MZe786 Rescues Cardiac Mitochondrial Activity in High sFlt-1 and Low HO-1 Environment

 , , , ,
, , , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Adenovirus Preparation
2.2. Animal Studies
2.3. Drug Preparation
2.4. Animal Experimental Protocol
2.5. Isolation of Mitochondria
2.6. Mitochondrial Respiration Assays
2.7. Quantitative Real-Time PCR
2.8. Statistical Analysis
3. Results
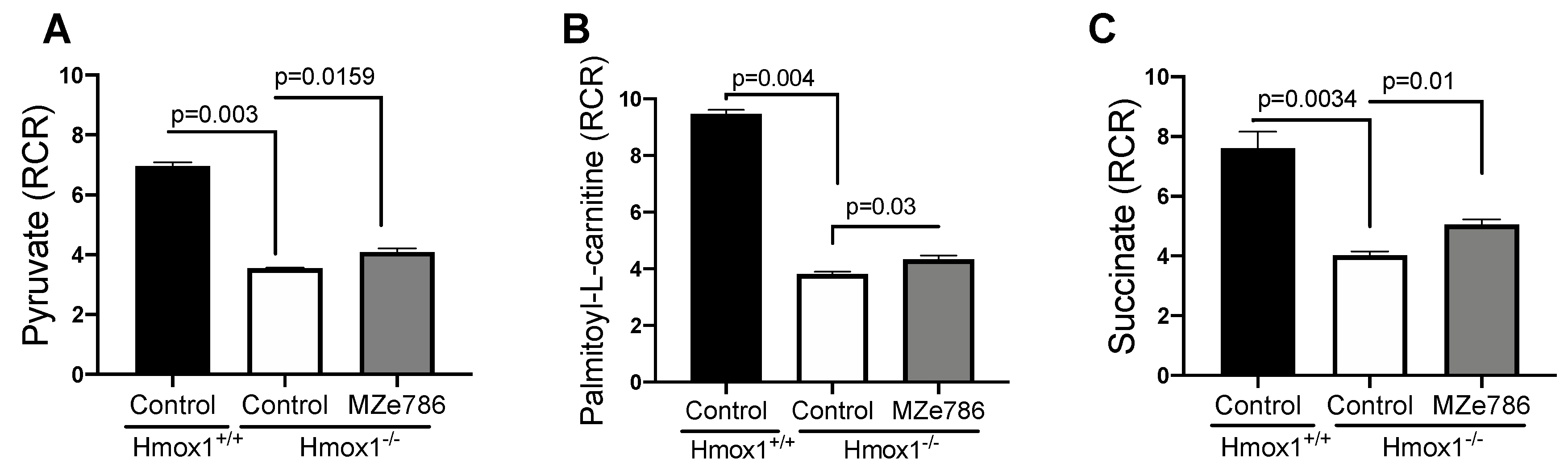
3.1. Loss of HO-1 Disturb the Cardiac Mitochondrial Respiration
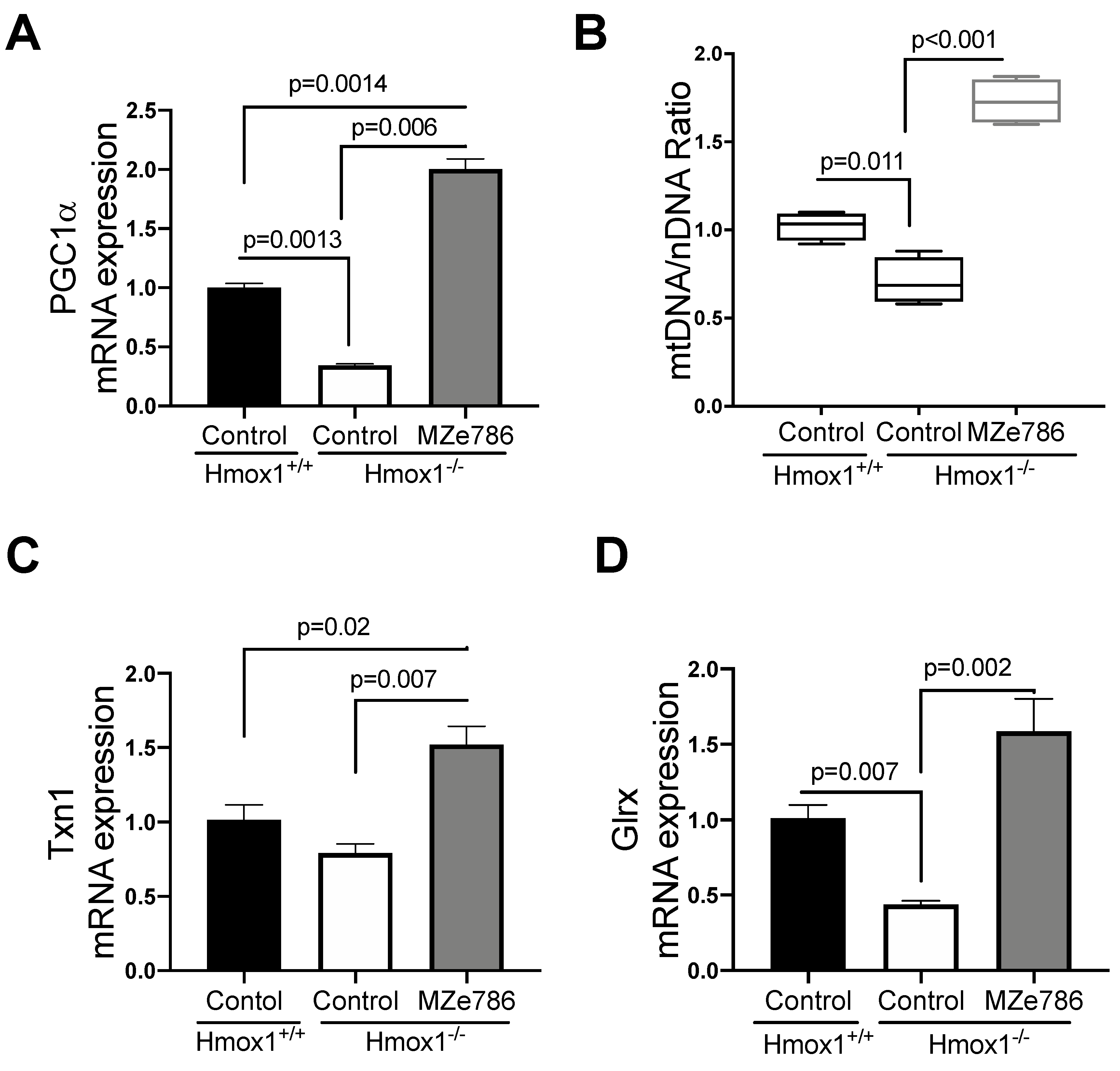
3.2. MZe786 Improves Cardiac Mitochondrial Biogenesis Signal and Stimulates Antioxidant Gene Transcription in Hmox1 Knockout Mice
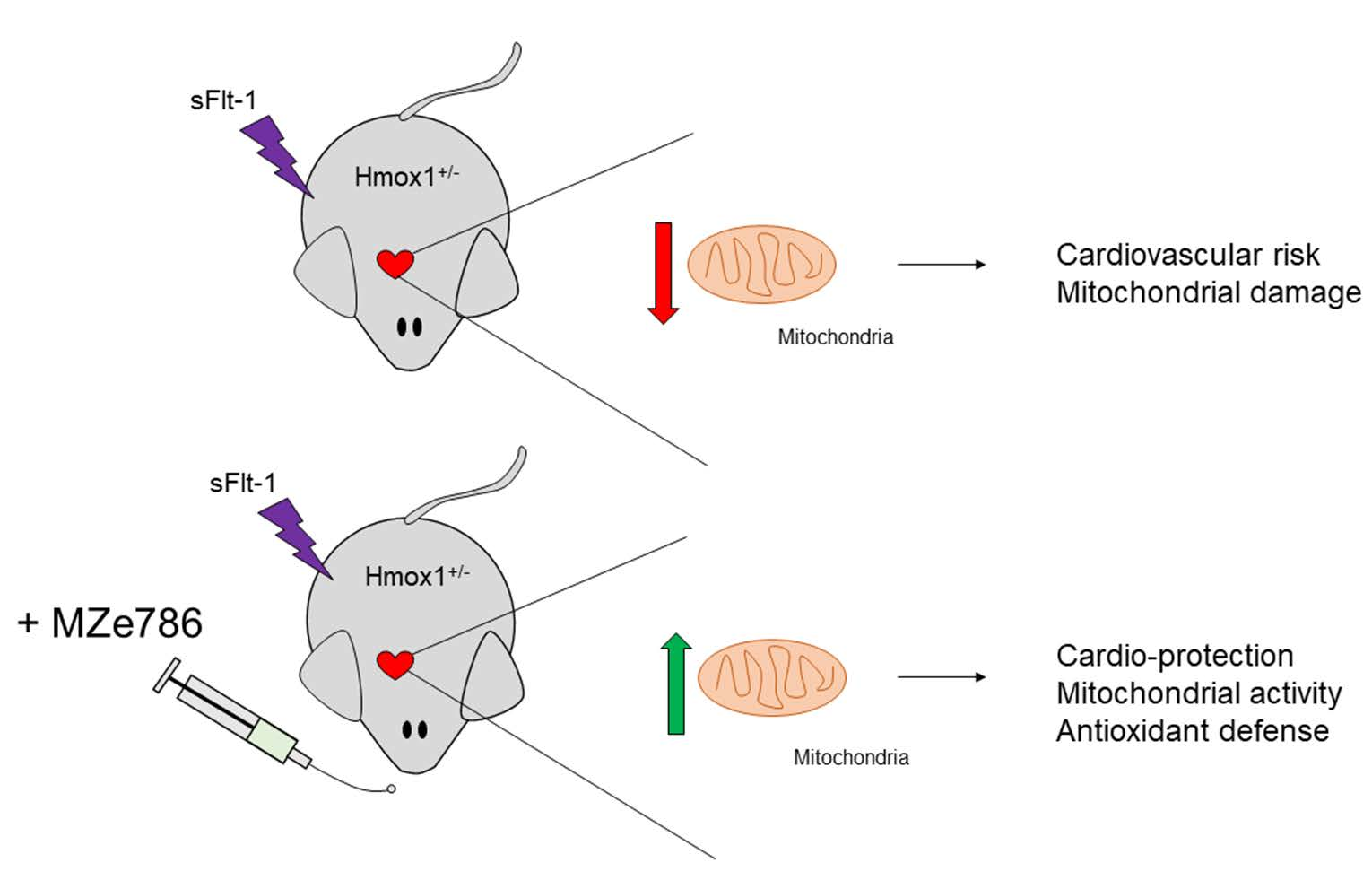
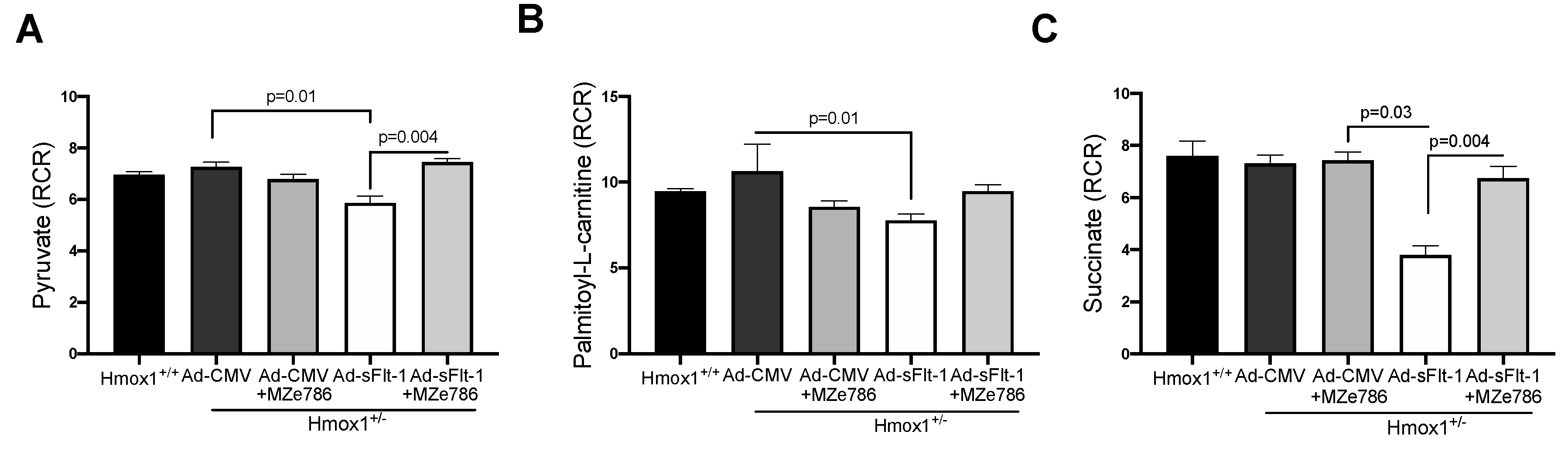
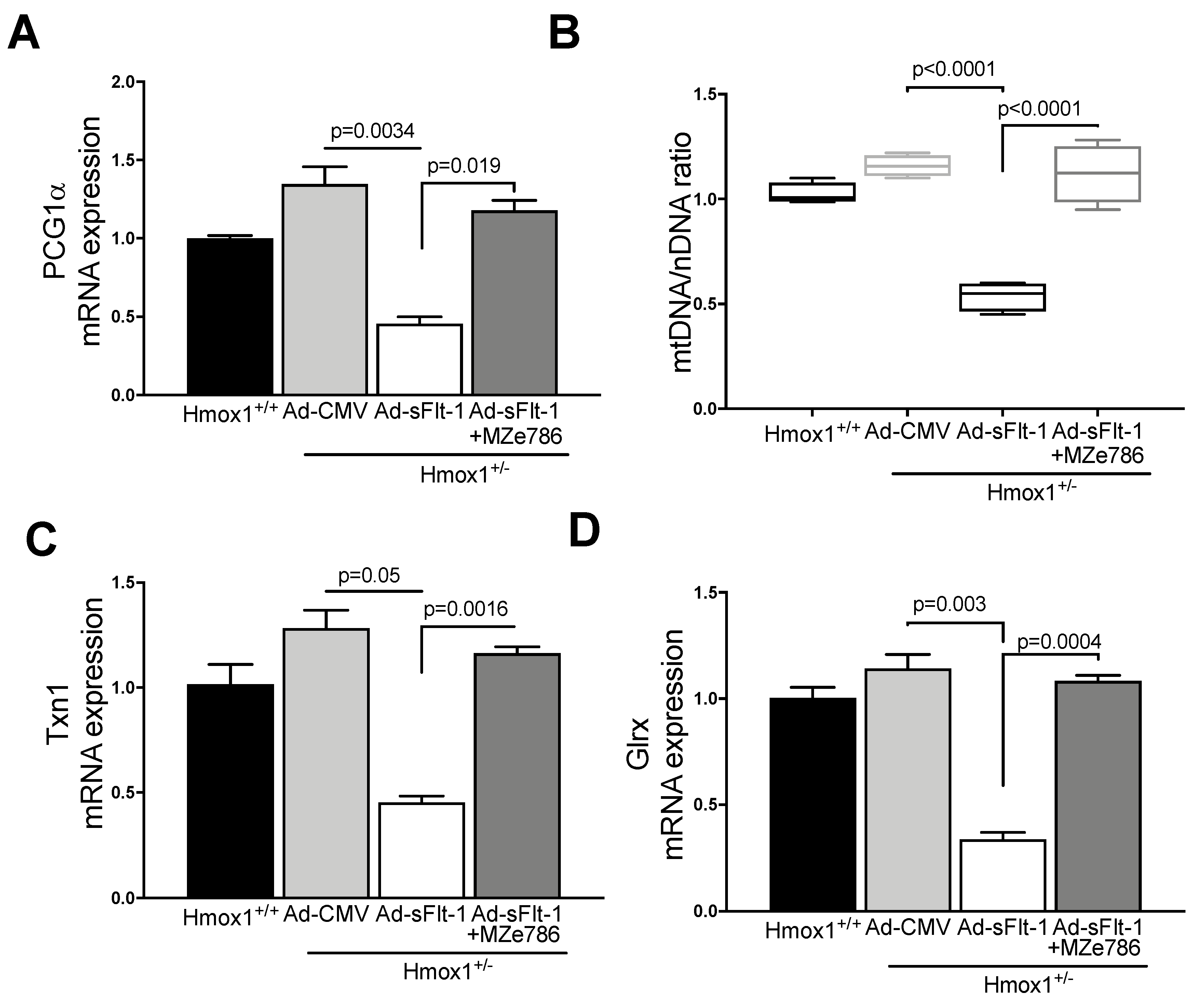
3.3. MZe786 Rescues the sFlt-1-Induced Inhibition of the Cardiac Mitochondrial Activity in Hmox1+/− Mice
3.4. MZe786 Stimulates the Cardiac Mitochondrial Biogenesis and Antioxidant Defence in Hmox1+/− Mice Exposed to High sFlt-1 Environment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schindler, A.E. New data about preeclampsia: Some possibilities of prevention. Gynecol. Endocrinol. 2018, 34, 636–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuklina, E.V.; Ayala, C.; Callaghan, W.M. Hypertensive disorders and severe obstetric morbidity in the United States. Obstet. Gynecol. 2009, 113, 1299–1306. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.L.; Goodwin, T.M.; Pan, V.L.; Ingles, S.A. Molecular epidemiology of preeclampsia. Obstet. Gynecol. Surv. 2003, 58, 39–66. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Tsigas, E.Z.; Callaghan, W.M. Health and economic burden of preeclampsia: No time for complacency. Am. J. Obstet. Gynecol. 2017, 217, 235–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, T.; Peneva, D.; Xu, X.; Sutton, A.; Triche, E.; Ehrenkranz, R.A.; Paidas, M.; Stevens, W. The Rising Burden of Preeclampsia in the United States Impacts Both Maternal and Child Health. Am. J. Perinatol. 2016, 33, 329–338. [Google Scholar] [PubMed]
- Kelly, B.B.; Narula, J.; Fuster, V. Recognizing global burden of cardiovascular disease and related chronic diseases. Mt. Sinai J. Med. 2012, 79, 632–640. [Google Scholar] [CrossRef]
- Trogdon, J.G.; Finkelstein, E.A.; Nwaise, I.A.; Tangka, F.K.; Orenstein, D. The economic burden of chronic cardiovascular disease for major insurers. Health Promot. Pract. 2007, 8, 234–242. [Google Scholar] [CrossRef]
- Finegold, J.A.; Asaria, P.; Francis, D.P. Mortality from ischaemic heart disease by country, region, and age: Statistics from World Health Organisation and United Nations. Int. J. Cardiol. 2013, 168, 934–945. [Google Scholar] [CrossRef] [Green Version]
- Bellamy, L.; Casas, J.P.; Hingorani, A.D.; Williams, D.J. Pre-eclampsia and risk of cardiovascular disease and cancer in later life: Systematic review and meta-analysis. BMJ 2007, 335, 974. [Google Scholar] [CrossRef] [Green Version]
- Honigberg, M.C.; Zekavat, S.M.; Aragam, K.; Klarin, D.; Bhatt, D.L.; Scott, N.S.; Peloso, G.M.; Natarajan, P. Long-Term Cardiovascular Risk in Women with Hypertension During Pregnancy. J. Am. Coll. Cardiol. 2019, 74, 2743–2754. [Google Scholar] [CrossRef]
- Leon, L.J.; McCarthy, F.P.; Direk, K.; Gonzalez-Izquierdo, A.; Prieto-Merino, D.; Casas, J.P.; Chappell, L. Preeclampsia and Cardiovascular Disease in a Large UK Pregnancy Cohort of Linked Electronic Health Records: A CALIBER Study. Circulation 2019, 140, 1050–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craici, I.; Wagner, S.; Garovic, V.D. Preeclampsia and future cardiovascular risk: Formal risk factor or failed stress test? Ther. Adv. Cardiovasc. Dis. 2008, 2, 249–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irgens, H.U.; Reisaeter, L.; Irgens, L.M.; Lie, R.T. Long term mortality of mothers and fathers after pre-eclampsia: Population based cohort study. BMJ 2001, 323, 1213–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, A. Heparin-binding angiogenic growth factors in pregnancy: A review. Placenta 1997, 18, 215–258. [Google Scholar] [CrossRef]
- Levine, R.J.; Maynard, S.E.; Qian, C.; Lim, K.H.; England, L.J.; Yu, K.F.; Schisterman, E.F.; Thadhani, R.; Sachs, D.P.; Epstein, F.H.; et al. Circulating angiogenic factors and the risk of preeclampsia. N. Engl. J. Med. 2004, 350, 672–683. [Google Scholar] [CrossRef] [Green Version]
- Maynard, S.E.; Venkatesha, S.; Thadhani, R.; Karumanchi, S.A. Soluble Fms-like tyrosine kinase 1 and endothelial dysfunction in the pathogenesis of preeclampsia. Pediatr Res. 2005, 57 Pt 2, 1R–7R. [Google Scholar] [CrossRef] [Green Version]
- Cindrova-Davies, T.; Sanders, D.A.; Burton, G.J.; Charnock-Jones, D.S. Soluble FLT1 sensitizes endothelial cells to inflammatory cytokines by antagonizing VEGF receptor-mediated signalling. Cardiovasc. Res. 2011, 89, 671–679. [Google Scholar] [CrossRef] [Green Version]
- Lou, W.Z.; Jiang, F.; Hu, J.; Chen, X.X.; Song, Y.N.; Zhou, X.Y.; Liu, J.; Bian, X.; Gao, J. Maternal Serum Angiogenic Factor sFlt-1 to PlGF Ratio in Preeclampsia: A Useful Marker for Differential Diagnosis and Prognosis Evaluation in Chinese Women. Dis. Markers 2019, 2019, 6270187. [Google Scholar] [CrossRef]
- Lehnen, H.; Mosblech, N.; Reineke, T.; Puchooa, A.; Menke-Mollers, I.; Zechner, U.; Gembruch, U. Prenatal Clinical Assessment of sFlt-1 (Soluble fms-like Tyrosine Kinase-1)/PlGF (Placental Growth Factor) Ratio as a Diagnostic Tool for Preeclampsia, Pregnancy-induced Hypertension, and Proteinuria. Geburtshilfe Frauenheilkd. 2013, 73, 440–445. [Google Scholar] [CrossRef] [Green Version]
- Palmer, K.R.; Kaitu’u-Lino, T.J.; Hastie, R.; Hannan, N.J.; Ye, L.; Binder, N.; Cannon, P.; Tuohey, L.; Johns, T.G.; Shub, A.; et al. Placental-Specific sFLT-1 e15a Protein Is Increased in Preeclampsia, Antagonizes Vascular Endothelial Growth Factor Signaling, and Has Antiangiogenic Activity. Hypertension 2015, 66, 1251–1259. [Google Scholar] [CrossRef]
- Cudmore, M.; Ahmad, S.; Al-Ani, B.; Fujisawa, T.; Coxall, H.; Chudasama, K.; Devey, L.R.; Wigmore, S.J.; Abbas, A.; Hewett, P.W.; et al. Negative regulation of soluble Flt-1 and soluble endoglin release by heme oxygenase-1. Circulation 2007, 115, 1789–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, A.; Rezai, H.; Broadway-Stringer, S. Evidence-Based Revised View of the Pathophysiology of Preeclampsia. Adv. Exp. Med. Biol. 2017, 956, 355–374. [Google Scholar] [PubMed]
- Wikstrom, A.K.; Larsson, A.; Eriksson, U.J.; Nash, P.; Olovsson, M. Early postpartum changes in circulating pro- and anti-angiogenic factors in early-onset and late-onset pre-eclampsia. Acta Obstet. Gynecol. Scand. 2008, 87, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Saleh, L.; Samantar, R.; Garrelds, I.M.; van den Meiracker, A.H.; Visser, W.; Danser, A.H.J. Low Soluble Fms-Like Tyrosine Kinase-1, Endoglin, and Endothelin-1 Levels in Women with Confirmed or Suspected Preeclampsia Using Proton Pump Inhibitors. Hypertension 2017, 70, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Akhter, T.; Wikstrom, A.K.; Larsson, M.; Larsson, A.; Wikstrom, G.; Naessen, T. Association between angiogenic factors and signs of arterial aging in women with pre-eclampsia. Ultrasound Obstet. Gynecol. 2017, 50, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Yet, S.F.; Layne, M.D.; Liu, X.; Chen, Y.H.; Ith, B.; Sibinga, N.E.; Perrella, M.A. Absence of heme oxygenase-1 exacerbates atherosclerotic lesion formation and vascular remodeling. FASEB J. 2003, 17, 1759–1761. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wei, J.; Peng, D.H.; Layne, M.D.; Yet, S.F. Absence of heme oxygenase-1 exacerbates myocardial ischemia/reperfusion injury in diabetic mice. Diabetes 2005, 54, 778–784. [Google Scholar] [CrossRef] [Green Version]
- Yet, S.F.; Tian, R.; Layne, M.D.; Wang, Z.Y.; Maemura, K.; Solovyeva, M.; Ith, B.; Melo, L.G.; Zhang, L.; Ingwall, J.S.; et al. Cardiac-specific expression of heme oxygenase-1 protects against ischemia and reperfusion injury in transgenic mice. Circ. Res. 2001, 89, 168–173. [Google Scholar] [CrossRef] [Green Version]
- Hull, T.D.; Boddu, R.; Guo, L.; Tisher, C.C.; Traylor, A.M.; Patel, B.; Joseph, R.; Prabhu, S.D.; Suliman, H.D.; Piantadosi, C.A.; et al. Heme oxygenase-1 regulates mitochondrial quality control in the heart. JCI Insight 2016, 1, e85817. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.W.; Moore, P.K. H2S Synthesizing Enzymes: Biochemistry and Molecular Aspects. Handb. Exp. Pharmacol. 2015, 230, 3–25. [Google Scholar]
- Suzuki, K.; Olah, G.; Modis, K.; Coletta, C.; Kulp, G.; Gero, D.; Szoleczky, P.; Chang, T.; Zhou, Z.; Wu, L.; et al. Hydrogen sulfide replacement therapy protects the vascular endothelium in hyperglycemia by preserving mitochondrial function. Proc. Natl. Acad. Sci. USA 2011, 108, 13829–13834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yong, Q.C.; Choo, C.H.; Tan, B.H.; Low, C.M.; Bian, J.S. Effect of hydrogen sulfide on intracellular calcium homeostasis in neuronal cells. Neurochem. Int. 2010, 56, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Cheung, S.H.; Lau, J.Y.W. Hydrogen sulfide mediates athero-protection against oxidative stress via S-sulfhydration. PLoS ONE 2018, 13, e0194176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, M.; Zhuo, C.; Jiang, R.; Chen, G.; Shan, J.; Ping, J.; Tian, H.; Wang, L.; Lin, C.; Hu, L. Protective effect of hydrogen sulphide against myocardial hypertrophy in mice. Oncotarget 2017, 8, 22344–22352. [Google Scholar] [CrossRef]
- Zhao, W.; Zhang, J.; Lu, Y.; Wang, R. The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 2001, 20, 6008–6016. [Google Scholar] [CrossRef] [Green Version]
- Zanardo, R.C.; Brancaleone, V.; Distrutti, E.; Fiorucci, S.; Cirino, G.; Wallace, J.L. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006, 20, 2118–2120. [Google Scholar] [CrossRef]
- Elrod, J.W.; Calvert, J.W.; Morrison, J.; Doeller, J.E.; Kraus, D.W.; Tao, L.; Jiao, X.; Scalia, R.; Kiss, L.; Szabo, C.; et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. USA 2007, 104, 15560–15565. [Google Scholar] [CrossRef] [Green Version]
- Papapetropoulos, A.; Pyriochou, A.; Altaany, Z.; Yang, G.; Marazioti, A.; Zhou, Z.; Jeschke, M.G.; Branski, L.K.; Herndon, D.N.; Wang, R.; et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 21972–21977. [Google Scholar] [CrossRef] [Green Version]
- Giuffre, A.; Vicente, J.B. Hydrogen Sulfide Biochemistry and Interplay with Other Gaseous Mediators in Mammalian Physiology. Oxid. Med. Cell Longev. 2018, 2018, 6290931. [Google Scholar] [CrossRef]
- D’Araio, E.; Shaw, N.; Millward, A.; Demaine, A.; Whiteman, M.; Hodgkinson, A. Hydrogen sulfide induces heme oxygenase-1 in human kidney cells. Acta Diabetol. 2014, 51, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Y.; Li, X.H.; Zhang, T.; Fu, J.; Cui, X.D. Hydrogen sulfide upregulates heme oxygenase-1 expression in rats with volume overload-induced heart failure. Biomed. Rep. 2013, 1, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Sparatore, A.; Perrino, E.; Tazzari, V.; Giustarini, D.; Rossi, R.; Rossoni, G.; Erdmann, K.; Schröder, H.; Del Soldato, P. Pharmacological profile of a novel H(2)S-releasing aspirin. Free Radic. Biol. Med. 2009, 46, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Rossoni, G.; Manfredi, B.; Tazzari, V.; Sparatore, A.; Trivulzio, S.; Del Soldato, P.; Berti, F. Activity of a new hydrogen sulfide-releasing aspirin (ACS14) on pathological cardiovascular alterations induced by glutathione depletion in rats. Eur. J. Pharmacol. 2010, 648, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Giustarini, D.; Del Soldato, P.; Sparatore, A.; Rossi, R. Modulation of thiol homeostasis induced by H2S-releasing aspirin. Free Radic. Biol. Med. 2010, 48, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Aranguren, L.C.; Espinosa-Gonzalez, C.T.; Gonzalez-Ortiz, L.M.; Sanabria-Barrera, S.M.; Riano-Medina, C.E.; Nunez, A.F.; Ahmed, A.; Vasquez-Vivar, J.; López, M. Soluble Fms-Like Tyrosine Kinase-1 Alters Cellular Metabolism and Mitochondrial Bioenergetics in Preeclampsia. Front. Physiol. 2018, 9, 83. [Google Scholar] [CrossRef] [Green Version]
- Ky, B.; French, B.; Ruparel, K.; Sweitzer, N.K.; Fang, J.C.; Levy, W.C.; Sawyer, D.B.; Cappola, T.P. The vascular marker soluble fms-like tyrosine kinase 1 is associated with disease severity and adverse outcomes in chronic heart failure. J. Am. Coll. Cardiol. 2011, 58, 386–394. [Google Scholar] [CrossRef] [Green Version]
- Gruson, D.; Hermans, M.P.; Ferracin, B.; Ahn, S.A.; Rousseau, M.F. Sflt-1 in heart failure: Relation with disease severity and biomarkers. Scand. J. Clin. Lab. Investig. 2016, 76, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Sakamuri, S.; Sperling, J.A.; Sure, V.N.; Dholakia, M.H.; Peterson, N.R.; Rutkai, I.; Mahalingam, P.S.; Satou, R.; Katakam, P.V.G. Measurement of respiratory function in isolated cardiac mitochondria using Seahorse XFe24 Analyzer: Applications for aging research. Geroscience 2018, 40, 347–356. [Google Scholar] [CrossRef]
- Boutagy, N.E.; Rogers, G.W.; Pyne, E.S.; Ali, M.M.; Hulver, M.W.; Frisard, M.I. Using Isolated Mitochondria from Minimal Quantities of Mouse Skeletal Muscle for High throughput Microplate Respiratory Measurements. J. Vis. Exp. 2015, 104, e53216. [Google Scholar] [CrossRef] [Green Version]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef] [Green Version]
- Quiros, P.M.; Goyal, A.; Jha, P.; Auwerx, J. Analysis of mtDNA/nDNA Ratio in Mice. Curr. Protoc. Mouse Biol. 2017, 7, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siasos, G.; Tsigkou, V.; Kosmopoulos, M.; Theodosiadis, D.; Simantiris, S.; Tagkou, N.M.; Tsimpiktsioglou, A.; Stampouloglou, P.K.; Oikonomou, E.; Mourouzis, K.; et al. Mitochondria and cardiovascular diseases-from pathophysiology to treatment. Ann. Transl. Med. 2018, 6, 256. [Google Scholar]
- Arany, Z.; He, H.; Lin, J.; Hoyer, K.; Handschin, C.; Toka, O.; Ahmad, F.; Matsui, T.; Chin, S.; Wu, H.; et al. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005, 1, 259–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riehle, C.; Abel, E.D. PGC-1 proteins and heart failure. Trends Cardiovasc. Med. 2012, 22, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Xu, X.; Hu, X.; Fassett, J.; Zhu, G.; Tao, Y.; Li, J.; Huang, Y.; Zhang, P.; Zhao, B.; et al. PGC-1 alpha regulates expression of myocardial mitochondrial antioxidants and myocardial oxidative stress after chronic systolic overload. Antioxid. Redox Signal. 2010, 13, 1011–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Azuma, J.; Kalish, F.; Wong, R.J.; Stevenson, D.K. Maternal heme oxygenase 1 regulates placental vasculature development via angiogenic factors in mice. Biol. Reprod. 2011, 85, 1005–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushnell, C.; McCullough, L.D.; Awad, I.A.; Chireau, M.V.; Fedder, W.N.; Furie, K.L.; Howard, V.J.; Lichtman, J.H.; Lisabeth, L.D.; Piña, I.L.; et al. Guidelines for the prevention of stroke in women: A statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2014, 45, 1545–1588. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.C.; Best, K.E.; Pearce, M.S.; Waugh, J.; Robson, S.C.; Bell, R. Cardiovascular disease risk in women with pre-eclampsia: Systematic review and meta-analysis. Eur. J. Epidemiol. 2013, 28, 1–19. [Google Scholar] [CrossRef]
- Lykke, J.A.; Langhoff-Roos, J.; Sibai, B.M.; Funai, E.F.; Triche, E.W.; Paidas, M.J. Hypertensive pregnancy disorders and subsequent cardiovascular morbidity and type 2 diabetes mellitus in the mother. Hypertension 2009, 53, 944–951. [Google Scholar] [CrossRef]
- Wu, P.; Haththotuwa, R.; Kwok, C.S.; Babu, A.; Kotronias, R.A.; Rushton, C. Preeclampsia and Future Cardiovascular Health: A Systematic Review and Meta-Analysis. Circ. Cardiovasc. Qual. Outcomes 2017, 10, e003497. [Google Scholar] [CrossRef]
- McDonald, S.D.; Malinowski, A.; Zhou, Q.; Yusuf, S.; Devereaux, P.J. Cardiovascular sequelae of preeclampsia/eclampsia: A systematic review and meta-analyses. Am. Heart, J. 2008, 156, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Rahman, M.; Zhang, X.; Acevedo, C.H.; Nijjar, S.; Rushton, I.; Bussolati, B.; St. John, J. Induction of placental heme oxygenase-1 is protective against TNFalpha-induced cytotoxicity and promotes vessel relaxation. Mol. Med. 2000, 6, 391–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenclussen, M.L.; Linzke, N.; Schumacher, A.; Fest, S.; Meyer, N.; Casalis, P.A. Heme oxygenase-1 is critically involved in placentation, spiral artery remodeling, and blood pressure regulation during murine pregnancy. Front. Pharmacol. 2014, 5, 291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaartokallio, T.; Utge, S.; Klemetti, M.M.; Paananen, J.; Pulkki, K.; Romppanen, J. Fetal Microsatellite in the Heme Oxygenase 1 Promoter Is Associated With Severe and Early-Onset Preeclampsia. Hypertension 2018, 71, 95–102. [Google Scholar] [CrossRef]
- Lancel, S.; Hassoun, S.M.; Favory, R.; Decoster, B.; Motterlini, R.; Neviere, R. Carbon monoxide rescues mice from lethal sepsis by supporting mitochondrial energetic metabolism and activating mitochondrial biogenesis. J. Pharmacol. Exp. Ther. 2009, 329, 641–648. [Google Scholar] [CrossRef] [Green Version]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef]
- Wisneski, J.A.; Gertz, E.W.; Neese, R.A.; Gruenke, L.D.; Morris, D.L.; Craig, J.C. Metabolic fate of extracted glucose in normal human myocardium. J. Clin. Investig. 1985, 76, 1819–1827. [Google Scholar] [CrossRef]
- McCommis, K.S.; Finck, B.N. Mitochondrial pyruvate transport: A historical perspective and future research directions. Biochem. J. 2015, 466, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Fillmore, N.; Mori, J.; Lopaschuk, G.D. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br. J. Pharmacol. 2014, 171, 2080–2090. [Google Scholar] [CrossRef] [Green Version]
- Pfleger, J.; He, M.; Abdellatif, M. Mitochondrial complex II is a source of the reserve respiratory capacity that is regulated by metabolic sensors and promotes cell survival. Cell Death Dis. 2015, 6, e1835. [Google Scholar] [CrossRef] [Green Version]
- Maynard, S.E.; Min, J.Y.; Merchan, J.; Lim, K.H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Investig. 2003, 111, 649–658. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Zou, Y.; Ge, Z.; Zuo, Q.; Huang, S.Y.; Sun, L. A Role of sFlt-1 in Oxidative Stress and Apoptosis in Human and Mouse Pre-Eclamptic Trophoblasts. Biol. Reprod. 2015, 93, 73. [Google Scholar] [CrossRef]
- Nandi, S.; Ravindran, S.; Kurian, G.A. Role of endogenous hydrogen sulfide in cardiac mitochondrial preservation during ischemia reperfusion injury. Biomed. Pharmacother. 2018, 97, 271–279. [Google Scholar] [CrossRef]
- Shimizu, Y.; Polavarapu, R.; Eskla, K.L.; Nicholson, C.K.; Koczor, C.A.; Wang, R.; Lewis, W.; Shiva, S.; Lefer, D.J.; Calvert, J.W. Hydrogen sulfide regulates cardiac mitochondrial biogenesis via the activation of AMPK. J. Mol. Cell Cardiol. 2018, 116, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Shen, Z.; Luo, S.; Guo, W.; Zhu, Y.Z. The Cardioprotective Effects of Hydrogen Sulfide in Heart Diseases: From Molecular Mechanisms to Therapeutic Potential. Oxid. Med. Cell Longev. 2015, 2015, 925167. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Kan, J.T.; Cheng, Z.Y.; Chen, J.F.; Shen, Y.Q.; Xu, J.; Wu, D.; Zhu, Y. Hydrogen sulfide as an endogenous modulator in mitochondria and mitochondria dysfunction. Oxid. Med. Cell Longev. 2012, 2012, 878052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Gu, Y.; Wen, M.; Zhao, S.; Wang, W.; Ma, Y.; Meng, G.; Han, Y.; Wang, Y.; Liu, G.; et al. Hydrogen Sulfide Induces Keap1 S-sulfhydration and Suppresses Diabetes-Accelerated Atherosclerosis via Nrf2 Activation. Diabetes 2016, 65, 3171–3184. [Google Scholar] [CrossRef] [Green Version]
- Shin, I.; Hong, J.; Jeon, C.M.; Shin, N.R.; Kwon, O.K.; Kim, H.S.; Kim, J.C.; Oh, S.R.; Ahn, K.S. Diallyl-disulfide, an organosulfur compound of garlic, attenuates airway inflammation via activation of the Nrf-2/HO-1 pathway and NF-kappaB suppression. Food Chem. Toxicol. 2013, 62, 506–513. [Google Scholar] [CrossRef]
- Wang, R.; Szabo, C.; Ichinose, F.; Ahmed, A.; Whiteman, M.; Papapetropoulos, A. The role of H2S bioavailability in endothelial dysfunction. Trends Pharmacol. Sci. 2015, 36, 568–578. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Ahmad, S.; Cai, M.; Rennie, J.; Fujisawa, T.; Crispi, F.; Baily, J.; Miller, M.R.; Cudmore, M.; Hadoke, P.W.F.; et al. Dysregulation of hydrogen sulfide producing enzyme cystathionine gamma-lyase contributes to maternal hypertension and placental abnormalities in preeclampsia. Circulation 2013, 127, 2514–2522. [Google Scholar] [CrossRef] [Green Version]
- Lu, F.; Xing, J.; Zhang, X.; Dong, S.; Zhao, Y.; Wang, L.; Li, H.; Yang, F.; Xu, C.; Zhang, W. Exogenous hydrogen sulfide prevents cardiomyocyte apoptosis from cardiac hypertrophy induced by isoproterenol. Mol. Cell Biochem. 2013, 381, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Karwi, Q.G.; Bornbaum, J.; Boengler, K.; Torregrossa, R.; Whiteman, M.; Wood, M.E.; Schulz, R.; Baxter, G.F. AP39, a mitochondria-targeting hydrogen sulfide (H2 S) donor, protects against myocardial reperfusion injury independently of salvage kinase signalling. Br. J. Pharmacol. 2017, 174, 287–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holwerda, K.M.; Burke, S.D.; Faas, M.M.; Zsengeller, Z.; Stillman, I.E.; Kang, P.M.; van Goor, H.; McCurley, A.; Jaffe, I.Z.; Karumanchi, S.A.; et al. Hydrogen sulfide attenuates sFlt1-induced hypertension and renal damage by upregulating vascular endothelial growth factor. J. Am. Soc. Nephrol. 2014, 25, 717–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bos, E.M.; van Goor, H.; Joles, J.A.; Whiteman, M.; Leuvenink, H.G. Hydrogen sulfide: Physiological properties and therapeutic potential in ischaemia. Br. J. Pharmacol. 2015, 172, 1479–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, C.K.; Lambert, J.P.; Molkentin, J.D.; Sadoshima, J.; Calvert, J.W. Thioredoxin 1 is essential for sodium sulfide-mediated cardioprotection in the setting of heart failure. Arterioscler Arterioscler. Thromb. Vasc. Biol. 2013, 33, 744–751. [Google Scholar] [CrossRef] [Green Version]
- Matsui, R.; Ferran, B.; Oh, A.; Croteau, D.; Shao, D.; Han, J.; Pimentel, D.R.; Bachschmid, M.M. Redox Regulation via Glutaredoxin-1 and Protein S-Glutathionylation. Antioxid Redox Signal. 2020, 32, 677–700. [Google Scholar] [CrossRef]
- Gallogly, M.M.; Shelton, M.D.; Qanungo, S.; Pai, H.V.; Starke, D.W.; Hoppel, C.L.; Lesnefsky, E.J.; Mieyal, J.J. Glutaredoxin regulates apoptosis in cardiomyocytes via NFkappaB targets Bcl-2 and Bcl-xL: Implications for cardiac aging. Antioxid Redox Signal. 2010, 12, 1339–1353. [Google Scholar] [CrossRef] [Green Version]
- Pai, H.V.; Starke, D.W.; Lesnefsky, E.J.; Hoppel, C.L.; Mieyal, J.J. What is the functional significance of the unique location of glutaredoxin 1 (GRx1) in the intermembrane space of mitochondria? Antioxid Redox Signal. 2007, 9, 2027–2033. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanchez-Aranguren, L.C.; Rezai, H.; Ahmad, S.; Alzahrani, F.A.; Sparatore, A.; Wang, K.; Ahmed, A. MZe786 Rescues Cardiac Mitochondrial Activity in High sFlt-1 and Low HO-1 Environment. Antioxidants 2020, 9, 598. https://doi.org/10.3390/antiox9070598
Sanchez-Aranguren LC, Rezai H, Ahmad S, Alzahrani FA, Sparatore A, Wang K, Ahmed A. MZe786 Rescues Cardiac Mitochondrial Activity in High sFlt-1 and Low HO-1 Environment. Antioxidants. 2020; 9(7):598. https://doi.org/10.3390/antiox9070598
Chicago/Turabian StyleSanchez-Aranguren, Lissette Carolina, Homira Rezai, Shakil Ahmad, Faisal A. Alzahrani, Anna Sparatore, Keqing Wang, and Asif Ahmed. 2020. "MZe786 Rescues Cardiac Mitochondrial Activity in High sFlt-1 and Low HO-1 Environment" Antioxidants 9, no. 7: 598. https://doi.org/10.3390/antiox9070598