

The Development of Surface-Modified Liposomes as an Intranasal Delivery System for Group A Streptococcus Vaccines
 , ,
, ,  , , , ,
, , , ,  ,
,  , ,
, , 
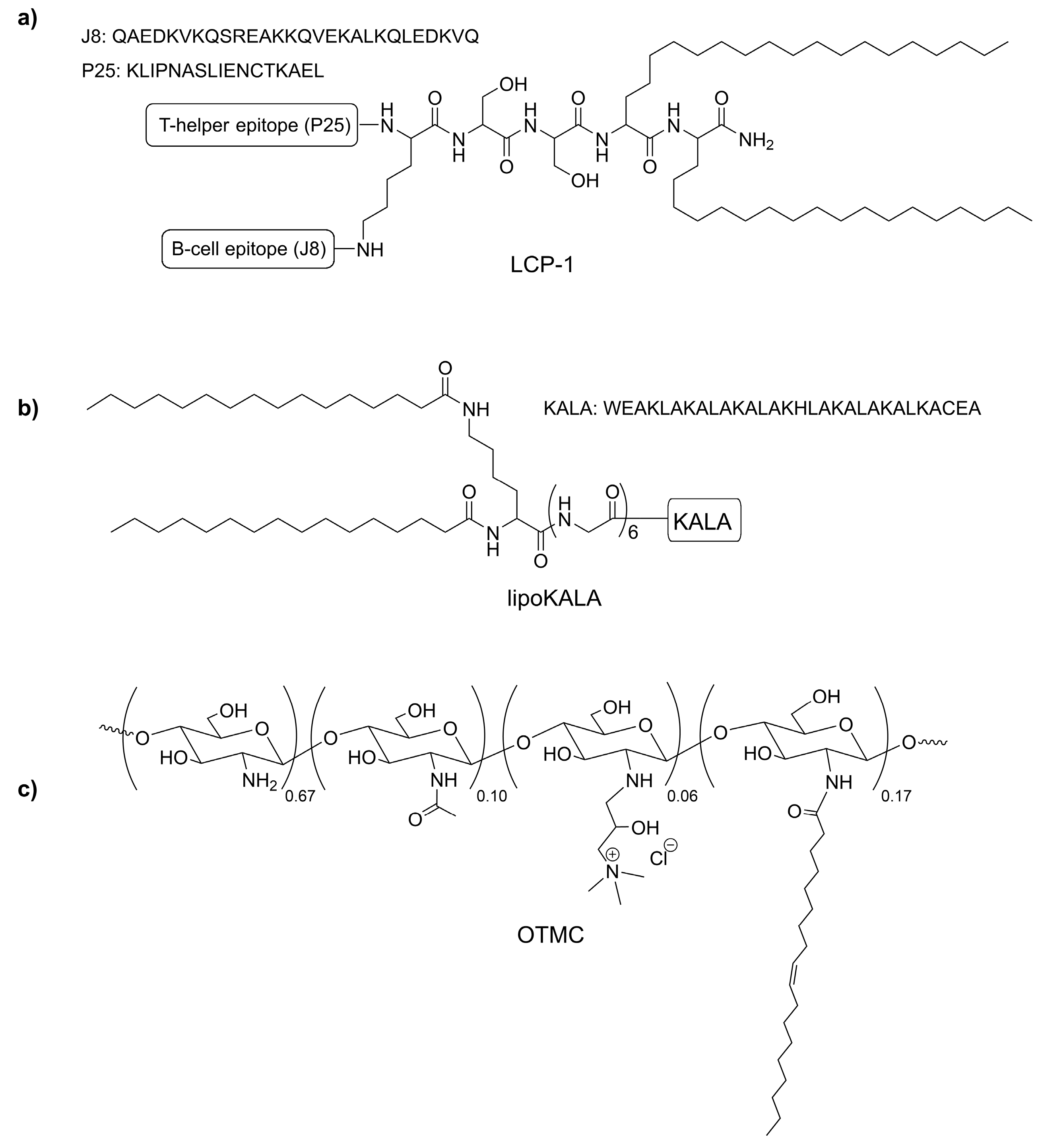{kind=link}
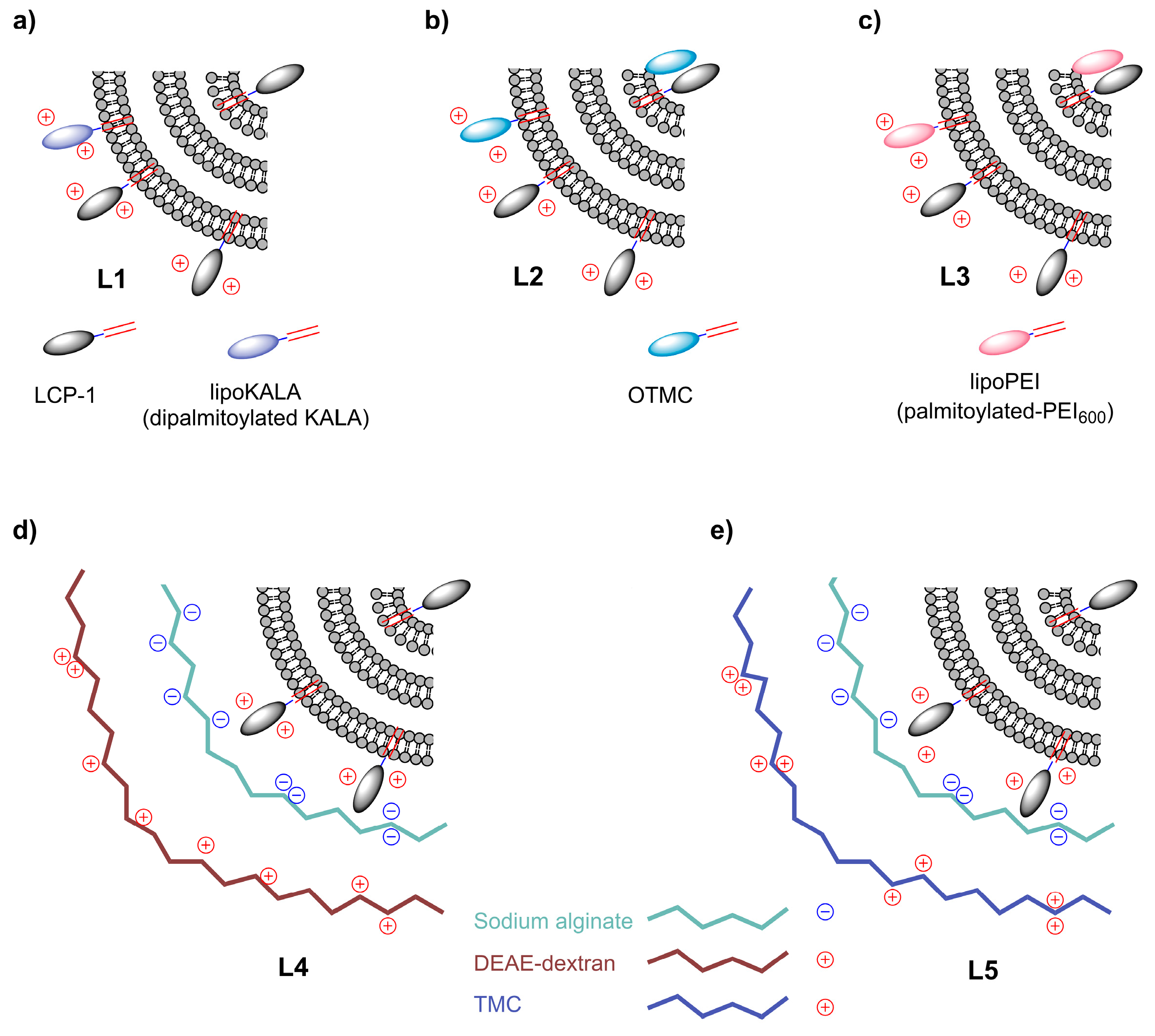{kind=link}
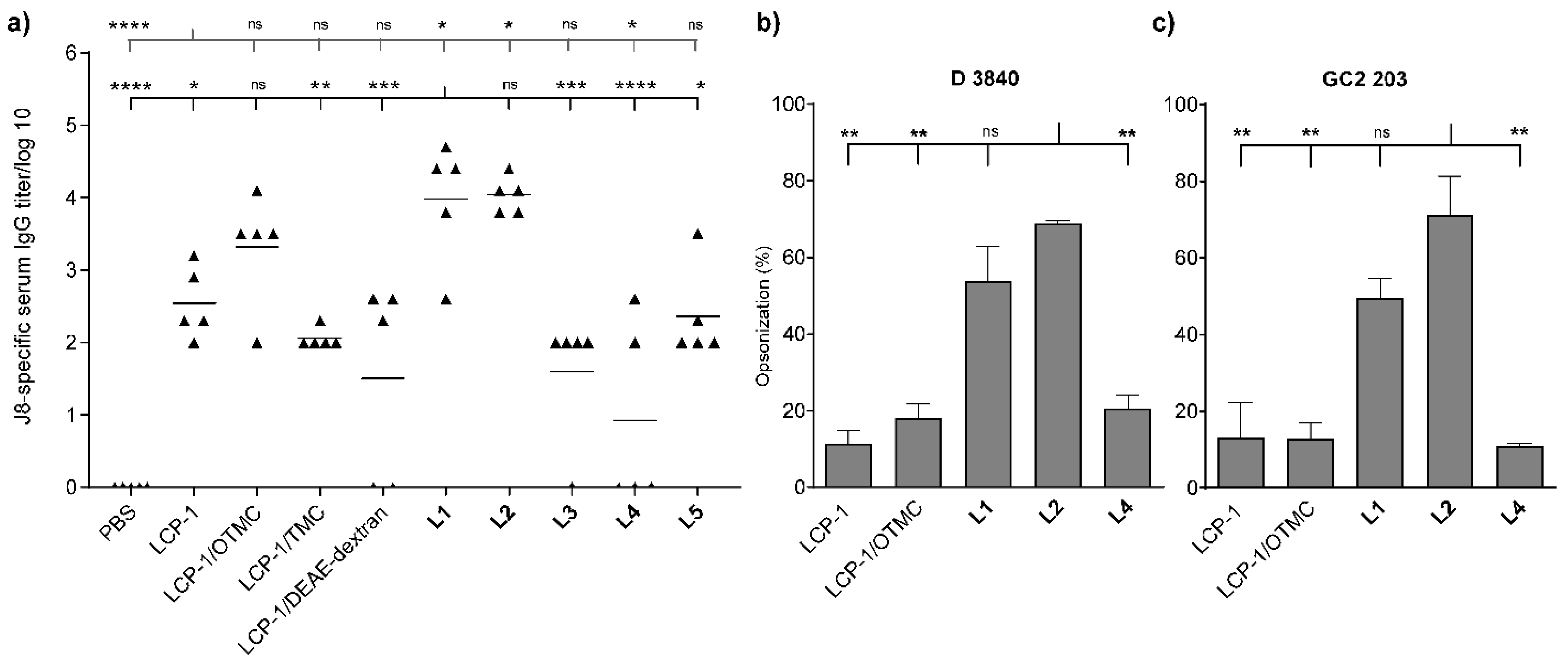{kind=link}
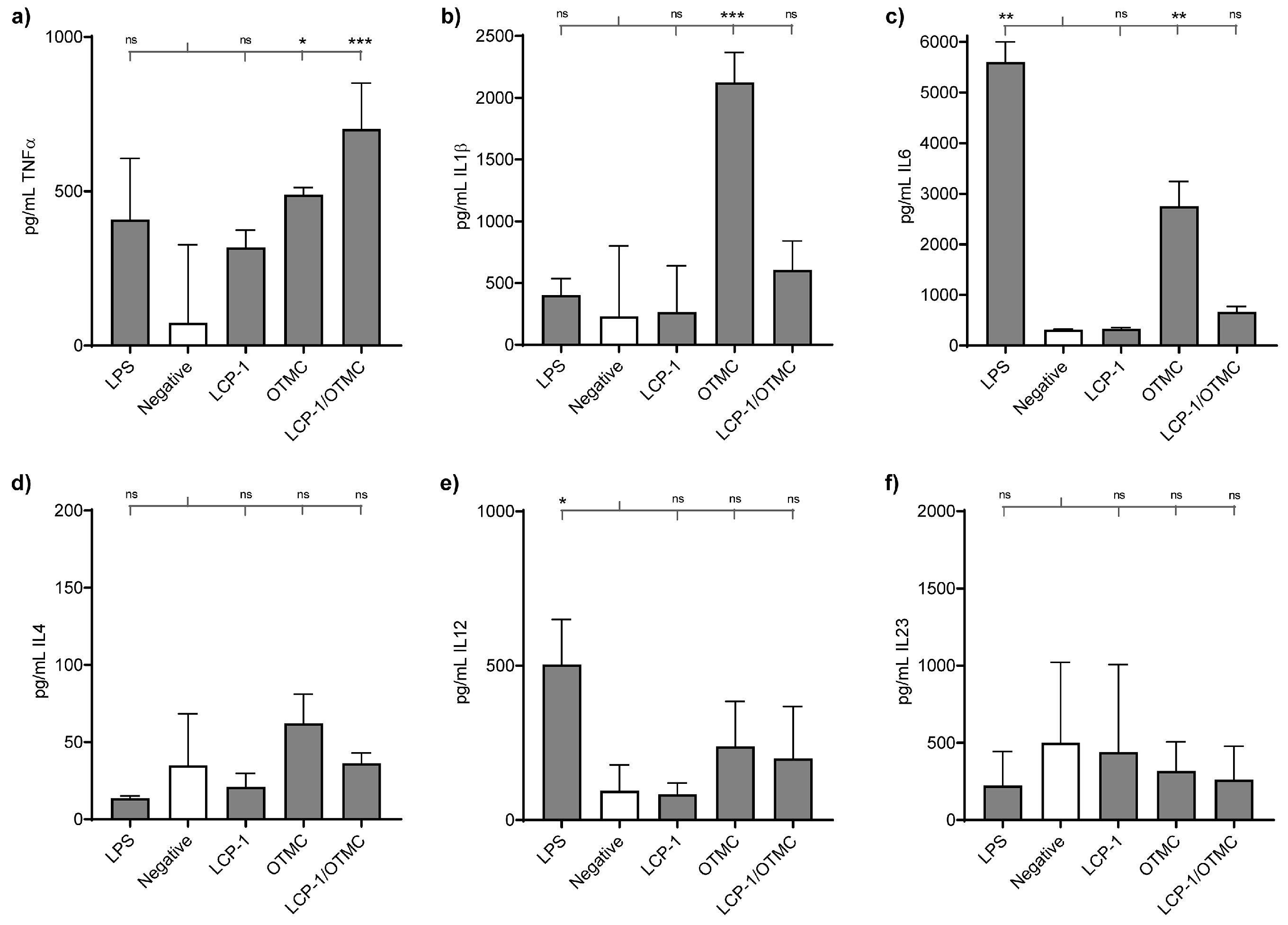{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of LCP−1-Loaded Multilamellar Liposomes
2.2. Characterization of Vaccine Candidates by Dynamic Light Scattering
2.3. Immunization Study
2.4. Determination of IgG Titres
2.5. Opsonization Assays
2.6. Ex vivo Cytokine Profiling in Dendritic Cells
2.7. Ethics Statement
2.8. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Skwarczynski, M.; Toth, I. Non-invasive mucosal vaccine delivery: Advantages, challenges and the future. Expert Opin. Drug Deliv. 2020, 17, 435–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marasini, N.; Skwarczynski, M.; Toth, I. Intranasal delivery of nanoparticle-based vaccines. Ther. Deliv. 2017, 8, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, H.; Kett, V. Current prospects and future challenges for nasal vaccine delivery. Hum. Vaccin. Immunother. 2017, 13, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Oscherwitz, J.; Hankenson, F.C.; Yu, F.; Cease, K.B. Low-dose intraperitoneal Freund’s adjuvant: Toxicity and immunogenicity in mice using an immunogen targeting amyloid-beta peptide. Vaccine 2006, 24, 3018–3025. [Google Scholar] [CrossRef]
- Powers, J.G.; Nash, P.B.; Rhyan, J.C.; Yoder, C.A.; Miller, L.A. Comparison of immune and adverse effects induced by AdjuVac and Freund’s complete adjuvant in New Zealand white rabbits (Oryctolagus cuniculus). Lab. Anim. 2007, 36, 51–58. [Google Scholar] [CrossRef]
- Petrovsky, N. Comparative Safety of Vaccine Adjuvants: A Summary of Current Evidence and Future Needs. Drug Saf. 2015, 38, 1059–1074. [Google Scholar] [CrossRef]
- Sanyahumbi, A.S.; Colquhoun, S.; Wyber, R.; Carapetis, J.R. Global disease burden of group A Streptococcus. In Streptococcus pyogenes: Basic Biology to Clinical Manifestations; University of Oklahoma Health Sciences Center: Oklahoma City, OK, USA, 2016. [Google Scholar]
- Watkins, D.A.; Johnson, C.O.; Colquhoun, S.M.; Karthikeyan, G.; Beaton, A.; Bukhman, G.; Forouzanfar, M.H.; Longenecker, C.T.; Mayosi, B.M.; Mensah, G.A.; et al. Global, Regional, and National Burden of Rheumatic Heart Disease, 1990–2015. N. Engl. J. Med. 2017, 377, 713–722. [Google Scholar] [CrossRef]
- Azuar, A.; Jin, W.; Mukaida, S.; Hussein, W.M.; Toth, I.; Skwarczynski, M. Recent advances in the development of peptide vaccines and their delivery systems against group a streptococcus. Vaccines 2019, 7, 58. [Google Scholar] [CrossRef] [Green Version]
- Sekuloski, S.; Batzloff, M.R.; Griffin, P.; Parsonage, W.; Elliott, S.; Hartas, J.; O’Rourke, P.; Marquart, L.; Pandey, M.; Rubin, F.A. Evaluation of safety and immunogenicity of a group A streptococcus vaccine candidate (MJ8VAX) in a randomized clinical trial. PLoS ONE 2018, 13, e0198658. [Google Scholar] [CrossRef] [Green Version]
- Alharbi, N.; Skwarczynski, M.; Toth, I. The influence of component structural arrangement on peptide vaccine immunogenicity. Biotechnol. Adv. 2022, 60, 108029. [Google Scholar] [CrossRef]
- Marasini, N.; Ghaffar, K.A.; Giddam, A.K.; Batzloff, M.R.; Good, M.F.; Skwarczynski, M.; Toth, I. Highly Immunogenic Trimethyl Chitosan-based Delivery System for Intranasal Lipopeptide Vaccines against Group A Streptococcus. Curr. Drug Deliv. 2017, 14, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Azuar, A.; Toth, I.; Skwarczynski, M. Liposomes for the Delivery of Lipopeptide Vaccines. In Vaccine Design: Methods and Protocols, Volume 3. Resources for Vaccine Development; Thomas, S., Ed.; Springer: New York, NY, USA, 2022; pp. 295–307. [Google Scholar]
- Marasini, N.; Ghaffar, K.A.; Skwarczynski, M.; Toth, I. Liposomes as a Vaccine Delivery System. In Micro- and Nanotechnology in Vaccine Development; Skwarczynski, M., Toth, I., Eds.; William Andrew Inc.: Norwich, UK, 2017; pp. 221–239. [Google Scholar]
- Zhao, L.; Skwarczynski, M.; Toth, I. Polyelectrolyte-Based Platforms for the Delivery of Peptides and Proteins. ACS Biomater. Sci. Eng. 2019, 5, 4937–4950. [Google Scholar] [CrossRef] [PubMed]
- Volodkin, D.V.; Schaaf, P.; Mohwald, H.; Voegel, J.-C.; Ball, V. Effective embedding of liposomes into polyelectrolyte multilayered films: The relative importance of lipid-polyelectrolyte and interpolyelectrolyte interactions. Soft Matter 2009, 5, 1394–1405. [Google Scholar] [CrossRef]
- Ghaffar, K.A.; Marasini, N.; Giddam, A.K.; Batzloff, M.R.; Good, M.F.; Skwarczynski, M.; Toth, I. Liposome-based intranasal delivery of lipopeptide vaccine candidates against group A streptococcus. Acta Biomater. 2016, 41, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Marasini, N.; Giddam, A.K.; Khalil, Z.G.; Hussein, W.M.; Capon, R.J.; Batzloff, M.R.; Good, M.F.; Toth, I.; Skwarczynski, M. Double adjuvanting strategy for peptide-based vaccines: Trimethyl chitosan nanoparticles for lipopeptide delivery. Nanomedicine 2016, 11, 3223–3235. [Google Scholar] [CrossRef] [PubMed]
- Houston, W.E.; Crabbs, C.L.; Kremer, R.J.; Springer, J.W. Adjuvant effects of diethylaminoethyl-dextran. Infect. Immun. 1976, 13, 1559–1562. [Google Scholar] [CrossRef] [Green Version]
- Piedrafita, D.; Preston, S.; Kemp, J.; de Veer, M.; Sherrard, J.; Kraska, T.; Elhay, M.; Meeusen, E. The Effect of Different Adjuvants on Immune Parameters and Protection following Vaccination of Sheep with a Larval-Specific Antigen of the Gastrointestinal Nematode, Haemonchus contortus. PLoS ONE 2013, 8, e78357. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Firdaus, F.; Azuar, A.; Khalil, Z.G.; Marasini, N.; Capon, R.J.; Hussein, W.M.; Toth, I.; Skwarczynski, M. Cell-Penetrating Peptides-Based Liposomal Delivery System Enhanced Immunogenicity of Peptide-Based Vaccine against Group A Streptococcus. Vaccines 2021, 9, 499. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.C.; Yang, J.; Hussein, W.M.; Zhao, L.; Wang, X.; Khalil, Z.G.; Capon, R.J.; Toth, I.; Stephenson, R.J. Polyethylenimine: An Intranasal Adjuvant for Liposomal Peptide-Based Subunit Vaccine against Group A Streptococcus. ACS Infect. Dis. 2020, 6, 2502–2512. [Google Scholar] [CrossRef] [PubMed]
- Yostawonkul, J.; Surassmo, S.; Iempridee, T.; Pimtong, W.; Suktham, K.; Sajomsang, W.; Gonil, P.; Ruktanonchai, U.R. Surface modification of nanostructure lipid carrier (NLC) by oleoyl-quaternized-chitosan as a mucoadhesive nanocarrier. Colloids Surf. B Biointerfaces 2017, 149, 301–311. [Google Scholar] [CrossRef]
- Bartlett, S.; Skwarczynski, M.; Toth, I. Lipids as Activators of Innate Immunity in Peptide Vaccine Delivery. Curr. Med. Chem. 2020, 27, 2887–2901. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.L.; Flanagan, K.L.; Prakash, M.D.; Plebanski, M. Malaria vaccines in the eradication era: Current status and future perspectives. Expert Rev. Vaccines 2019, 18, 133–151. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Jin, W.; Cruz, J.G.; Marasini, N.; Khalil, Z.G.; Capon, R.J.; Hussein, W.M.; Skwarczynski, M.; Toth, I. Development of polyelectrolyte complexes for the delivery of peptide-based subunit vaccines against group A streptococcus. Nanomaterials 2020, 10, 823. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Bashiri, S.; Toth, I.; Skwarczynski, M. Preparation of Trimethyl Chitosan-Based Polyelectrolyte Complexes for Peptide Subunit Vaccine Delivery. In Bacterial Vaccines: Methods and Protocols; Bidmos, F., Bossé, J., Langford, P., Eds.; Springer: New York, NY, USA, 2022; pp. 141–149. [Google Scholar]
- Marasini, N.; Giddam, A.K.; Ghaffar, K.A.; Batzloff, M.R.; Good, M.F.; Skwarczynski, M.; Toth, I. Multilayer engineered nanoliposomes as a novel tool for oral delivery of lipopeptide-based vaccines against group A Streptococcus. Nanomedicine 2016, 11, 1223–1236. [Google Scholar] [CrossRef] [PubMed]
- Zaman, M.; Abdel-Aal, A.B.; Phillipps, K.S.; Fujita, Y.; Good, M.F.; Toth, I. Structure-activity relationship of lipopeptide Group A streptococcus (GAS) vaccine candidates on toll-like receptor 2. Vaccine 2010, 28, 2243–2248. [Google Scholar] [CrossRef]
- Tenchov, R.; Bird, R.; Curtze, A.E.; Zhou, Q. Lipid Nanoparticles—From Liposomes to mRNA Vaccine Delivery, a Landscape of Research Diversity and Advancement. ACS Nano 2021, 15, 16982–17015. [Google Scholar] [CrossRef]
- Yang, J.R.; Luo, Y.C.; Shibu, M.A.; Toth, I.; Skwarczynski, M. Cell-Penetrating Peptides: Efficient Vectors for Vaccine Delivery. Curr. Drug Deliv. 2019, 16, 430–443. [Google Scholar] [CrossRef]
- Zhao, L.; Yang, J.; Nahar, U.J.; Khalil, Z.G.; Capon, R.J.; Hussein, W.M.; Skwarczynski, M.; Toth, I. A dual-adjuvanting strategy for peptide-based subunit vaccines against group A Streptococcus: Lipidation and polyelectrolyte complexes. Bioorg. Med. Chem. 2020, 28, 115823. [Google Scholar] [CrossRef]
- Bashiri, S.; Koirala, P.; Toth, I.; Skwarczynski, M. Carbohydrate Immune Adjuvants in Subunit Vaccines. Pharmaceutics 2020, 12, 965. [Google Scholar] [CrossRef]
- Nahar, U.J.; Toth, I.; Skwarczynski, M. Mannose in vaccine delivery. J. Control. Release 2022, 351, 284–300. [Google Scholar] [CrossRef]
- Reintjens, N.R.M.; Tondini, E.; de Jong, A.R.; Meeuwenoord, N.J.; Chiodo, F.; Peterse, E.; Overkleeft, H.S.; Filippov, D.V.; van der Marel, G.A.; Ossendorp, F.; et al. Self-Adjuvanting Cancer Vaccines from Conjugation-Ready Lipid A Analogues and Synthetic Long Peptides. J. Med. Chem. 2020, 63, 11691–11706. [Google Scholar] [CrossRef] [PubMed]
- Norpi, A.S.M.; Nordin, M.L.; Ahmad, N.; Katas, H.; Fuaad, A.A.A.; Sukri, A.; Marasini, N.; Azmi, F. New modular platform based on multi-adjuvanted amphiphilic chitosan nanoparticles for efficient lipopeptide vaccine delivery against group A streptococcus. Asian J. Pharm. Sci. 2022, 17, 435–446. [Google Scholar] [CrossRef]
- Stanisic, D.I.; Ho, M.-F.; Nevagi, R.; Cooper, E.; Walton, M.; Islam, M.T.; Hussein, W.M.; Skwarczynski, M.; Toth, I.; Good, M.F. Development and Evaluation of a Cryopreserved Whole-Parasite Vaccine in a Rodent Model of Blood-Stage Malaria. mBio 2021, 12, e0265721. [Google Scholar] [CrossRef]
- Al-Nazal, H.A.; Cooper, E.; Ho, M.F.; Eskandari, S.; Majam, V.; Giddam, A.K.; Hussein, W.M.; Islam, M.T.; Skwarczynski, M.; Toth, I.; et al. Pre-clinical evaluation of a whole-parasite vaccine to control human babesiosis. Cell Host Microbe 2021, 29, 894–903.e5. [Google Scholar] [CrossRef]
- Ghaffar, K.A.; Marasini, N.; Giddam, A.K.; Batzloff, M.R.; Good, M.F.; Skwarczynski, M.; Toth, I. The Role of Size in Development of Mucosal Liposome-Lipopeptide Vaccine Candidates against Group A Streptococcus. Med. Chem. 2017, 13, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kong, X.; Shi, S.; Zheng, X.; Guo, G.; Wei, Y.; Qian, Z. Preparation of alginate coated chitosan microparticles for vaccine delivery. BMC Biotechnol. 2008, 8, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slütter, B.; Plapied, L.; Fievez, V.; Sande, M.A.; des Rieux, A.; Schneider, Y.-J.; Van Riet, E.; Jiskoot, W.; Préat, V. Mechanistic study of the adjuvant effect of biodegradable nanoparticles in mucosal vaccination. J. Control. Release 2009, 138, 113–121. [Google Scholar] [CrossRef]
- Fernández-Ruiz, M.; Humar, A.; Baluch, A.; Keshwani, S.; Husain, S.; Kumar, D. Baseline serum interleukin-6 to interleukin-2 ratio is associated with the response to seasonal trivalent influenza vaccine in solid organ transplant recipients. Vaccine 2015, 33, 7176–7182. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Wang, J.; Wang, X.; Jin, H.; Zhao, G.; Ding, Z.; Kang, Y.; Wang, B. The effects of IL-6 and TNF-alpha as molecular adjuvants on immune responses to FMDV and maturation of dendritic cells by DNA vaccination. Vaccine 2008, 26, 5111–5122. [Google Scholar] [CrossRef]
- Ovsyannikova, I.G.; Reid, K.C.; Jacobson, R.M.; Oberg, A.L.; Klee, G.G.; Poland, G.A. Cytokine production patterns and antibody response to measles vaccine. Vaccine 2003, 21, 3946–3953. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, N.; Morita, R.; Bourdery, L.; Bentebibel, S.E.; Zurawski, S.M.; Banchereau, J.; Ueno, H. Human dendritic cells induce the differentiation of interleukin-21-producing T follicular helper-like cells through interleukin-12. Immunity 2009, 31, 158–169. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, N.; Bustamante, J.; Bourdery, L.; Bentebibel, S.E.; Boisson-Dupuis, S.; Hamlin, F.; Tran, M.V.; Blankenship, D.; Pascual, V.; Savino, D.A.; et al. IL-12 receptor β1 deficiency alters in vivo T follicular helper cell response in humans. Blood 2013, 121, 3375–3385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassese, G.; Arce, S.; Hauser, A.E.; Lehnert, K.; Moewes, B.; Mostarac, M.; Muehlinghaus, G.; Szyska, M.; Radbruch, A.; Manz, R.A. Plasma cell survival is mediated by synergistic effects of cytokines and adhesion-dependent signals. J. Immunol. 2003, 171, 1684–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plebanski, M.; Elson, C.J.; Billington, W.D. Dependency on interleukin-1 of primary human in vitro T cell responses to soluble antigens. Eur. J. Immunol. 1992, 22, 2353–2358. [Google Scholar] [CrossRef]
- Schurich, A.; Raine, C.; Morris, V.; Ciurtin, C. The role of IL-12/23 in T cell–related chronic inflammation: Implications of immunodeficiency and therapeutic blockade. Rheumatology 2018, 57, 246–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junttila, I.S. Tuning the Cytokine Responses: An Update on Interleukin (IL)-4 and IL-13 Receptor Complexes. Front. Immunol. 2018, 9, 888. [Google Scholar] [CrossRef] [Green Version]
- Ren, K.; Torres, R. Role of interleukin-1beta during pain and inflammation. Brain Res. Rev. 2009, 60, 57–64. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Boer, J.C.; Khongkow, M.; Phunpee, S.; Khalil, Z.G.; Bashiri, S.; Deceneux, C.; Goodchild, G.; Hussein, W.M.; Capon, R.J.; et al. The Development of Surface-Modified Liposomes as an Intranasal Delivery System for Group A Streptococcus Vaccines. Vaccines 2023, 11, 305. https://doi.org/10.3390/vaccines11020305
Yang J, Boer JC, Khongkow M, Phunpee S, Khalil ZG, Bashiri S, Deceneux C, Goodchild G, Hussein WM, Capon RJ, et al. The Development of Surface-Modified Liposomes as an Intranasal Delivery System for Group A Streptococcus Vaccines. Vaccines. 2023; 11(2):305. https://doi.org/10.3390/vaccines11020305
Chicago/Turabian StyleYang, Jieru, Jennifer C. Boer, Mattaka Khongkow, Sarunya Phunpee, Zeinab G. Khalil, Sahra Bashiri, Cyril Deceneux, Georgia Goodchild, Waleed M. Hussein, Robert J. Capon, and et al. 2023. "The Development of Surface-Modified Liposomes as an Intranasal Delivery System for Group A Streptococcus Vaccines" Vaccines 11, no. 2: 305. https://doi.org/10.3390/vaccines11020305