NoxO1 Knockout Promotes Longevity in Mice


Abstract
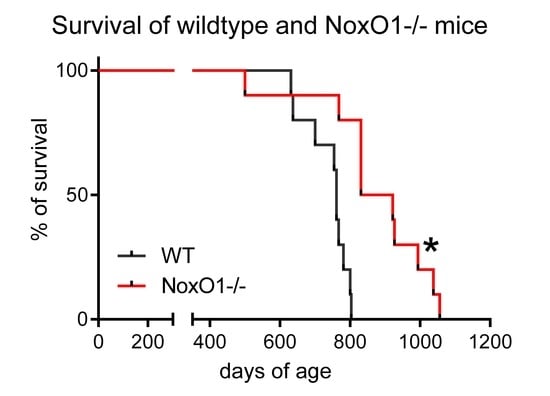
:
1. Introduction
2. Materials and Methods
2.1. Animal Studies
2.2. Organ Chamber Experiments
2.3. Real-Time PCR
2.4. DNA-damage Detection (Comet-Assay)
2.5. Amplex Red Assay for H2O2 Formation
2.6. Statistics
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Maser, R.S.; DePinho, R.A. Connecting chromosomes, crisis, and cancer. Science 2002, 297, 565–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass, D.; Viñuela, A.; Davies, M.N.; Ramasamy, A.; Parts, L.; Knowles, D.; Brown, A.A.; Hedman, Å.K.; Small, K.S.; Buil, A.; et al. Gene expression changes with age in skin, adipose tissue, blood and brain. Genome Biol. 2013, 14, R75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Schröder, K. NADPH oxidase-derived reactive oxygen species: Dosis facit venenum. Exp. Physiol. 2019, 104, 447–452. [Google Scholar] [CrossRef] [Green Version]
- Hahner, F.; Moll, F.; Schröder, K. NADPH Oxidases in the differentiation of endothelial cells. Cardiovasc. Res. 2019. [Google Scholar] [CrossRef] [Green Version]
- Schröder, K.; Weissmann, N.; Brandes, R.P. Organizers and activators: Cytosolic Nox proteins impacting on vascular function. Free Radic. Biol. Med. 2017, 109, 22–32. [Google Scholar] [CrossRef]
- Rezende, F.; Moll, F.; Walter, M.; Helfinger, V.; Hahner, F.; Janetzko, P.; Ringel, C.; Weigert, A.; Fleming, I.; Weissmann, N.; et al. The NADPH organizers NoxO1 and p47phox are both mediators of diabetes-induced vascular dysfunction in mice. Redox Biol. 2018, 15, 12–21. [Google Scholar] [CrossRef]
- Moll, F.; Walter, M.; Rezende, F.; Helfinger, V.; Vasconez, E.; de Oliveira, T.; Greten, F.R.; Olesch, C.; Weigert, A.; Radeke, H.H.; et al. NoxO1 Controls Proliferation of Colon Epithelial Cells. Front. Immunol. 2018, 9, 973. [Google Scholar] [CrossRef] [Green Version]
- Schwerd, T.; Bryant, R.V.; Pandey, S.; Capitani, M.; Meran, L.; Cazier, J.-B.; Jung, J.; Mondal, K.; Parkes, M.; Mathew, C.G.; et al. NOX1 loss-of-function genetic variants in patients with inflammatory bowel disease. Mucosal. Immunol. 2018, 11, 562–574. [Google Scholar] [CrossRef]
- Khoshnevisan, R.; Anderson, M.; Babcock, S.; Anderson, S.; Illig, D.; Marquardt, B.; Sherkat, R.; Schröder, K.; Moll, F.; Hollizeck, S.; et al. NOX1 Regulates Collective and Planktonic Cell Migration: Insights From Patients With Pediatric-Onset IBD and NOX1 Deficiency. Inflamm. Bowel Dis. 2020. [Google Scholar] [CrossRef]
- Brandes, R.P.; Harenkamp, S.; Schürmann, C.; Josipovic, I.; Rashid, B.; Rezende, F.; Löwe, O.; Moll, F.; Epah, J.; Eresch, J.; et al. The Cytosolic NADPH Oxidase Subunit NoxO1 Promotes an Endothelial Stalk Cell Phenotype. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1558–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiss, P.J.; Knisz, J.; Zhang, Y.; Baltrusaitis, J.; Sigmund, C.D.; Thalmann, R.; Smith, R.J.H.; Verpy, E.; Bánfi, B. Inactivation of NADPH oxidase organizer 1 results in severe imbalance. Curr. Biol. 2006, 16, 208–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benkhoff, S.; Loot, A.E.; Pierson, I.; Sturza, A.; Kohlstedt, K.; Fleming, I.; Shimokawa, H.; Grisk, O.; Brandes, R.P.; Schröder, K. Leptin potentiates endothelium-dependent relaxation by inducing endothelial expression of neuronal NO synthase. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helfinger, V.; von Gall, F.F.; Henke, N.; Kunze, M.M.; Schmid, T.; Heidler, J.; Wittig, I.; Radeke, H.H.; Marschall, V.; Anderson, K.; et al. Hydrogen peroxide formation by Nox4 limits malignant transformation. bioRxiv 2017. [Google Scholar] [CrossRef] [Green Version]
- de Magalhaes, J.P.; Costa, J.; Church, G.M. An analysis of the relationship between metabolism, developmental schedules, and longevity using phylogenetic independent contrasts. J. Gerontol. A Biol. Sci. Med. Sci. 2007, 62, 149–160. [Google Scholar] [CrossRef]
- Zullo, A.; Simone, E.; Grimaldi, M.; Musto, V.; Mancini, F.P. Sirtuins as Mediator of the Anti-Ageing Effects of Calorie Restriction in Skeletal and Cardiac Muscle. Int. J. Mol. Sci. 2018, 19, 928. [Google Scholar] [CrossRef] [Green Version]
- Xia, N.; Strand, S.; Schlufter, F.; Siuda, D.; Reifenberg, G.; Kleinert, H.; Förstermann, U.; Li, H. Role of SIRT1 and FOXO factors in eNOS transcriptional activation by resveratrol. Nitric Oxide 2013, 32, 29–35. [Google Scholar] [CrossRef]
- Villa, F.; Carrizzo, A.; Spinelli, C.C.; Ferrario, A.; Malovini, A.; Maciąg, A.; Damato, A.; Auricchio, A.; Spinetti, G.; Sangalli, E.; et al. Genetic Analysis Reveals a Longevity-Associated Protein Modulating Endothelial Function and Angiogenesis. Circ. Res. 2015, 117, 333–345. [Google Scholar] [CrossRef] [Green Version]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef]
- Syeda, A.H.; Hawkins, M.; McGlynn, P. Recombination and replication. Cold Spring Harb. Perspect. Biol. 2014, 6, a016550. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Shamanna, R.A.; Keijzers, G.; Anand, R.; Rasmussen, L.J.; Cejka, P.; Croteau, D.L.; Bohr, V.A. RECQL4 Promotes DNA End Resection in Repair of DNA Double-Strand Breaks. Cell Rep. 2016, 16, 161–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungvari, Z.; Parrado-Fernandez, C.; Csiszar, A.; de Cabo, R. Mechanisms Underlying Caloric Restriction and Lifespan Regulation. Circ. Res. 2008, 102, 519–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, H.Y.; Miller, C.; Bitterman, K.J.; Wall, N.R.; Hekking, B.; Kessler, B.; Howitz, K.T.; Gorospe, M.; de Cabo, R.; Sinclair, D.A. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 2004, 305, 390–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattison, J.A.; Colman, R.J.; Beasley, T.M.; Allison, D.B.; Kemnitz, J.W.; Roth, G.S.; Ingram, D.K.; Weindruch, R.; de Cabo, R.; Anderson, R.M. Caloric restriction improves health and survival of rhesus monkeys. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Berrington de Gonzalez, A.; Hartge, P.; Cerhan, J.R.; Flint, A.J.; Hannan, L.; MacInnis, R.J.; Moore, S.C.; Tobias, G.S.; Anton-Culver, H.; Freeman, L.B.; et al. Body-mass index and mortality among 1.46 million white adults. N. Engl. J. Med. 2010, 363, 2211–2219. [Google Scholar] [CrossRef] [Green Version]
- Flegal, K.M.; Kit, B.K.; Orpana, H.; Graubard, B.I. Association of all-cause mortality with overweight and obesity using standard body mass index categories: A systematic review and meta-analysis. JAMA 2013, 309, 71–82. [Google Scholar] [CrossRef] [Green Version]
- Headey, B.; Yong, J. Happiness and Longevity: Unhappy People Die Young, Otherwise Happiness Probably Makes No Difference. Soc. Indic. Res. 2019, 142, 713–732. [Google Scholar] [CrossRef]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [Green Version]
- Rezende, F.; Schürmann, C.; Schütz, S.; Harenkamp, S.; Herrmann, E.; Seimetz, M.; Weißmann, N.; Schröder, K. Knock out of the NADPH oxidase Nox4 has no impact on life span in mice. Redox Biol. 2017, 11, 312–314. [Google Scholar] [CrossRef]
- Guo, Z.; Yang, H.; Hamilton, M.L.; VanRemmen, H.; Richardson, A. Effects of age and food restriction on oxidative DNA damage and antioxidant enzyme activities in the mouse aorta. Mech. Ageing Dev. 2001, 122, 1771–1786. [Google Scholar] [CrossRef]
- Stirling, P.C.; Chan, Y.A.; Minaker, S.W.; Aristizabal, M.J.; Barrett, I.; Sipahimalani, P.; Kobor, M.S.; Hieter, P. R-loop-mediated genome instability in mRNA cleavage and polyadenylation mutants. Genes Dev. 2012, 26, 163–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, R.W.; Setlow, R.B. Correlation between deoxyribonucleic acid excision-repair and life-span in a number of mammalian species. Proc. Natl. Acad. Sci. USA 1974, 71, 2169–2173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeijmakers, J.H.J. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- di Fagagna, F.D.A. Living on a break: Cellular senescence as a DNA-damage response. Nat. Rev. Cancer 2008, 8, 512–522. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene. | Forward/Reverse | 5′-->3′ |
|---|---|---|
| mOGG1 | fw | GGAGCTGGAAACCCTACACAA |
| rev | GGGTCTTGTCTCAGCAGTCT | |
| mLig1 | fw | AGAGCTGGGTGTTGGTGATG |
| rev | TCCCCCTTCTCAGCTACCTC | |
| mPARP1 | fw | GCGGAGAAGACATTGGGTGA |
| rev | ACCATCTTCTTGGACAGGCG | |
| mRAD50 | fw | TCCCTCCTGGAACCAAAGGA |
| rev | TCGAAACTGCAGGCGAATCT | |
| mMSH2 | fw | GGCCCAGGATGCCATTGTTA |
| rev | AAGTGAGCCAGCACATCGTT | |
| mSIRT1 | fw | ATGCTGGCCTAATAGACTTG |
| rev | AGCACCGTGGAATATGTAAC | |
| mPGC1α | fw | ACAGCTTTCTGGGTGGATTG |
| rev | TGTCTCTGTGAGAACCGCTA | |
| hmr-EF2 | fw | GACATCACCAAGGGTGTGCAG |
| rev | GCGGTCAGCACACTGGCATA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schader, T.; Reschke, C.; Spaeth, M.; Wienstroer, S.; Wong, S.; Schröder, K. NoxO1 Knockout Promotes Longevity in Mice. Antioxidants 2020, 9, 226. https://doi.org/10.3390/antiox9030226
Schader T, Reschke C, Spaeth M, Wienstroer S, Wong S, Schröder K. NoxO1 Knockout Promotes Longevity in Mice. Antioxidants. 2020; 9(3):226. https://doi.org/10.3390/antiox9030226
Chicago/Turabian StyleSchader, Tim, Christina Reschke, Manuela Spaeth, Susanne Wienstroer, Szeka Wong, and Katrin Schröder. 2020. "NoxO1 Knockout Promotes Longevity in Mice" Antioxidants 9, no. 3: 226. https://doi.org/10.3390/antiox9030226