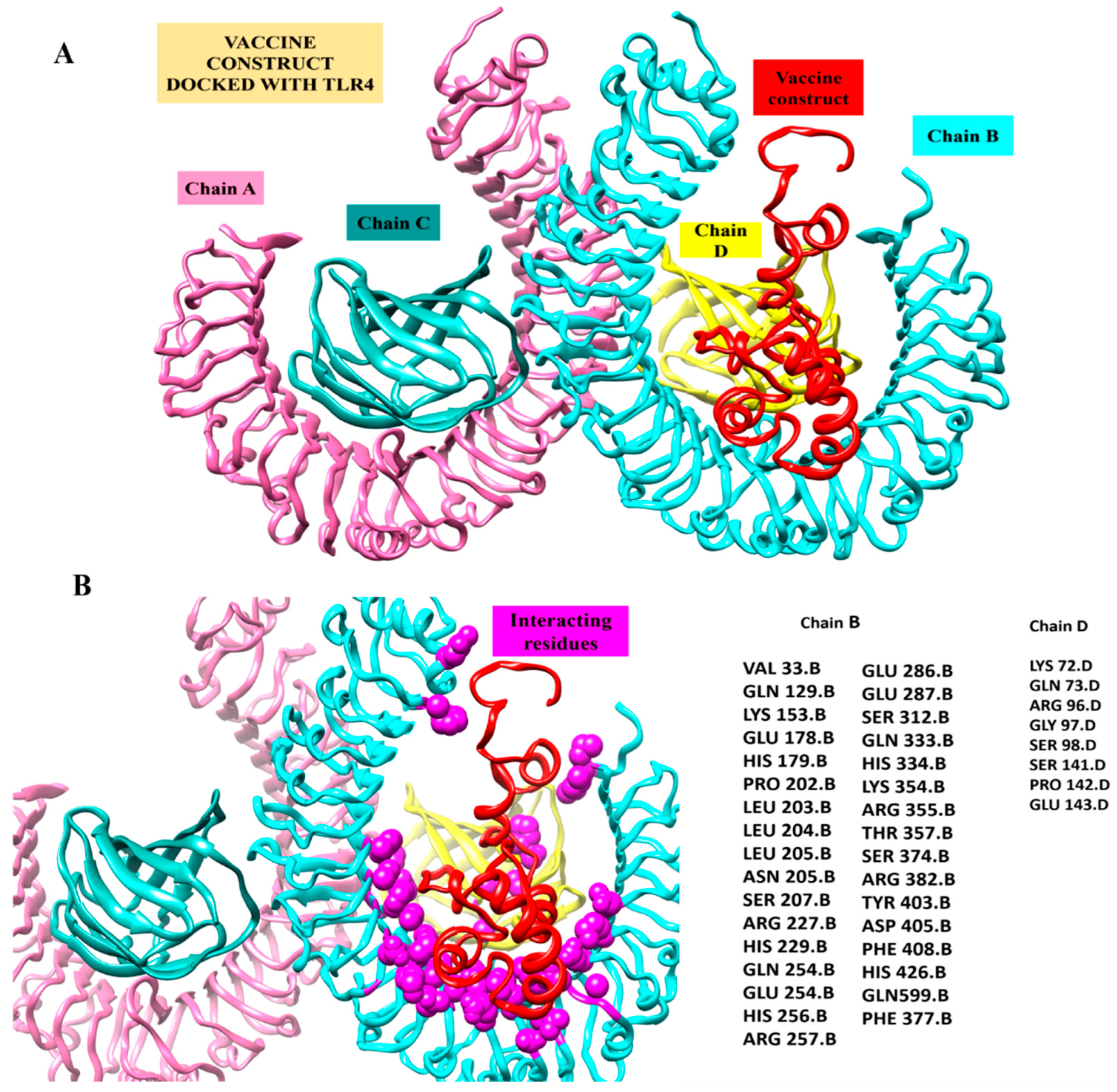
3.9. Docking Interaction of MEPVC and TLR4 Receptor
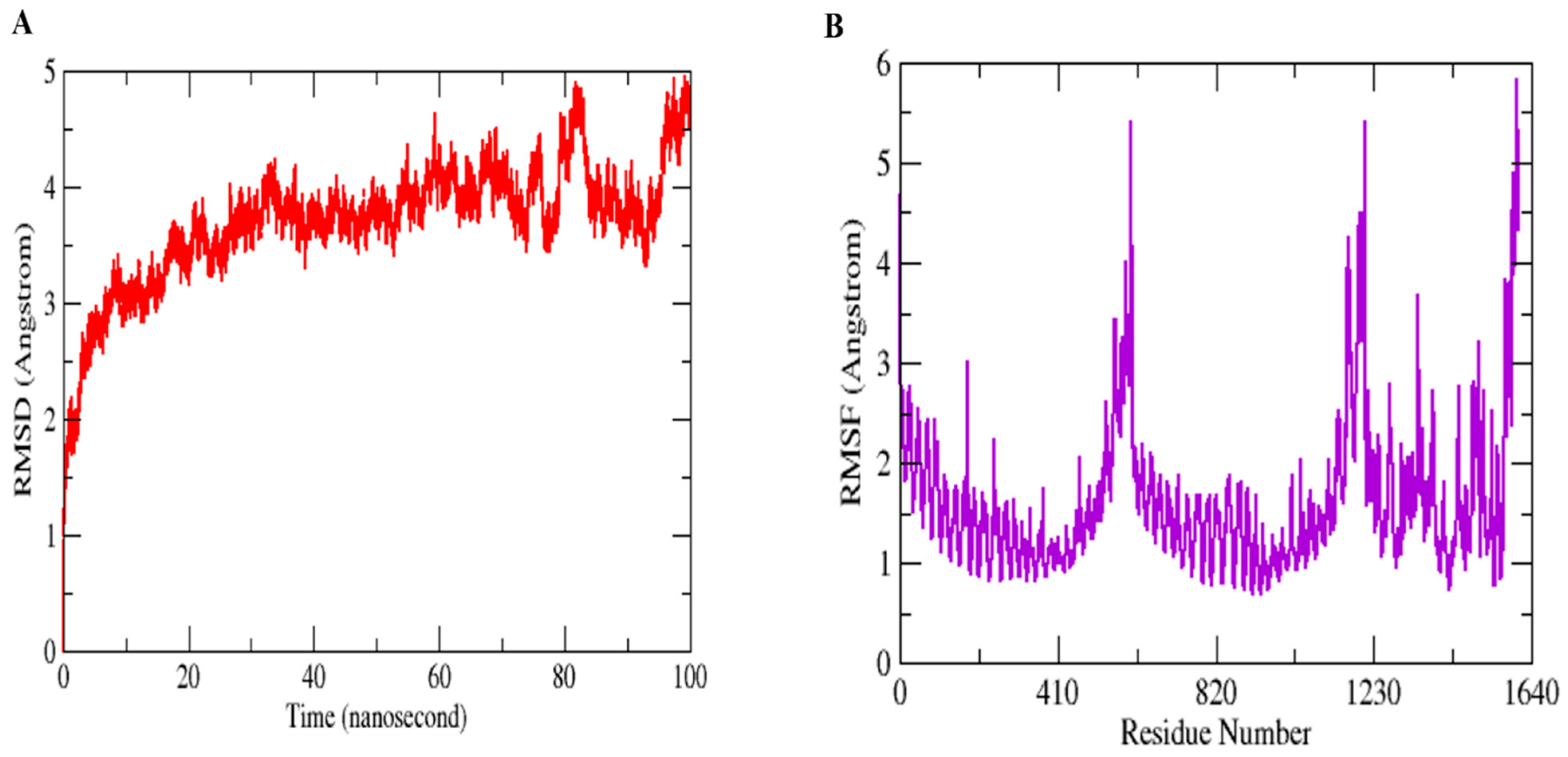
3.10. MD Simulation Assays to Study Conformational Stability and Residual Flexibility
3.11. Determination of the Binding Free Energy of TLR4-Vaccie Ensemble Complexes
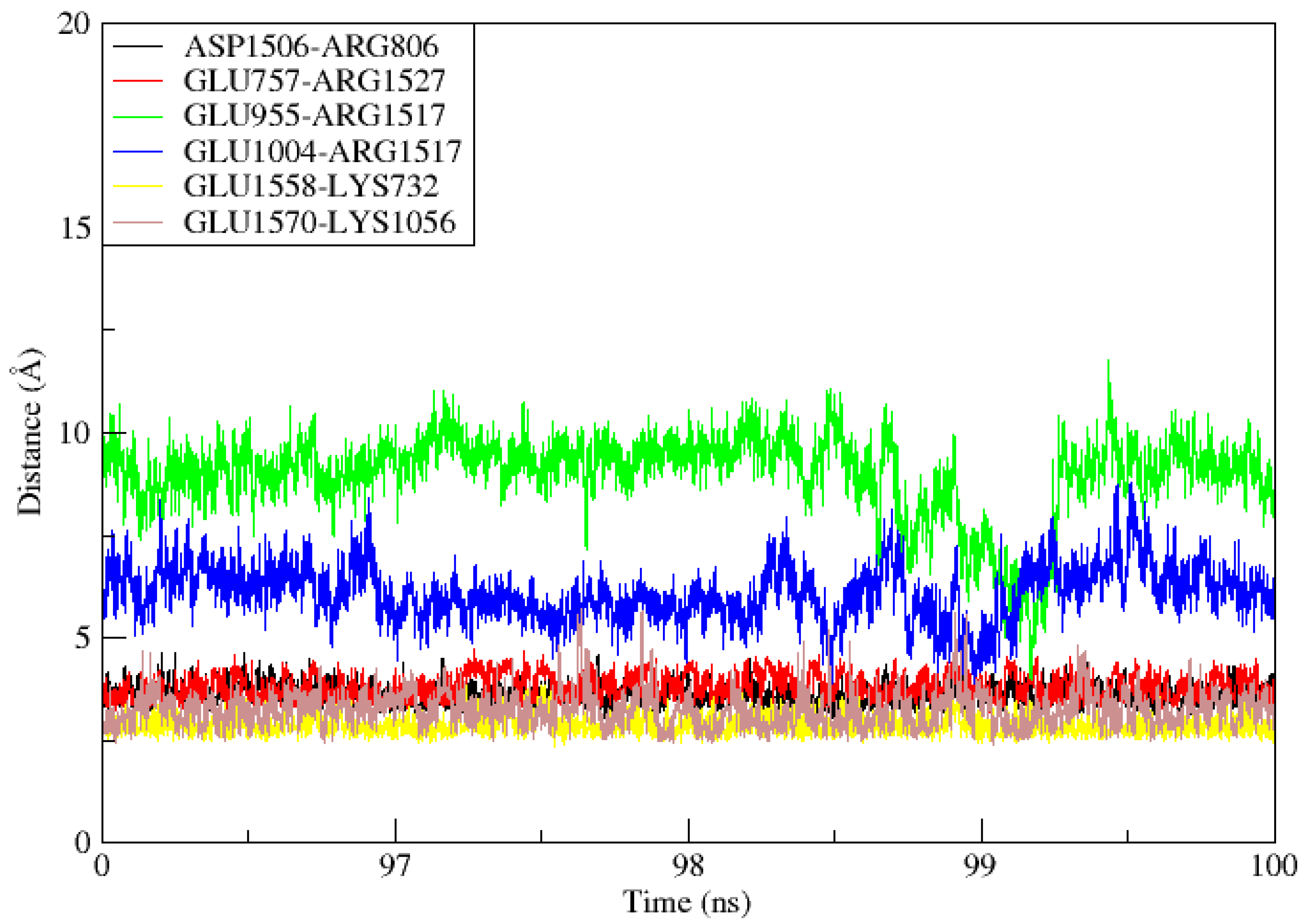
3.12. TLR4-MEPVC Stability and Salt Bridges
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Elmahallawy, E.K.; Martínez, A.S.; Rodriguez-Granger, J.; Hoyos-Mallecot, Y.; Agil, A.; Mari, J.M.N.; Fernández, J.G. Diagnosis of leishmaniasis. JIDC 2014, 8, 961–972. [Google Scholar] [CrossRef] [PubMed]
- De Freitas, E.O.; Leoratti, F.M.D.S.; Freire-de-Lima, C.G.; Morrot, A.; Feijó, D.F. The contribution of immune evasive mechanisms to parasite persistence in visceral leishmaniasis. Front. Immunol. 2016, 7, 153. [Google Scholar] [CrossRef] [PubMed]
- Sunyoto, T.; Potet, J.; Boelaert, M. Why miltefosine—A life-saving drug for leishmaniasis—Is unavailable to people who need it the most. BMJ Glob. Health 2018, 3, e000709. [Google Scholar] [CrossRef] [PubMed]
- Desjeux, P. Leishmaniasis: Current situation and new perspectives. Comp. Immunol. Microbiol. Infect. Dis. 2004, 27, 305–318. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. World Health Statistics; World Health Organization: Geneva, Switzerland, 2010. [Google Scholar]
- Pashaei, A.; Ghatee, M.; Sajedi, H. Convolution neural network joint with mixture of extreme learning machines for feature extraction and classification of accident images. J. Real Time Image Process. 2020, 17, 1051–1066. [Google Scholar] [CrossRef]
- Alvar, J.; Vélez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; den Boer, M.; Team, W.L.C. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef]
- Ashour, A.; Atia, A.; Akash, N.; Jumaa, E.; Alkhishrabi, A. Cutaneous Leishmaniasis in Al-Jabal Al-Gharbi, Libya: Incidence and Epidemiology. Khalij-Libya J. Dent. Med. Res. 2022, 6, 81–85. [Google Scholar] [CrossRef]
- de Souza Machado, A.A.; Kloas, W.; Zarfl, C.; Hempel, S.; Rillig, M.C. Microplastics as an emerging threat to terrestrial ecosystems. Glob. Chang. Biol. 2018, 24, 1405–1416. [Google Scholar] [CrossRef]
- Zijlstra, E.E. Biomarkers in post-kala-azar dermal leishmaniasis. Front. Cell. Infect. Microbiol. 2019, 9, 228. [Google Scholar] [CrossRef]
- Akhtari, J.; Soosaraei, M.; Ziaei, H.; Fakhar, M. Last decade developments on Leishmania vaccines with emphasis on nanovaccines. J. Maz. Univ. Med. Sci. 2017, 26, 232–253. [Google Scholar]
- Abu Ammar, A.; Nasereddin, A.; Ereqat, S.; Dan-Goor, M.; Jaffe, C.L.; Zussman, E.; Abdeen, Z. Amphotericin B-loaded nanoparticles for local treatment of cutaneous leishmaniasis. Drug Deliv. Transl. Res. 2019, 9, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Kip, A.E.; Schellens, J.H.; Beijnen, J.H.; Dorlo, T.P. Clinical pharmacokinetics of systemically administered antileishmanial drugs. Clin. Pharmacokinet. 2018, 57, 151–176. [Google Scholar] [CrossRef] [PubMed]
- Alexandrino-Junior, F.; Silva, K.G.D.H.E.; Freire, M.C.L.C.; Lione, V.D.O.F.; Cardoso, E.A.; Marcelino, H.R.; Genre, J.; Oliveira, A.G.D.; Egito, E.S.T.D. A functional wound dressing as a potential treatment for cutaneous leishmaniasis. Pharmaceutics 2019, 11, 200. [Google Scholar] [CrossRef] [PubMed]
- Minodier, P.; Parola, P. Cutaneous leishmaniasis treatment. Travel Med. Infect. Dis. 2007, 5, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; Chakravarty, J. An update on pharmacotherapy for leishmaniasis. Expert Opin. Pharmacother. 2015, 16, 237–252. [Google Scholar] [CrossRef]
- dos Santos Nogueira, F.; Avino, V.C.; Galvis-Ovallos, F.; Pereira-Chioccola, V.L.; Moreira, M.A.B.; Romariz, A.P.P.L.; Molla, L.M.; Menz, I. Use of miltefosine to treat canine visceral leishmaniasis caused by Leishmania infantum in Brazil. Parasites Vectors 2019, 12, 79. [Google Scholar] [CrossRef]
- Osorio, Y.; Travi, B.L.; Renslo, A.R.; Peniche, A.G.; Melby, P.C. Identification of small molecule lead compounds for visceral leishmaniasis using a novel ex vivo splenic explant model system. PLoS Negl. Trop. Dis. 2011, 5, e962. [Google Scholar] [CrossRef]
- Lieke, T.; Nylen, S.; Eidsmo, L.; McMaster, W.; Mohammadi, A.; Khamesipour, A.; Berg, L.; Akuffo, H. Leishmania surface protein gp63 binds directly to human natural killer cells and inhibits proliferation. Clin. Exp. Immunol. 2008, 153, 221–230. [Google Scholar] [CrossRef]
- Mangoni, M.L.; Saugar, J.M.; Dellisanti, M.; Barra, D.; Simmaco, M.; Rivas, L. Temporins, small antimicrobial peptides with leishmanicidal activity. J. Biol. Chem. 2005, 280, 984–990. [Google Scholar] [CrossRef]
- Isnard, A.; Shio, M.; Olivier, M. Impact of Leishmania metalloprotease GP63 on macrophage signaling. Front. Cell. Infect. Microbiol. 2012, 2, 72. [Google Scholar] [CrossRef]
- Berberich, C.; Ramírez-Pineda, J.R.; Hambrecht, C.; Alber, G.; Skeiky, Y.A.; Moll, H. Dendritic cell (DC)-based protection against an intracellular pathogen is dependent upon DC-derived IL-12 and can be induced by molecularly defined antigens. J. Immunol. Res. 2003, 170, 3171–3179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacks, D.L. Vaccines against tropical parasitic diseases: A persisting answer to a persisting problem. Nat. Immunol. 2014, 15, 403–405. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; Chakravarty, J.; Meena, L.P. Leishmaniasis: Treatment, drug resistance and emerging therapies. Expert Opin. Orphan Drugs. 2019, 7, 1–10. [Google Scholar] [CrossRef]
- Bahrami, A.A.; Payandeh, Z.; Khalili, S.; Zakeri, A.; Bandehpour, M. Immunoinformatics: In silico approaches and computational design of a multi-epitope, immunogenic protein. Int. Rev. Immunol. 2019, 38, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Raoufi, E.; Hemmati, M.; Eftekhari, S.; Khaksaran, K.; Mahmodi, Z.; Farajollahi, M.M.; Mohsenzadegan, M. Epitope prediction by novel immunoinformatics approach: A state-of-the-art review. Int. J. Pept. Res. Ther. 2020, 26, 1155–1163. [Google Scholar] [CrossRef]
- Xu, C.; Ye, B.; Han, Z.; Huang, M.; Zhu, Y. Comparison of transcriptional profiles between CD4+ and CD8+ T cells in HIV type 1-infected patients. AIDS Res. Hum. Retrovir. 2014, 30, 134–141. [Google Scholar] [CrossRef]
- Mansueto, P.; Vitale, G.; Di Lorenzo, G.; Rini, G.; Mansueto, S.; Cillari, E. Immunopathology of leishmaniasis: An update. Int. J. Immunopathol. Pharmacol. 2007, 20, 435–445. [Google Scholar] [CrossRef]
- Rhaiem, R.B.; Houimel, M. Targeting Leishmania major parasite with peptides derived from a combinatorial phage display library. Acta Trop. 2016, 159, 11–19. [Google Scholar] [CrossRef]
- Skwarczynski, M.; Toth, I. Peptide-based synthetic vaccines. Chem. Sci. 2016, 7, 842–854. [Google Scholar] [CrossRef]
- Conceicao, J.; Davis, R.; Carneiro, P.P.; Giudice, A.; Muniz, A.C.; Wilson, M.E.; Carvalho, E.M.; Bacellar, O. Characterization of neutrophil function in human cutaneous leishmaniasis caused by Leishmania braziliensis. PLoS Negl. Trop. Dis. 2016, 10, e0004715. [Google Scholar] [CrossRef]
- Sachdeva, R.; Banerjea, A.C.; Malla, N.; Dubey, M.L. Immunogenicity and efficacy of single antigen Gp63, polytope and polytopeHSP70 DNA vaccines against visceral Leishmaniasis in experimental mouse model. PLoS ONE 2009, 4, e7880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashemzadeh, P.; Ghorbanzadeh, V.; Lashgarian, H.E.; Kheirandish, F.; Dariushnejad, H. Harnessing Bioinformatic approaches to design novel multi-epitope subunit vaccine against Leishmania infantum. Int. J. Pept. Res. Ther. 2020, 26, 1417–1428. [Google Scholar] [CrossRef]
- Firouzmand, H.; Sahranavard, M.; Badiee, A.; Khamesipour, A.; Alavizadeh, S.H.; Samiei, A.; Soroush, D.; Tavassoti Kheiri, M.; Mahboudi, F.; Jaafari, M.R. The role of LPD-nanoparticles containing recombinant major surface glycoprotein of Leishmania (rgp63) in protection against leishmaniasis in murine model. Immunopharmacol. Immunotoxicol. 2018, 40, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Castro Neto, A.L.; Brito, A.N.; Rezende, A.M.; Magalhães, F.B.; de Melo Neto, O.P. In silico characterization of multiple genes encoding the GP63 virulence protein from Leishmania braziliensis: Identification of sources of variation and putative roles in immune evasion. BMC Genom. 2019, 20, 118. [Google Scholar] [CrossRef]
- Foroutan, M.; Ghaffarifar, F.; Sharifi, Z.; Dalimi, A.; Pirestani, M. Bioinformatics analysis of ROP8 protein to improve vaccine design against Toxoplasma gondii. Infect. Genet. Evol. 2018, 62, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Rasti, S.; Ghorbanzadeh, B.; Kheirandish, F.; Mousavi, S.G.; Pirozmand, A.; Hooshyar, H.; Abani, B. Comparison of molecular, microscopic, and culture methods for diagnosis of cutaneous leishmaniasis. J. Clin. Lab. Anal. 2016, 30, 610–615. [Google Scholar] [CrossRef]
- Moafi, M.; Rezvan, H.; Sherkat, R.; Taleban, R. Leishmania vaccines entered in clinical trials: A review of literature. Int. J. Prev. Med. 2019, 10, 95. [Google Scholar]
- Tahir ul Qamar, M.; Rehman, A.; Tusleem, K.; Ashfaq, U.A.; Qasim, M.; Zhu, X.; Fatima, I.; Shahid, F.; Chen, L.-L. Designing of a next generation multiepitope based vaccine (MEV) against SARS-COV-2: Immunoinformatics and in silico approaches. PLoS ONE 2020, 15, e0244176. [Google Scholar] [CrossRef]
- Esboei, B.R.; Mohebali, M.; Mousavi, P.; Fakhar, M.; Akhoundi, B. Potent antileishmanial activity of chitosan against Iranian strain of Leishmania major (MRHO/IR/75/ER): In vitro and in vivo assay. J. Vector Borne Dis. 2018, 55, 111. [Google Scholar]
- Mahendran, R.; Jeyabaskar, S.; Sitharaman, G.; Michael, R.D.; Paul, A.V. Computer-aided vaccine designing approach against fish pathogens Edwardsiella tarda and Flavobacterium columnare using bioinformatics softwares. Drug Des. Dev. Ther. 2016, 10, 1703. [Google Scholar] [CrossRef]
- Caro-Gomez, E.; Gazi, M.; Goez, Y.; Valbuena, G. Discovery of novel cross-protective Rickettsia prowazekii T-cell antigens using a combined reverse vaccinology and in vivo screening approach. Vaccine 2014, 32, 4968–4976. [Google Scholar] [CrossRef]
- Gaafar, B.; Ali, S.A.; Abd-Elrahman, K.A.; Almofti, Y.A. Immunoinformatics approach for multiepitope vaccine prediction from H, M, F, and N proteins of Peste des Petits ruminants virus. J. Immunol. Res. 2019, 2019, 6124030. [Google Scholar] [CrossRef] [PubMed]
- Carrillo, E.; Crusat, M.; Nieto, J.; Chicharro, C.; del Carmen Thomas, M.; Martínez, E.; Valladares, B.; Cañavate, C.; Requena, J.M.; López, M.C. Immunogenicity of HSP-70, KMP-11 and PFR-2 leishmanial antigens in the experimental model of canine visceral leishmaniasis. Vaccine 2008, 26, 1902–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafati, S.; Gholami, E.; Hassani, N.; Ghaemimanesh, F.; Taslimi, Y.; Taheri, T.; Soong, L. Leishmania major heat shock protein 70 (HSP70) is not protective in murine models of cutaneous leishmaniasis and stimulates strong humoral responses in cutaneous and visceral leishmaniasis patients. Vaccine 2007, 25, 4159–4169. [Google Scholar] [CrossRef]
- Mazumder, S.; Maji, M.; Das, A.; Ali, N. Potency, efficacy and durability of DNA/DNA, DNA/protein and protein/protein based vaccination using gp63 against Leishmania donovani in BALB/c mice. PLoS ONE 2011, 6, e14644. [Google Scholar] [CrossRef] [PubMed]
- Elfaki, M.E.; Khalil, E.A.; De Groot, A.S.; Musa, A.M.; Gutierrez, A.; Younis, B.M.; Salih, K.A.; El-Hassan, A.M. Immunogenicity and immune modulatory effects of in silico predicted L. donovani candidate peptide vaccines. Hum. Vaccin. Immunother. 2012, 8, 1769–1774. [Google Scholar] [CrossRef]
- Lang, T.; de Chastellier, C.; Frehel, C.; Hellio, R.; Metezeau, P.; Leao, S.; Antoine, J.-C. Distribution of MHC class I and of MHC class II molecules in macrophages infected with Leishmania amazonensis. J. Cell Sci. 1994, 107, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Khatoon, N.; Pandey, R.K.; Prajapati, V.K. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci. Rep. 2017, 7, 8285. [Google Scholar] [CrossRef] [PubMed]
- Groot, A.S.D.; Moise, L.; McMurry, J.A.; Martin, W. Epitope-based immunome-derived vaccines: A strategy for improved design and safety. Clin. Appl. Immunol. 2009, 2, 39–69. [Google Scholar]
- Reed, S.G.; Coler, R.N.; Campos-Neto, A. Development of a leishmaniasis vaccine: The importance of MPL. Expert Rev. Vaccines 2003, 2, 239–252. [Google Scholar] [CrossRef]
- Duthie, M.S.; Windish, H.P.; Fox, C.B.; Reed, S.G. Use of defined TLR ligands as adjuvants within human vaccines. Immunol. Rev. 2011, 239, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Kim, W.S.; Choi, H.G.; Jang, B.; Lee, K.; Park, J.H.; Kim, H.J.; Cho, S.N.; Shin, S.J. Mycobacterium tuberculosis RpfB drives Th1-type T cell immunity via a TLR4-dependent activation of dendritic cells. J. Leukoc. Biol. 2013, 94, 733–749. [Google Scholar] [CrossRef] [PubMed]
- Le-Barillec, K.; Magalhaes, J.G.; Corcuff, E.; Thuizat, A.; Sansonetti, P.J.; Phalipon, A.; Di Santo, J.P. Roles for T and NK cells in the innate immune response to Shigella flexneri. J. Immunol. 2005, 175, 1735–1740. [Google Scholar] [CrossRef]
- Magnan, C.N.; Randall, A.; Baldi, P. SOLpro: Accurate sequence-based prediction of protein solubility. Bioinformatics 2009, 25, 2200–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setrerrahmane, S.; Yu, J.; Hao, J.; Zheng, H.; Xu, H. Novel production method of innovative antiangiogenic and antitumor small peptides in Escherichia coli. Drug Des. Dev. Ther. 2017, 11, 3207. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef]
- Ismail, S.; Ahmad, S.; Azam, S.S. Immunoinformatics characterization of SARS-CoV-2 spike glycoprotein for prioritization of epitope based multivalent peptide vaccine. J. Mol. Liq. 2020, 314, 113612. [Google Scholar] [CrossRef]
- Calis, J.J.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef]
- Yadav, S.; Prakash, J.; Shukla, H.; Das, K.C.; Tripathi, T.; Dubey, V.K. Design of a multi-epitope subunit vaccine for immune-protection against Leishmania parasite. Pathog. Glob. Health 2020, 114, 471–481. [Google Scholar] [CrossRef]
- ul Qamar, M.T.; Ahmad, S.; Fatima, I.; Ahmad, F.; Shahid, F.; Naz, A.; Abbasi, S.W.; Khan, A.; Mirza, M.U.; Ashfaq, U.A. Designing multi-epitope vaccine against Staphylococcus aureus by employing subtractive proteomics, reverse vaccinology and immuno-informatics approaches. Comput. Biol. Med. 2021, 132, 104389. [Google Scholar] [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Consortium, O.S.D.D.; Raghava, G.P. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [PubMed]
- Hoque, H.; Islam, R.; Ghosh, S.; Rahaman, M.M.; Jewel, N.A.; Miah, M.A. Implementation of in silico methods to predict common epitopes for vaccine development against Chikungunya and Mayaro viruses. Heliyon 2021, 7, e06396. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.A.; Ami, J.Q.; Faisal, K.; Chowdhury, R.; Ghosh, P.; Hossain, F.; Abd El Wahed, A.; Mondal, D. An immunoinformatic approach driven by experimental proteomics: In silico design of a subunit candidate vaccine targeting secretory proteins of Leishmania donovani amastigotes. Parasites Vectors 2020, 13, 196. [Google Scholar] [CrossRef]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A protein structure and structural feature prediction server. Nucleic Acids Res. 2005, 33, 72–76. [Google Scholar] [CrossRef] [Green Version]
- Giardine, B.; Riemer, C.; Hardison, R.C.; Burhans, R.; Elnitski, L.; Shah, P.; Zhang, Y.; Blankenberg, D.; Albert, I.; Taylor, J.; et al. Galaxy: A platform for interactive large-scale genome analysis. Genome Res. 2005, 15, 1451–1455. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, 384–388. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. Geometry-based flexible and symmetric protein docking. Proteins Struct. Funct. Genet 2005, 60, 224–231. [Google Scholar] [CrossRef]
- Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: Fast interaction refinement in molecular docking. Proteins Struct. Funct. Genet. 2007, 69, 139–159. [Google Scholar] [CrossRef]
- Heinzelmann, G.; Gilson, M.K. Automated docking refinement and virtual compound screening with absolute binding free energy calculations. BioRxiv 2020. [Google Scholar] [CrossRef]
- Naz, S.; Ahmad, S.; Walton, S.; Abbasi, S.W. Multi-epito.pe based vaccine design against Sarcoptes scabiei paramyosin using immunoinformatics approach. J. Mol. Liq. 2020, 319, 114105. [Google Scholar] [CrossRef]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA. py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Start | End | Peptides | Length | Antigenicity Score |
|---|---|---|---|---|---|
| 1 | 40 | 48 | HAGALQHRC | 9 | 0.7469 |
| 2 | 54 | 106 | QARVRQSVADHHKAPGAVSAVGLPYVTLDAAHTAAAADPRPGSARSVVRDVNW | 53 | 0.7005 |
| 3 | 166 | 206 | QLHTERLKVQQVQGKWKVTDMVGDICGDFKVPQAHITEGFS | 41 | 0.5666 |
| 4 | 277 | 292 | FEDARIVANVPNVRGK | 16 | 0.6676 |
| 5 | 321 | 342 | EVEDQGGAGSAGSHIKMRNAQD | 22 | 2.1090 |
| 6 | 428 | 452 | TRHPGLPPYWQYFTDPSLAGVSAFM | 25 | 0.4103 |
| 7 | 460 | 489 | PYSDGSCTQRASEAHASLLPFNVFSDAARC | 30 | 0.8693 |
| 8 | 492 | 507 | GAFRPKATDGIVKSYA | 16 | 0.6250 |
| 9 | 565 | 585 | CQGNVQAAKDGGNTAAGRRGP | 21 | 1.4080 |
| T Cell Epitopes | Percentile Score | MHCPred Score (nM) | Allergenicity | Antigenicity | Solubility | IFN-γ | Toxicity | Virulency | |
|---|---|---|---|---|---|---|---|---|---|
| MHCI | MHCII | ||||||||
| RVRQSVADH | 0.4 | 19 | 50.12 | Non-allergen | 0.6 | Good soluble | + | Non-toxin | 0.6586 |
| AADPRPGSA | 1.3 | 6.4 | 55.72 | Non-allergen | 0.8052 | Good soluble | + | Non-toxin | 0.6586 |
| RSVVRDVNW | 0.1 | 14 | 24.27 | Non-allergen | 0.9752 | Good soluble | + | Non-toxin | 0.6586 |
| RLKVQQVQG | 0.08 | 0.81 | 26.61 | Non-allergen | 0.7406 | Good soluble | + | Non-toxin | 0.6586 |
| LHTERLKVQ | 20 | 0.81 | 97.5 | Non-allergen | 0.8995 | Good soluble | + | Non-toxin | 0.6586 |
| FEDARIVAN | 1.3 | 0.73 | 5.93 | Non-allergen | 1.1664 | Good soluble | + | Non-toxin | 0.6586 |
| NVFSDAARC | 1.6 | 25 | 3.24 | Non-allergen | 1.0135 | Good soluble | + | Non-toxin | 0.6586 |
| YSDGSCTQR | 0.94 | 75 | 10.38 | Non-allergen | 0.8450 | Good soluble | + | Non-toxin | 0.6586 |
| DGGNTAAGR | 2.5 | 75.38 | 31.12 | Non-allergen | 0.9705 | Good soluble | + | Non-toxin | 0.6586 |
| Criteria | Score |
|---|---|
| No. of amino acids | 119 |
| Molecular Weight | 11,825.08 |
| Total number of negatively charged residues | 09 |
| Total number of positively charged residues | 13 |
| Theoretical pI | 9.68 |
| Estimated half-life in mammalian reticulocytes in vitro | 4.4 h |
| Instability Index (II) | 25.96 |
| Aliphatic Index | 51.68 |
| Grand average of hydrophaticity (GRAVY) | −0.682 |
| Solubility | 0.71, 0.903 |
| Solution Rank | Solution Number | Docking Global Energy | Attractive van der Waals Energy | Repulsive van der Waals Energy | Atomic Contact Energy | Hydrogen Bonding Energy |
|---|---|---|---|---|---|---|
| 1 | 5 | 8.12 | −1.18 | 0.16 | 1.74 | −0.57 |
| 2 | 7 | 8.12 | −23.52 | 13.15 | 18.17 | −4.27 |
| 3 | 9 | 12.76 | −2.38 | 0.64 | 1.29 | −0.48 |
| 4 | 4 | 31.29 | −16.52 | 25.75 | 13.70 | −3.23 |
| 5 | 6 | 51.16 | −11.81 | 6.06 | 7.43 | −0.65 |
| 6 | 1 | 127.18 | −54.05 | 242.23 | 4.95 | −7.53 |
| 7 | 10 | 170.66 | −50.85 | 292.63 | −2.23 | −5.51 |
| 8 | 2 | 867.90 | −66.81 | 1157.25 | 12.48 | −10.11 |
| 9 | 3 | 3487.64 | −80.82 | 4489.93 | 24.81 | −15.97 |
| 10 | 8 | 6092.43 | −127.20 | 7856.27 | 16.37 | −34.99 |
| Generalized Born | |||
|---|---|---|---|
| Complex: | |||
| Energy Component | Average | Std. Dev. | Err. of Mean |
| VDWAALS | −13,331.9017 | 51.5473 | 5.1547 |
| EEL | −113,422.7858 | 113.5974 | 11.3597 |
| EGB | −18,564.5203 | 85.5735 | 8.5573 |
| ESURF | 471.6862 | 2.6545 | 0.2654 |
| G gas | −126,754.6874 | 115.0917 | 11.5092 |
| G solv | −18,092.8341 | 85.3047 | 8.5305 |
| TOTAL | −144,847.5215 | 87.4204 | 8.742 |
| Receptor: | |||
| Energy Component | Average | Std. Dev. | Err. of Mean |
| VDWAALS | −10,091.7946 | 45.7379 | 4.5738 |
| EEL | −81,782.776 | 118.6831 | 11.8683 |
| EGB | −17,803.7552 | 89.6395 | 8.9639 |
| ESURF | 374.3564 | 2.2659 | 0.2266 |
| G gas | −91,874.5705 | 119.4938 | 11.9494 |
| G solv | −17,429.3988 | 88.5926 | 8.8593 |
| TOTAL | −109,303.9694 | 82.1018 | 8.2102 |
| Ligand: | |||
| Energy Component | Average | Std. Dev. | Err. of Mean |
| VDWAALS | −2773.0544 | 20.5908 | 2.0591 |
| EEL | −27,196.8614 | 85.3765 | 8.5376 |
| EGB | −5466.5473 | 65.2155 | 6.5216 |
| ESURF | 165.1464 | 1.205 | 0.1205 |
| G gas | −29,969.9158 | 83.196 | 8.3196 |
| G solv | −5301.4009 | 65.3738 | 6.5374 |
| TOTAL | −35,271.3167 | 42.7365 | 4.2737 |
| Differences (Complex-Receptor—Ligand): | |||
| Energy Component | Average | Std. Dev. | Err. of Mean |
| VDWAALS | −467.0527 | 10.8948 | 1.0895 |
| EEL | −4443.1483 | 64.8796 | 6.488 |
| EGB | 4705.7822 | 56.3535 | 5.6354 |
| ESURF | −67.8165 | 0.8586 | 0.0859 |
| DELTA G gas | −4910.2011 | 62.5925 | 6.2592 |
| DELTA G solv | 4637.9657 | 55.8599 | 5.586 |
| DELTA TOTAL | −272.2354 | 12.1577 | 1.2158 |
| Poisson Boltzmann | |||
|---|---|---|---|
| Complex: | |||
| Energy Component | Average | Std. Dev. | Err. of Mean |
| VDWAALS | −13,331.9017 | 51.5473 | 5.1547 |
| EEL | −113,422.7858 | 113.5974 | 11.3597 |
| EPB | −18,084.3392 | 74.7642 | 7.4764 |
| ENPOLAR | 324.2708 | 0.9841 | 0.0984 |
| G gas | −126,754.6874 | 115.0917 | 11.5092 |
| G solv | −17,760.0684 | 74.5556 | 7.4556 |
| TOTAL | −144,514.7558 | 91.3773 | 9.1377 |
| Receptor: | |||
| Energy Component | Average | Std. Dev. | Err. of Mean |
| VDWAALS | −10,091.7946 | 45.7379 | 4.5738 |
| EEL | −81,782.776 | 118.6831 | 11.8683 |
| EPB | −17,279.2394 | 91.9939 | 9.1994 |
| ENPOLAR | 256.9962 | 0.7679 | 0.0768 |
| G gas | −91,874.5705 | 119.4938 | 11.9494 |
| G solv | −17,022.2432 | 91.7489 | 9.1749 |
| TOTAL | −108,896.8137 | 82.893 | 8.2893 |
| Ligand: | |||
| Energy Component | Average | Std. Dev. | Err. of Mean |
| VDWAALS | −2773.0544 | 20.5908 | 2.0591 |
| EEL | −27,196.8614 | 85.3765 | 8.5376 |
| EPB | −5357.5329 | 61.3195 | 6.132 |
| ENPOLAR | 120.0538 | 0.6479 | 0.0648 |
| G gas | −29,969.9158 | 83.196 | 8.3196 |
| G solv | −5237.4791 | 61.554 | 6.1554 |
| TOTAL | −35,207.3949 | 46.5701 | 4.657 |
| Differences (Complex-Receptor—Ligand) | |||
| Energy Component | Average | Std. Dev. | Err. of Mean |
| VDWAALS | −467.0527 | 10.8948 | 1.0895 |
| EEL | −4443.1483 | 64.8796 | 6.488 |
| EPB | 4552.4332 | 57.0836 | 5.7084 |
| ENPOLAR | −52.7792 | 0.5371 | 0.0537 |
| EDISPER | 0 | 0 | 0 |
| DELTA G gas | −4910.2011 | 62.5925 | 6.2592 |
| DELTA G solv | 4499.654 | 56.8086 | 5.6809 |
| DELTA TOTAL | −410.5471 | 14.1814 | 1.4181 |
| GB | PB | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | Sidechain | Backbone | Total | Sidechain | Backbone | ||||||
| MET15 | −2.08462 | MET15 | −2.40965 | PHE237 | −1.06161 | MET15 | −1.44671 | MET15 | −1.60689 | SER60 | −0.77127 |
| GLU16 | −1.17813 | GLU16 | −1.05225 | LEU782 | −1.66875 | GLU16 | −2.13704 | GLU16 | −1.92609 | THR84 | −0.8626 |
| ASP34 | −5.04397 | ASP34 | −5.29744 | LEU783 | −1.58457 | ASP34 | −5.13311 | ASP34 | −4.97831 | GLY85 | −0.74245 |
| PHE37 | −5.19558 | SER36 | −1.51823 | PHE842 | −1.14768 | PHE37 | −3.32421 | PHE37 | −3.20194 | PHE237 | −1.31058 |
| ASP58 | −1.43186 | PHE37 | −4.89467 | LYS981 | −1.69243 | ARG61 | −4.5124 | ARG61 | −4.35588 | ARG238 | −1.8331 |
| ARG61 | −3.40484 | ASP58 | −1.70757 | TYR982 | −0.15898 | VAL108 | −1.37047 | VAL108 | −1.32842 | LEU782 | −2.03181 |
| THR84 | −2.70391 | ARG61 | −4.11754 | ASP984 | −0.10635 | HIE133 | −3.00269 | HIE133 | −2.95278 | LEU783 | −1.1302 |
| VAL108 | −1.40956 | THR84 | −2.12421 | SER995 | −0.10725 | ASP155 | −5.04159 | ASP155 | −4.86819 | PHE842 | −1.28279 |
| HIE133 | −2.04865 | VAL108 | −1.38103 | ASN996 | −0.05226 | LYS204 | −2.44477 | LYS204 | −2.36699 | ARG843 | −1.5378 |
| ASP155 | −6.43927 | HIE133 | −2.03865 | Residue | ARG208 | −2.46066 | ARG208 | −2.46258 | LYS981 | −1.28934 | |
| LYS204 | −1.98953 | ASP155 | −6.65367 | ARG231 | −5.28115 | ARG231 | −5.18781 | ||||
| ARG208 | −1.22748 | LYS204 | −2.1196 | PHE237 | −4.21487 | PHE237 | −2.90432 | ||||
| ARG231 | −4.5883 | ARG208 | −1.62838 | ARG238 | −8.6338 | ARG238 | −6.80086 | ||||
| VAL233 | −0.92789 | ARG231 | −4.66365 | ASN239 | −3.62178 | ASN239 | −3.18728 | ||||
| PHE237 | −5.58086 | VAL233 | −1.0402 | ARG263 | −2.76684 | ARG263 | −2.66258 | ||||
| ARG238 | −7.69397 | PHE237 | −4.51921 | TYR266 | −1.41979 | TYR266 | −1.18676 | ||||
| ASN239 | −3.91616 | ARG238 | −6.94139 | VAL290 | −2.20019 | VAL290 | −1.80646 | ||||
| ARG263 | −1.27341 | ASN239 | −3.97193 | LEU393 | −1.78254 | LEU393 | −1.92587 | ||||
| TYR266 | −1.13488 | ARG263 | −1.57907 | LEU418 | −2.08328 | LEU418 | −1.89394 | ||||
| VAL290 | −2.1697 | TYR266 | −1.14303 | PHE437 | −3.73036 | PHE437 | −3.53362 | ||||
| LEU393 | −2.0549 | VAL290 | −2.03742 | MET620 | −2.2787 | MET620 | −2.80336 | ||||
| LEU418 | −2.06666 | LEU393 | −2.26239 | GLU621 | −3.29011 | GLU621 | −3.11259 | ||||
| PHE437 | −4.37701 | PHE414 | −1.07004 | ASN637 | −3.3685 | ASN637 | −3.3096 | ||||
| MET620 | −3.03892 | LEU418 | −2.17538 | ASP639 | −5.13609 | ASP639 | −5.00157 | ||||
| GLU621 | −1.5327 | PHE437 | −4.36564 | PHE642 | −3.84124 | PHE642 | −3.71211 | ||||
| ASN637 | −3.12 | MET620 | −3.41881 | ASP663 | −1.4491 | ASP663 | −0.92414 | ||||
| ASP639 | −4.40471 | GLU621 | −1.50262 | ARG666 | −3.2697 | ARG666 | −3.41851 | ||||
| PHE642 | −5.54916 | ASN637 | −3.09983 | VAL713 | −1.93046 | VAL713 | −1.73128 | ||||
| ASP663 | −2.19012 | ASP639 | −4.64608 | GLU714 | −1.81689 | GLU714 | −1.58249 | ||||
| SER665 | −1.68462 | SER641 | −1.18362 | LYS732 | −5.19682 | LYS732 | −5.02788 | ||||
| ARG666 | −2.60525 | PHE642 | −5.27163 | HIE738 | −2.23817 | HIE738 | −1.90826 | ||||
| THR689 | −2.49328 | ASP663 | −2.32401 | LEU782 | −3.40719 | LEU782 | −1.37525 | ||||
| VAL713 | −1.62158 | SER665 | −2.22498 | ARG806 | −10.0732 | ARG806 | −9.55822 | ||||
| GLU714 | −3.33969 | ARG666 | −3.3758 | HIE808 | −1.15354 | HIE808 | −0.93046 | ||||
| LYS732 | −3.27881 | THR689 | −2.1897 | HIE835 | −1.17941 | HIE835 | −1.06968 | ||||
| HIE738 | −1.19432 | VAL713 | −1.71425 | PHE842 | −4.72342 | PHE842 | −3.44065 | ||||
| HIE758 | −1.06866 | GLU714 | −2.94628 | ARG843 | −6.34089 | ARG843 | −4.80317 | ||||
| LEU782 | −3.33685 | LYS732 | −3.87176 | ASN844 | −4.46346 | ASN844 | −4.10789 | ||||
| LEU783 | −2.21718 | HIE738 | −1.20185 | ARG868 | −1.40767 | ARG868 | −1.24233 | ||||
| ARG806 | −7.8341 | HIE758 | −1.10525 | TYR871 | −1.50897 | TYR871 | −1.29594 | ||||
| HIE808 | −2.951 | LEU782 | −1.66809 | VAL895 | −1.67395 | VAL895 | −1.68745 | ||||
| HIE835 | −1.78933 | ASN784 | −1.06739 | PHE956 | −2.18221 | PHE956 | −2.07051 | ||||
| PHE842 | −6.00546 | ARG806 | −7.91147 | LYS981 | −1.76858 | LYS981 | −2.28329 | ||||
| ARG843 | −6.33914 | HIE808 | −3.27018 | TYR982 | −1.39534 | TYR982 | −1.03322 | ||||
| ASN844 | −5.59359 | ARG813 | −1.28889 | ASN996 | −2.69019 | ASN996 | −2.59746 | ||||
| ARG868 | −1.71646 | HIE835 | −1.95128 | LEU998 | −1.85412 | LEU998 | −1.80163 | ||||
| ALA870 | −1.02429 | PHE842 | −4.85791 | ||||||||
| TYR871 | −1.17867 | ARG843 | −5.3531 | ||||||||
| VAL895 | −2.11224 | ASN844 | −5.27324 | ||||||||
| HIE913 | −1.32297 | ARG868 | −2.16871 | ||||||||
| THR936 | −1.09677 | TYR871 | −1.13235 | ||||||||
| PHE956 | −2.67546 | VAL895 | −1.88138 | ||||||||
| LYS981 | −2.23869 | HIE913 | −1.44291 | ||||||||
| TYR982 | −2.93645 | ARG934 | −1.00448 | ||||||||
| ASN996 | −3.03567 | PHE956 | −2.74494 | ||||||||
| LEU998 | −2.06441 | TYR982 | −2.7774 | ||||||||
| ASN996 | −2.98316 | ||||||||||
| LEU998 | −2.14224 | ||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naz, S.; Aroosh, A.; Caner, A.; Şahar, E.A.; Toz, S.; Ozbel, Y.; Abbasi, S.W. Immunoinformatics Approach to Design a Multi-Epitope Vaccine against Cutaneous Leishmaniasis. Vaccines 2023, 11, 339. https://doi.org/10.3390/vaccines11020339
Naz S, Aroosh A, Caner A, Şahar EA, Toz S, Ozbel Y, Abbasi SW. Immunoinformatics Approach to Design a Multi-Epitope Vaccine against Cutaneous Leishmaniasis. Vaccines. 2023; 11(2):339. https://doi.org/10.3390/vaccines11020339
Chicago/Turabian StyleNaz, Shumaila, Aiman Aroosh, Ayse Caner, Esra Atalay Şahar, Seray Toz, Yusuf Ozbel, and Sumra Wajid Abbasi. 2023. "Immunoinformatics Approach to Design a Multi-Epitope Vaccine against Cutaneous Leishmaniasis" Vaccines 11, no. 2: 339. https://doi.org/10.3390/vaccines11020339