(Ascorb)ing Pb Neurotoxicity in the Developing Brain


Department of Anatomy, University of Otago, Dunedin 9016, New Zealand
*
Author to whom correspondence should be addressed.
Antioxidants 2020, 9(12), 1311; https://doi.org/10.3390/antiox9121311
Submission received: 29 November 2020
/
Revised: 16 December 2020
/
Accepted: 17 December 2020
/
Published: 21 December 2020
(This article belongs to the Special Issue Role of Natural Antioxidants on Neuroprotection and Neuroinflammation)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Lead (Pb) neurotoxicity is a major concern, particularly in children. Developmental exposure to Pb can alter neurodevelopmental trajectory and has permanent neuropathological consequences, including an increased vulnerability to further stressors. Ascorbic acid is among most researched antioxidant nutrients and has a special role in maintaining redox homeostasis in physiological and physio-pathological brain states. Furthermore, because of its capacity to chelate metal ions, ascorbic acid may particularly serve as a potent therapeutic agent in Pb poisoning. The present review first discusses the major consequences of Pb exposure in children and then proceeds to present evidence from human and animal studies for ascorbic acid as an efficient ameliorative supplemental nutrient in Pb poisoning, with a particular focus on developmental Pb neurotoxicity. In doing so, it is hoped that there is a revitalization for further research on understanding the brain functions of this essential, safe, and readily available vitamin in physiological states, as well to justify and establish it as an effective neuroprotective and modulatory factor in the pathologies of the nervous system, including developmental neuropathologies.
Keywords:
lead; ascorbate; neuronal; antioxidant; chelation; nutrient; vitamin C; dehydroascorbate; blood lead level (BLL)1. Introduction
Lead (Pb) is a metallic contaminant. Though it is not a particularly abundant element, Pb contamination is prevalent in all phases of the environment (air, water, soil, and food chain). Hence, its exposure can be mediated through all these sources. Pb is among the oldest known occupational toxins. However, due to its widespread employment in the manufacturing of batteries, paint pigments, alloys for solder, ammunition, and plastics; the commercial usage of Pb has exceeded all other previous periods in the 20th century. This has resulted in a significant mobilization of free Pb in the environmental and biological spheres, with a consequent increase in its potency as a toxicant. Of note, almost all forms (metallic, organic, and inorganic) of Pb can be equally potent in their toxicities. Unfortunately, Pb is also nonbiodegradable and as such has a prolonged persistence. Because it detrimentally effects a broad-range of physiological, biochemical, and behavioral functions, Pb is a well-studied toxicant [1]. In fact, it affects almost all tissues and organ systems, such as the respiratory, hematopoietic, renal, cardiovascular, urinary, and urogenital systems, as well as bones [2,3,4,5]. Moreover, apart from its widely characterized effects in humans [4,5,6,7,8] and animals [3], the detrimental effects of Pb on the physiology of plants [9] and fish [10,11,12] have also been well-documented.
2. Pb Neurotoxicity in Children
The brain is a prominent target of Pb toxicity. The developing brain is particularly vulnerable to exposure to environmental neurotoxicants such as Pb [13,14,15,16]. Interestingly, contemporary researchers have repeatedly documented no safe blood lead level (BLL) in children [15,17,18], indicating that any early-life exposure to Pb, even in minute quantities, leads to sustained neuropathological alterations [19,20,21,22]. Because of the disproportionate incidence of Pb exposure in children, it is not surprising that the majority of epidemiological research on the health effects of Pb has been focused on children [13,15,23,24]. There are a number of factors responsible for this. First, children exhibit an increased gastrointestinal absorption of Pb [7,25]. Second, their behavior and lifestyle (more hand-to-mouth activities and outdoor time, as well as physical proximity to ground level) increase the likelihood of Pb exposure from contaminated soil and dust [19]. Moreover, children’s immature nervous system is particularly vulnerable to Pb toxicity because of a developing blood–brain barrier (BBB) [6,26]. The neurodevelopmental phenomena of cell division, migration, synaptogenesis, synapse pruning, and the formation of neuronal–glial interactions occur during critical periods in fetal and early childhood, and Pb can suppress all these processes [7,26]. It is therefore not surprising that early-life Pb exposure can have detrimental effects on neurodevelopmental trajectory and long-lasting implications for neurocognition [7,26,27]. It should be noted that Pb neurotoxicity is exacerbated by concomitant exposure to other neurotoxicants [28]. For the purpose of this review, however, we solely concentrate on the Pb-mediated effects. Interestingly, Pb exposure in children can also reduce the intake of essential nutrients, as indicated by a strong negative correlation between BBLs and dietary iron intake [29,30], signifying the potency of the multiple and sometimes indirect effects of Pb exposure.
2.1. Sources of Pb Exposure in Children
Sources of Pb exposure are mainly environment-dependent but are highly varied. The exposure of children from mothers who have been exposed occupationally [31], for example, can be through the placenta in utero and through breast milk [32,33,34,35,36]. Dwelling in geographical locations, such as mines and smelters [37,38], industrial sites [39,40], and waste sites [41], have also been known to result in significant elevations in BLLs in children. Socioeconomic status is another key determinant that increases the likelihood of early-life exposure to Pb [40,42,43]. This might be due to low nutritional status and increased likelihood of exposure to Pb, e.g., from the air or in low income accommodation containing lead paints [7,44]. In particular, a poor diet and deficiency of essential nutrients, such as milk products, are critical predictors of the Pb burden and BLLs [45]. Parental and community factors, such as parental education and occupation, number of siblings, standard of living indices, living in a crowded neighborhood, exposure to tobacco smoke, and playing outdoors, might also explain the association between socioeconomic status and Pb burden in children [37,46,47,48]. Interestingly, recreational activities like shooting have also been shown to result in Pb exposure and elevated BLLs in humans, including high school students [49]. Moreover, dietary exposure to Pb in wild gamebirds shot with lead ammunition significantly increases BLLs in children concomitantly with reductions in intelligence quotient (IQ) levels [50]. Lastly, man-made disasters, such as wars and military assaults on civilian populations, contribute tremendously to the likelihood of heavy metal neurotoxicity in children. This is particularly concerning for the besieged Palestinian population in the Gaza Strip who have endured three successive wars in the recent years [51,52,53,54].
It is of interest to note that genetic factors can predispose children to Pb-mediated effects on neurodevelopment and behavioral outcomes. Polymorphisms in the δ-aminolaevulinic acid dehydratase (ALAD) and vitamin D receptor (VDR) genes may potentially modify the effects of Pb exposure [55]. Gender is another critical factor that might contribute to the Pb burden and its effects in events of early-life exposure [56,57].
Conclusively, while this means that Pb neurotoxicity in children is dependent on a number of environmental factors (and genetic factors to some extent), considerable efforts are needed to reduce the risk of exposure at several levels [14,28,36,58,59]. Legislative measures, particularly concerning leaded gasoline, have indeed aided in reducing the incidences of Pb exposure [60,61,62]. Other factors such as improved protocols for pediatric screening by primary care givers might also prove to be helpful in this regard [63].
2.2. Neurobehavioral Effects of Early-Life Pb Exposure
Widespread studies in diverse environments have been conducted to understand the physiological implications of Pb exposure and elevations in BLLs in children. Multiple effects have been found to underlie the pathophysiology of Pb intoxication in children that manifest early in the subjects’ lives and predisposes them to development of neurological and psychological diseases much later in adulthood [7,26].
General cognition outcomes, as well as IQs, have been found to be correlated with BLLs in children [7,19], and the severity of deficits as measured by decrements in IQ points parallels the elevations in BLLs [22,64]. The steepest decrements in IQs have been observed to occur at BLLs in excess of <100 μg/L [19]. Fetal exposure to Pb has also been observed to induce an adverse effect on neurodevelopment, with observable deficits even at two years of age [35]. Similarly, a prospective study showed that a low exposure to Pb early in the life at 18–30 months of age resulted in a persistent decrease in IQ up to four-to-six years of age [65]. Consistent with the notion that early-life Pb exposure can have detrimental effects on neurodevelopmental trajectory and long-lasting implications for neurocognition, Baghurst et al. reported prenatal Pb exposure to have a lingering inverse effect on IQ levels for children up to seven years old [66] and further effects at up to 11–13 years [67] in children of the lead smelting community of Port Pirie, Australia. In a nine-year prospective study conducted in children based in Taiwan, BLLs were found to be significantly correlated with decreasing IQs, as well as delayed cognitive development, at five-to-eight years of age [68]. An inverse association of intellect function in children measured by an IQ-based assessment with BLL was found in a population of Italian adolescents [69]. Apart from general cognition, a strong inverse relationship between postnatal Pb exposure and executive functioning and goal-oriented problem solving (involving working memory, cognitive flexibility, attention/inhibition, and unitary executive functions) in children has been observed [21,70]. Even infants aged less than six months with prenatal exposure to Pb elicited strong neurobehavioral abnormalities [28]. Other aspects of child intelligence that are adversely affected by pre- and post-natal exposure to Pb include reading/language and arithmetic skills [56,71,72,73], nonverbal reasoning [32], reaction times [72], visual–auditory integration, visual–motor integration, and fine motor skills [71,74,75], as well as short term memory [7,70,71,72]. Lastly, a prospective cohort study in a New Zealand population born in 1972–1973 with possibly the longest follow-up period (four decades) to assess the effects of early-life Pb exposure confirmed the persistent effects of childhood Pb exposure, as indicated by a strong correlation between childhood BLLs with deficits in cognitive functions (perceptual reasoning, working memory, and IQ) and decline in socioeconomic status at 38 years [76].
Exposure to environmental and dietary Pb has been proposed to be a significant risk factor for attention deficit hyperactivity disorder (ADHD) in children, which is characterized by inattention, impulsivity, and hyperactivity [77,78]. Thus, using a child behavior assessment rating scale, Boucher et al. found that low levels of childhood Pb exposure were associated with ADHD behavior [79]. Other studies have also proposed a significant link between Pb burden, clinical ADHD cases, and AHDH-like behavior in children [28,70,80,81]. Interestingly, children with medically diagnosed AHDH conditions and controls dwelling near the Pb investigation area of a former refinery showed an association of BLL (but not blood Hg and Cd levels) with the disorder, indicating an elevated risk due to postnatal Pb, but not Hg or Cd, exposure [82]. Consistent with these findings, a questionnaire- and performance-based assessment of Romanian children found significant associations of ADHD-like behavior with BLL but not with other toxic metals, notably Hg and Al [83]. Interestingly, animal studies have also provided evidence for similar hyperactivity and attention deficits in rodents exposed to Pb [84,85,86,87].
ADHD-like behavioral attributes are considered as major risk factors for delinquent behavior including drug abuse later on in life. Indeed, childhood Pb exposure has been observed to be associated with substance abuse as adolescents [88,89]. Elevated Pb burden, as assessed by bone [90] and blood [89] Pb levels, has been associated with aggression and antisocial behavior in arrested juveniles. An association of delinquency, as measured by hostility, disruptive behavior, and difficulty in emotional regulation and processing of emotional cues with low BLLs, has also been observed in 9–11-years-old children from the Environmental Exposures and Child Health Outcomes (EECHO) project in the US [91]. An association of emotional problems in children with BLLs was also proposed in an independent study [92]. Predisposition of children to neuropsychiatric stress and anxiety by Pb exposure has been confirmed in a study conducted on young children in Chennai, India; wherein higher incidence of anxiety was observed as a function of BLL [70]. Delinquent behavior and emotional problems are often found to be associated with disruptions in signaling at the hypothalamus pituitary adrenal (HPA) axis [93,94]. Not surprisingly, models of developmental Pb exposure in animals have supported the hypothesis of neurodevelopmental alterations in emotional regulation via functional alterations in the HPA axis response system [95,96,97,98].
The incidence of autism spectrum disorders (ASD) in children is also often associated with early-life Pb exposure and elevations in BLLs [28,58,99]. In fact, the degree of autism severity may strongly correlate with Pb levels in hair and nail samples [100]. Schizophrenia is another disease that is heavily influenced by gene–environment interactions. There is evidence that early-life exposure to Pb can predispose individuals to develop psychiatric conditions like schizophrenia later in the life [101,102,103]. Indeed, developmental Pb exposure in experimental animals recapitulates certain behavioral and neuropathological characteristics of schizophrenic conditions, such as the disruption of the ontogenetic switch of N-methyl-D-aspartate receptor (NMDAR) subunits, the selective loss of parvalbumin-positive γ-aminobutyric acid (GABA)ergic interneurons, and the hyperactivity of subcortical dopaminergic system [104,105]. Moreover, Pb exposure in mutant disrupted-in-schizophrenia 1 (mDISC1) has provided further electrophysiological and behavioral evidence in support of developmental Pb as a risk factor for neuropsychiatric conditions [106].
Alzheimer’s disease (AD) is among the most common causes of dementia in the aged population. Noteworthy, most AD cases are sporadic in nature and influenced by a range of genetic and environmental factors. Recent epidemiological studies suggest an association of Pb burden and AD dementia (reviewed in [107,108]). Childhood Pb exposure, particularly in the critical period, leads to permanent alterations in nervous system functions, and this might extend to predisposing the brain to ageing-induced neurodegeneration, such as AD [109]. Indeed, evidence from studies conducted in zebrafish, rodent, and non-human primate models by Zawia’s and other research groups has supported the proposition that developmental exposure to Pb can result in an increased risk of developing AD pathology later on in life. Importantly, developmental Pb can result in alterations (at protein and gene levels) of the major players involved in the regulation of both of the major pathogenic species of AD (amyloid-beta and hyperphosphorylated tau) [110,111,112,113,114,115,116,117,118,119,120,121,122].
3. Molecular and Cellular Mechanisms of Developmental Pb Neurotoxicity
The neuropathological pathways of Pb toxicity in the developing brain have been extensively studied. Pb2+ mimics and substitutes for essential divalent cations like Ca2+ and Zn2+. In fact, the cellular entry of Pb2+ is dependent on Ca2+-permeant channels [18]. Because most of its deleterious effects on cellular physiology stem from its ability to substitute essential divalent cations like Ca2+ and Zn2+, Pb can influence a plethora of wide-ranging aspects of cellular physiology [18]. In the brain, particularly the developing brain, Pb can affect almost all of the processes critical to neuronal functions, such as pre- and post-synaptic functions, redox homeostasis, calcium regulation, protein homeostasis and signaling, epigenetics, genotoxicity and gene expression regulation, neuroinflammation, lipid metabolism, mitochondrial functions, and metabolism. Because there are several excellent reviews extensively describing the molecular and cellular aspects of Pb neurotoxicity, both alone [8,123,124,125,126,127,128,129,130,131,132,133,134] and as a component of toxic metal mixtures [135,136,137], we do not attempt to summarize them again in this review.
4. Ascorbic Acid (AA): Structure–Function
Ascorbic acid (AA) or vitamin C is water-soluble hexanoic sugar acid required for the normal healthy functioning and repair of every tissue type, mainly due to its function as a key endogenous antioxidant and a cofactor for enzymatic reactions [138,139]. Many animals can produce AA from glucose in the liver. However, because of the lack of a functional enzyme in higher primates including humans, AA is not synthesized endogenously and, hence, is an essential vitamin [140]. Significant presence in many fruits and vegetables commonly consumed by humans means that the physiological requirement for AA can normally be met satisfactorily. However, things can get complicated in cases of pathophysiological insults, a befitting example being that of scurvy [141].
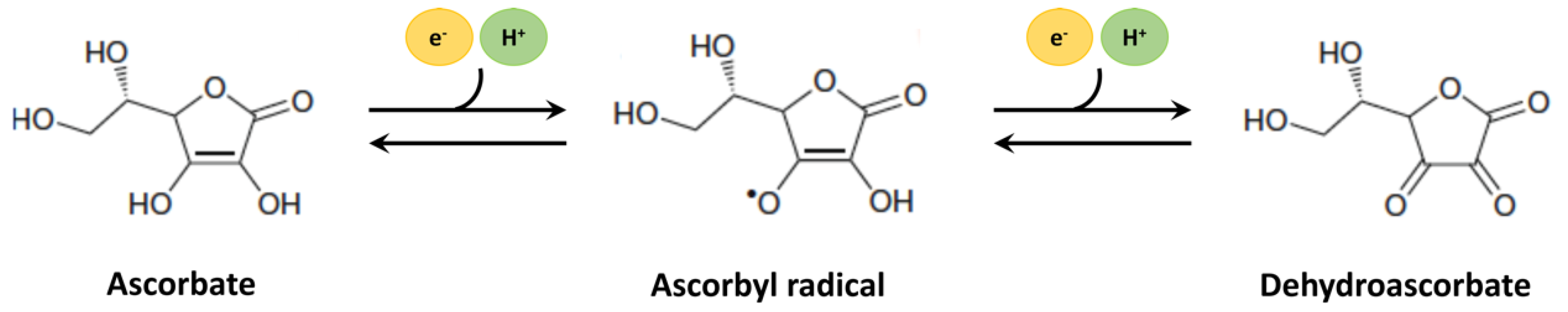
At physiological pH, AA occurs as a monovalent anion, ascorbate. As a lactone with an enediol group, it can donate electrons to convert to its neutral form, dehydroascorbate, which is formed upon the two-electron oxidation of ascorbate (Figure 1). Most biologically relevant oxidizing free radicals, however, result in one electron oxidation of ascorbate to form an ascorbyl radical or semi-dehydroascorbate. The ability to undergo transition to semi-dehydroascorbate by the action of free radicals actually forms the basis for the antioxidant property of AA and its capability to scavenge free radicals. Moreover, because ascorbate has a low redox potential, it can act as a broad-spectrum scavenger against free radicals such as peroxyl- and hydroxyl-radicals, superoxide, singlet oxygen, and peroxynitrite [142,143,144,145]. The recycling of both dehydroascorbate and semi-dehydroascorbate into ascorbate occurs by the action of glutathione and other intracellular thiol redox systems such as thioredoxin [146,147,148,149]. The antioxidant property of AA is singularly important for brain tissues because of a high rate of energy consumption, a high rate of metabolic activity, and a high polyunsaturated fatty acid content, all of which make it particularly vulnerable to oxidative damage. It is noteworthy that the antioxidant free scavenging activity of AA is not limited to the aqueous phase but also includes the protection of membranes and hydrophobic compartments through interaction with vitamin E [150,151], making it the first line of antioxidant defense [140].
The electron donating ability of ascorbate is also responsible for its ability to serve as an enzyme cofactor for redox-coupled reactions in collagen biosynthesis [152,153], noradrenaline–adrenaline synthesis [154] and biosynthesis of neuroendocrine peptides [155]. It should be noted that AA can act as a pro-oxidant in vitro and in the active sites of biosynthetic enzymes by virtue of acting as a reductant for redox-active transition metal ions such as ferric and cupric ions [156]. However, free ferric and cupric ions that are required for the pro-oxidant effects of AA are largely sequestered in forms unable to catalyze free radical reactions, at least in the healthy body [156,157]. Indeed, the in vivo pro-oxidant activities of AA, even in the presence of free, catalytically-active metal cations, have not been confirmed in physiological conditions [140,156,157]. In this respect, it should be noted the debate for in vivo pro-oxidant effects of AA have mainly relied upon a controversial study entitled “Vitamin C exhibits pro-oxidant properties” published in Nature, which reported elevated levels of DNA damage markers in human volunteers supplemented with AA [158]. However, serious questions have been raised regarding the results and their implications [159,160]. Furthermore, two major observations have supported the predominant role of AA as an antioxidant in vivo. First, AA is depleted in pathological states associated with an oxidative stress, and second, the lethality of glutathione deficiency in newborn rodents can be prevented by high doses of AA. Not surprisingly, animal and human supplementation studies have consistently supported an antioxidant function of AA with and without oxidative challenge, as evidenced by an evaluation of the markers of oxidative damage to DNA, lipids, and proteins [157]. Lastly, it should be noted here that in vitro pro-oxidant effects are not limited to AA. Some well-known reducing agents, such as glutathione and plant flavonoids, have often been found to exert damaging pro-oxidant effects when mixed with ferric or cupric ions [156,161,162].
5. Safety of AA
Upon the dietary intake of AA, its plasma concentration is tightly controlled at <100 μM in humans [163]. Increased amounts of ingested AA do not lead to a proportional increase in plasma AA levels; instead, it reaches a maximal upper limit due to a reduction in its intestinal absorption and an increase in its renal clearance [164,165]. While this may mean that there is no pharmacokinetic justification of using high doses of ascorbic acid, it also indicates that AA administration, even in megadoses, is practically harmless [164,166,167,168,169,170,171]. In fact, the safety of AA can be judged from the fact that the consumption of 10,000 mg AA daily for three years does not lead to any adverse health effects [172]. Moreover, claims of adverse effects of AA supplementation in higher doses have not been substantiated. For a more detailed comprehension of the pharmacokinetics of AA, readers are suggested to refer to an excellent recent review [173].
It should, however, be noted that recent studies have suggested that sustaining higher plasma levels of AA may be possible by using the oral infusion of liposomal AA [174]. Interestingly, recent studies have outlined the preparation of novel oral liposomal formulations of AA with increased bioavailability and improved antioxidant efficiency [175,176].
6. AA in the Brain
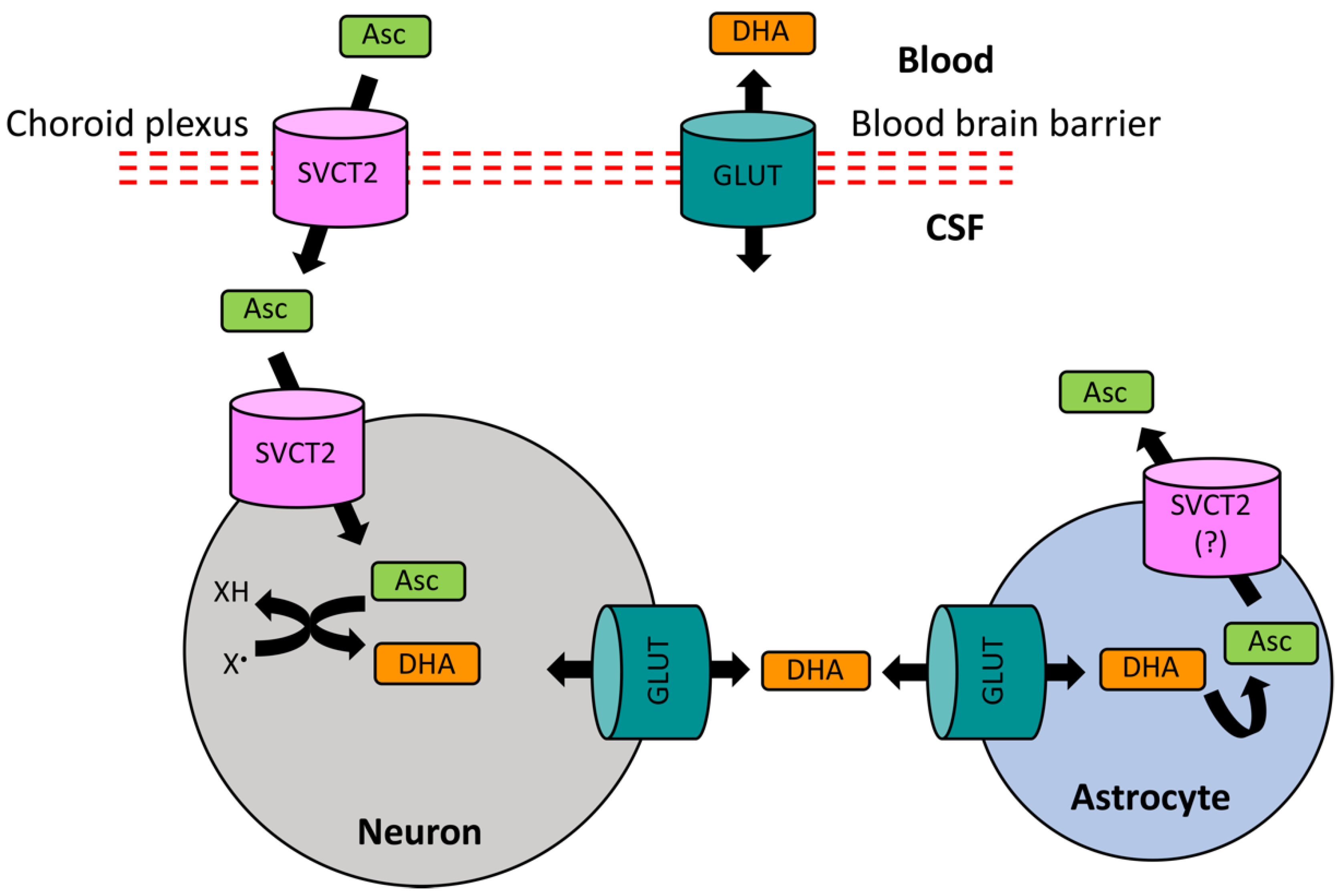
While AA is distributed throughout the body, the brain has the highest levels of and greatest retention capacity for AA [177,178,179,180], particularly the fetal brain [181]. Compared to the AA levels of 50 μM in plasma, AA is present at concentrations of 500 μM in cerebrospinal fluid (CSF) and 200–400 μM in extracellular fluid (ECF), highlighting presence of an active AA uptake in the brain. Indeed, the major carrier mechanisms for AA are the secondarily active specific transporters, the sodium-dependent ascorbate transporters (SVCT1 and SVCT2). While SVCT1 is responsible for the absorption of AA in the gut and its renal retention [138,140], SVCT2 has a high expression in the central nervous system (CNS), including the epithelial cells of the choroid plexus, and manages the uptake of AA from the blood into the CSF [182,183,184]. AA may also accumulate in extracellular fluid via simple diffusion across the BBB [185]. Moreover, ascorbate entry across the BBB is thought to occur by facilitative glucose transporter 1 (GLUT1), which requires the conversion into its neutral oxidation product, dehydroascorbate [186]. Dehydroascorbate is reconverted to ascorbate once inside the cells [187]. This route, however, may only be a minor pathway for ascorbate transport [183].
From the CSF, AA equilibrates with ECF and is accumulated in neurons and glia, where its concentration can reach up to 1 mM in glia and 10 mM in neurons [178,188]. The uptake of AA by brain cells is again mainly mediated by SVCT2 [184,189]. Neuronal and ECF levels of ascorbate can also be regulated by a glutamate–ascorbate hetero-exchange mechanism, possibly involving glutamate receptors [190,191]. In general, ascorbate content in neuron-rich grey matter is much higher than that in white matter, which is evident from its higher levels in anterior brain regions (cerebral cortex and hippocampus) and progressively lower levels in posterior regions (brain stem and spinal cord) [178]. There is also evidence that astrocytes rely on the GLUT-mediated transport of dehydroascorbate for regulating their intracellular AA levels [189,192]. Since, unlike the active SVCT-mediated ascorbate transport, the facilitated diffusion of dehydroascorbate via GLUTs is bidirectional, glial cells can regulate the levels of dehydroascorbate both intracellularly and in the vicinity of the synaptic contacts [138,193]. This is particularly important in the activity-induced brain state. Upon the stimulation of neuronal activity, AA is released from glial reservoirs into extracellular space [194,195]. AA in the synaptic cleft is taken up by neuronal terminals using SVCT2 (Figure 2) [196], where it is utilized for scavenging oxidizing free radicals. The consequently formed dehydroascorbate is then released in to the extracellular space by GLUTs [193,197], from where it is taken up by astrocytes through facilitated diffusion via GLUTs [193,197] and is then recycled back to ascorbate using glutathione and other antioxidant systems [140,198]. To this end, it seems logical to assume that a high concentration of AA in the brain (particularly in neurons) is an allostatic measure to match the higher oxidative metabolism rate, thus indicating that AA may act as a major endogenous antioxidant that maintains redox homeostasis [140,199]. Indeed, neurons have a much higher oxidative metabolic rate and lower level of redox resistance relative to glia [200]. In fact, neurons may be particularly sensitive to AA deficiency because of higher rates of metabolic activity [138,196].
From the neuronal perspective, AA can act both as a neuromodulator [201,202,203] and a neuroprotectant [139,178,180]. The modulation of neurotransmission by AA is known to occur at cholinergic, catecholaminergic, and glutamatergic synapses, wherein it can influence neurotransmitter release, binding, and uptake, and serves as a cofactor for their biosynthesis [204]. Neuroprotection mediated by AA in neuropathological states also occurs in a multimodal fashion. Apart from its described-above antioxidant functions, AA has anti-inflammatory and anti-depressive functions, as well as modulatory actions on epigenetic and gene expression mechanisms (reviewed in [139,140,199,204,205,206,207]).
Surprisingly, relatively little attention has been paid to the role of AA in CNS development and maturation [138]. AA is known to influence the myelination and differentiation of neuronal progenitors [208,209,210,211,212,213]. Equally important is the modulation exerted by AA at the DNA level during brain development [206,214,215]. It is of interest to note that the targeted deletion of SVCT2 (the major AA-specific transporter in the brain) and the consequent diminishment of brain AA to low-to-undetected levels result in death of newborn mice shortly after birth [138], thus indicating a major role of AA in the development of the brain and CNS function.
On the other hand of the developmental spectrum, plasma AA might be associated with improved memory and cognition in aged populations and, in fact, may reduce the incidence of AD, although this remains controversial [216]. Interestingly, the ex vivo uptake of AA is retarded in acute hippocampal slices in aged rats; consequently, even exogenous application of AA does not alleviate oxidative stress in these animals [217]. Further evidence for a relationship between brain AA uptake and metabolism in exacerbating ageing-related pathologies of the brain has come from two recent studies. The first study provided evidence for impaired SCVT2 functions and a consequent deficiency in the neuronal uptake of AA in cellular and mouse models of Huntington’s disease [196]. The second study illustrated increased susceptibility of a transgenic mouse model of AD to kainic acid-induced seizures by decrement in AA uptake mediated by the partial knockout of SCVT2 [218]. Data from these studies suggest that a disruption in AA uptake and metabolism in the brain may be a common feature of ageing-related neurodegeneration. It is of note that AA was found to concomitantly instigate hippocampal neurogenesis in an experimental rat model of d-galactose-induced brain ageing with the restoration of hippocampal-dependent memory functions [219]. Indeed, aged human subjects with dementia and AD-like symptoms have reduced plasma AA levels [220,221], and supplementation with AA may be beneficial in reducing the risk for the development of AD [222]. Interestingly, in a study conducted in hospitalized, acutely-ill aged patients, AA deficiency was found to be significantly associated with symptoms of depression [223], indicating that AA deficiency in ageing may elicit multiple neurocognitive consequences.
7. AA as a Potential Ameliorative Agent in Pb Neurotoxicity
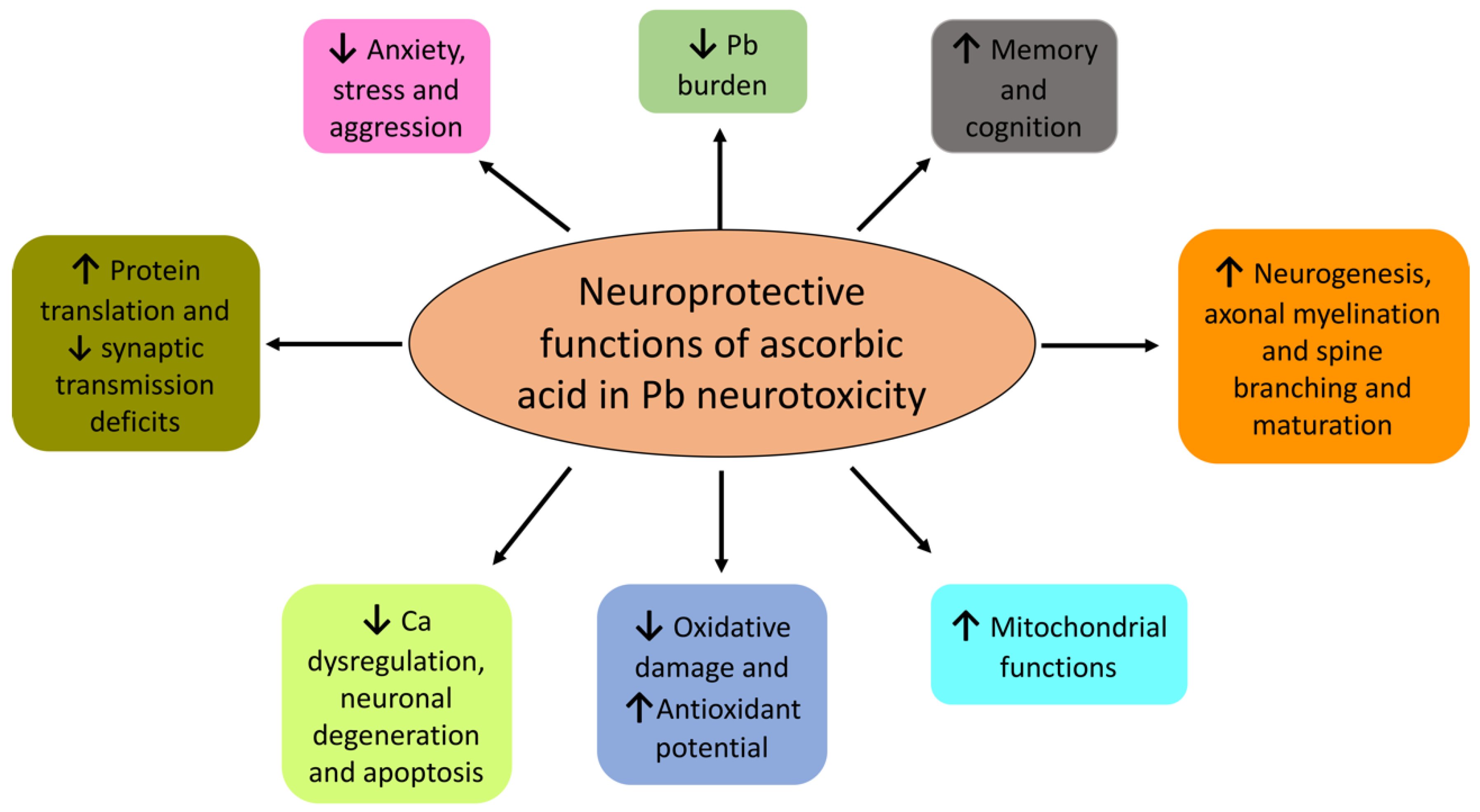
How effective is AA against Pb neurotoxicity? Mechanistically, AA can influence Pb-induced effects by multiple means, e.g., the chelation of Pb2+ ions that results in both reduced absorption in the gut and increased renal excretion. Is it of note that AA fulfils all the criteria for being an effective chelator against Pb2+ due to its (1) water solubility, (2) resistance to metabolic degradation, (3) ability to access metal storage sites, (4) prompt renal excretion, (5) retention of its chelation ability at a physiological pH, and (6) ability to reduce the toxicity of the metal upon formation of a complex with it [224]. Animal studies have supported the idea that orally administrated AA can chelate Pb and both increase its urinary clearance and reduce its intestinal absorption [225,226,227,228], as detailed in Section 7.2. Moreover, AA also reverses oxidative damage induced by Pb (see Section 4). In fact, AA also fulfils all the three criteria for acting as an effective antioxidant in Pb poisoning due to its abilities to (1) retard generation of free radicals and inactivate radical chain reaction; (2) chelate Pb2+ and prevent further radical generation; and (3) activate other endogenous antioxidant systems [229]. Indeed, animal studies have provided unequivocal evidence that AA both reduces oxidative damage and augments endogenous antioxidant potential in events of Pb exposure. Because of its multimodal effects, AA has unsurprisingly been proposed as a potential detoxifying agent in Pb toxicity (Figure 3) [4,229,230,231].
What are the advantages of AA over commonly used therapeutic agents against Pb neurotoxicity? The most commonly used antidotes for Pb toxicity are metal chelators, calcium disodium ethylene diamine tetra-acetic acid (CaNa2EDTA), and 2,3-meso-dimercaptosuccinic acid (succimer) [232,233]. However, the usage of these agents is rife with problems, such as delivery, bioavailability, and non-specific actions and toxicity. Hence, they may not be suitable in high-dose and long-term treatments [226,232,233,234,235,236,237]. This issue is especially important for cases where the subjects cannot be moved away from the sources of poisoning. Moreover, the use of chelators is not recommended in children, particularly for mild Pb exposure, which comprises majority of the cases [58,234]. In fact, chelation therapy in Pb-exposed children has not shown any benefits in reducing BLLs over time or stimulating cognitive, behavioral, and neuromotor attributes [24,238,239]. Interestingly, the use of succimer during pregnancy might actually enhance Pb-induced fetal developmental toxicity in mice, while Ca and AA reduce the maternal transfer of Pb to a fetus [240]. Hence, more attention should be given to safe and efficient natural dietary supplements. AA excellently fulfils the criteria in these respects.
7.1. Human Studies
Many human studies have provided evidence for the suitability of using AA as an ameliorative therapeutic agent in Pb exposure. Interestingly, the beneficial effects of AA in human Pb poisoning cases have been known since late 1930s. Thus, two case studies reported significant clinical improvements among a population of workers with occupational Pb exposure after the daily administration of AA [241,242]. Much later, Papaioannou et al. reported a reduction in BLLs in battery industry workers upon supplementation with 2 g of AA and 60 mg of zinc per day—importantly, while the workers were still on the job and constantly exposed to a Pb-rich environment [243]. Consistent with those studies, the amelioration of occupational Pb toxicity in a small sample of 36 Indian battery workers by AA supplementation (500 mg/day for a month) was observed, as evidenced by the reduction in the oxidative stress markers of lipid peroxides and nitrites in serum and the stimulation of antioxidant parameters, erythrocyte superoxide dismutase, and catalase; however and surprisingly, these effects occurred with no reduction in BLLs [244]. Ademuyiwa’s group conducted a series of studies on the effects of AA supplementation in Nigerian workers who were occupationally exposed to Pb. AA administration to auto-mechanics and attendants in petrol stations at a daily dose of 500 mg for two weeks resulted in marked decreases in BLLs, incidentally with increased urinary Pb excretion [245,246,247,248]. The AA-mediated reversal of the biochemical effects of Pb toxicity—such as reversal of elevated plasma, urine δ-aminolaevulinic acid (ALA), and glutathione levels; inhibition of ALAD; increased erythrocyte protoporphyrin; amelioration of ionoreglatory disruptions; inhibition of erythrocyte Ca2+-Mg2+-ATPase; and alterations in plasma Ca2+ and Mg2+ levels—have also been observed [245,246,247]. In a more recent study conducted in Mexican workers occupationally exposed to Pb, prolonged administration of AA significantly lowered BLL; restored blood ALAD activity, total antioxidant capacity, and the activities of antioxidant enzymes (such as superoxide dismutase (SOD), glutathione reductase (GR), and glutathione peroxidase (GPx)); and repressed blood oxidative damage, as assessed by lipid peroxidation products [249]. Pb-induced testicular dysfunction, as assessed by sperm parameters and sperm DNA fragmentation in workers from the battery-manufacturing industry from India, was found to be significantly reduced by AA supplementation at a dose of 1000 mg/day, five days per week, for a duration of three months [250]. Oral administration of 500 mg of AA daily for a month resulted in marked decreases in BLLs in Indian silver refiners who were occupationally exposed to Pb, in parallel with the reversal of the inhibition of the blood ALAD activity and increased blood Cu and Fe levels [251].
AA’s relationship with Pb in human subjects with nonoccupational and mild Pb exposure (including pregnant women and children) has also been characterized. For example, Sohler et al. conducted an uncontrolled trial and observed the effects of combined zinc and AA administration with a marked reduction of BLLs in a population of over 1000 psychiatric outpatients [252]. AA was also found to decrease BLLs in 85 subjects who volunteered to consume Pb-spiked drinks [253]. In another study conducted on heavy smokers, AA administration for a week had a pronounced (>80%) decrease in BLLs, but urinary Pb levels were not affected, compelling the authors to propose a reduced absorption of Pb [254]. Two other studies observed mild but nonsignificant decreases in BLLs upon the administration of AA [255,256], possibly due to the small sample size of their respective studies. After adjusting for age, education level, smoking, and alcohol consumption, the dietary intake of AA was found to be inversely correlated with BLLs in a population of over 700 male adults aged between 49 and 93 [257]. In one of the most prominent studies, Simon and Hudes conducted a large scale (over 19,000 subjects) population-based study in both American youths and adults, and they reported an independent inverse relationship between serum AA and BLLs among subjects [258], thus suggesting that a higher intake of AA may be effective in preventing Pb toxicity. Importantly, adults in the upper two quartiles of serum AA levels were found to have 65% and 68% less chances of eliciting high BLLs. Moreover, children in the highest serum AA quartile were found to be 89% less likely to have increased BLLs when compared to children in the lowest quartile for AA levels [258]. This study made the first significant contribution to the analysis of a relationship between AA and Pb with a large human sample size [259]. In another study with pregnant women, a significant reduction in BLLs was observed upon vitamin and mineral supplementation, with negative correlations between blood AA levels and BLLs in both supplemented and non-supplemented women [260]. It has been suggested that AA as a nutritional factor may be critical in the determination of BLLs in young children [259]. Indeed, AA supplementation has been shown to reduce the Pb burden in rural children, as assessed by Pb content in their hair samples, possibly by increasing the urinary excretion of the Pb–ascorbate complex [231]. In pregnant women, the supplementation of AA in combination with calcium phosphate was found to reduce the Pb burden in the placenta by 90% and in mothers’ milk by 15% [261]. In an urban population of Korean adults with the supplementation of dietary antioxidants, an increase in AA levels was significantly correlated with decrease in BLLs and the urinary oxidative stress marker 8-hydroxy-2′-deoxyguanosine (8-OHdG) [262]. In an interesting study combining clinical and animal data, Jin et al. observed significant ameliorative effects of AA (in combination with succimer and calcium supplementation) in early-life, mild Pb toxicity with significant reversals of Pb burdens in blood and bones and blood ALAD activity [263].
7.2. Animal Models: Reducing Pb Burden and Amelioration of Neuropathological Defects
Among the first studies that probed the effectiveness of AA as a prophylactic agent in reducing the Pb burden in experimental animals, Rudra et al. found partial rescue effects of AA supplementation in Pb-poisoned rats in terms of Pb burden and AA metabolism [264]. Suzuki and Yoshida found that dietary AA (upon co-administration with Fe) reduced the Pb burden in the liver, kidneys, and tibia bone, and it also reduced anemia and growth deficits in growing rats challenged with Pb for short [265] and moderately long terms [266], although the curative effects in the long term regime were only modest. An experimental model of chicks surprisingly did not show any interaction between AA and Pb [267]. Goyer and Cherian observed an increased urinary excretion of Pb and a reduced Pb burden in the blood, bone, brain, kidney, and liver in Pb-exposed, one-month-old rats treated with a combination of AA and EDTA [268]. An independent study also confirmed AA-mediated stimulation of the renal clearance of Pb and a consequent decrease in BLLs in rats [225], attesting to the proposed role of AA as a potential therapeutic agent in Pb poisoning. Consistently, a number of publications by Tondon and co-authors in the late 1980s also provided evidence supporting the effects of AA as a protective agent as part of a combinatorial supplementary regime on the reduction of the whole body Pb burden in multiple organs of experimental animals challenged with Pb [224,269,270]. In a study conducted in New Zealand, Dalley et al. found a strong prophylactic effect of AA supplementation in reducing the Pb burden in the femur bone, liver, kidney, and plasma of rats challenged with intravenous Pb [226]. A combinatorial therapeutic strategy against Pb exposure in drinking water in mice consisting of a supplementation of succimer, Ca, and AA was found to achieve significant ameliorative effects on the mobilization of blood, renal, hepatic, and brain Pb, as well as blood ALAD activity [271]. Collectively, data from these studies indicate that AA, particularly as part of a combinatorial therapeutic strategy, is efficient in reducing the Pb burden in multiple organs in animals challenged with Pb insults.
Recent studies have extended the knowledge base of AA–Pb interactions with respect to the ameliorative mechanisms involved at the behavioral, electrophysiological, and biochemical levels. For example, chronic intraperitoneal infusion of AA was found to rescue electrophysiological deficits in synaptic transmission (both spontaneous and evoked) in the neuromuscular junctions in the dorsiflexor skeletal muscle of mice exposed to Pb, thus indicating the protective roles of AA against Pb in peripheral nerves [272]. AA has also been shown to attenuate Pb-induced deficits in neurobehavior, such as memory-dependent cognitive functions including novel object recognition [273], anxiety, and aggression [274]. The assessment of a range of antioxidant nutrients for their effectiveness in reducing the Pb burden in adult mice identified AA as one of the promising chelating antioxidant nutrients with the capacity to reduce BBLs to almost 60% without any adverse effects on Ca, Fe, and Zn absorption [275]. The amelioration of Pb-induced oxidative stress in rat brains has also been proposed in other studies [230,274,276,277,278].
7.3. Animal Models: Developing Brain
Several animal studies have indicated that AA may serve as an effective ameliorative agent in events of gestational and lactational exposure to Pb. AA, as part of a dietary supplemental regime, was recently shown to reduce BLLs and reverse serum ALAD activity levels in juvenile mice pre-exposed to Pb with the consequent alleviation of deficits in behavior and redox homeostasis, importantly without eliciting any unwanted side-effects [233]. In juvenile rats co-exposed to lead and chlorpyrifos, an organophosphate insecticide, the administration of AA resulted in a marked reduction in oxidative damage concomitantly with the reversal of neurobehavioral and sensorimotor deficits [279]. An enriched milk formula consisting of AA and other antioxidants reduced BLLs and the Pb burden in organs such as the liver, kidney, bones, and brain in mouse pups exposed to Pb, along with ameliorative effects on blood ALAD, protoporphyrin, and thiobarbituric acid reactive substances (TBARS) levels [280]. The AA-mediated rescue of brain oxidative damage induced by perinatal exposure to Pb in rat pups was confirmed upon coadministration with a mixture of antioxidants including vitamin C and E during pregnancy and lactation [229]. In the follow-up study, the authors found further evidence for AA as a potential therapeutic agent with the partial reversal of deficits in ALAD activity, as well as the activity of catalase enzyme and levels of TBARS, in the same model of early-life Pb exposure [281]. In addition to lowering the Pb burden in the brain, Ghasemi et al. found neuroprotective effects of AA supplementation in juvenile rats exposed to Pb on oxidative stress markers, oxidized thiols, and TBARS, as well as spatial memory tested in a Morris water maze and passive avoidance learning [282,283]. Memory deficits in rat pups induced by chronic Pb exposure during gestation and lactation have been shown to be attenuated by AA and AA-rich traditional medicine lemon balm (Melissa officianlis) with comparable prophylactic effects, indicating AA as the major neuroprotective agent in Melissa [284]. A histopathological evaluation of young adult rats revealed that the intragastric intubation and oral supplementation of AA rescued Pb-induced effects on brain edema, satellitosis, monocytic aggregation, and encephalomalacia [285]. In an early-life Pb exposure model with gestational and lactational Pb exposure, AA in combination with fresh garlic juice extract was found to efficiently reduce blood and brain Pb levels, as well as to attenuate the detrimental effects of Pb-induced neurogenesis, as observed by doublecortin immunostaining [286]. Concomitant with a reduction in BLLs, retinal apoptosis, as assessed by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was found to be significantly attenuated by AA alone and with the coadministration with garlic juice extract in a developmental model of Pb toxicity in rat pups [287]. Moreover, there were AA-mediated protective effects on hippocampal long-term potentiation (LTP), with both population spike amplitude and slope of excitatory postsynaptic potentials), possibly due to reduced oxidative stress and an increase in antioxidant potential in rats challenged with Pb [288]. Consistently, AA alone or in combination with vitamin E was found to reduce oxidative damage and rescue deficits in endogenous antioxidant enzymes, such as GPx and SOD, in the hippocampi of rats challenged with Pb. In addition, the authors observed the attenuation of Pb-induced dysfunction in nitric oxide synthase (NOS) [289]. The preventive effects of AA in young adult rats challenged with Pb on biochemical antioxidant and signaling components (SOD, NOS, NO, Ca2+/calmodulin-dependent protein kinase II (CamKII), and cAMP response element binding protein (CREB)) in the hippocampus were reported and paralleled with the amelioration of spatial memory deficits in a Morris water maze test [290]. Other studies have also found ameliorative effects of AA alone and upon coadministration with a garlic juice extract on hippocampal apoptosis and neuronal degeneration in all the three major subregions (CA1, CA3, and dentate gyrus) examined and in a gestational and lactational model of Pb toxicity in rats [291,292].
Our own studies have provided evidence for the amelioration of early-life (perinatal and postnatal—from gestational day (GD) 15 to postnatal day (PND) 21) Pb-mediated effects on the nerve terminals of multiple brain regions, such as cerebral [293], hippocampal [293], and cerebellar synapses [294], by maternal AA supplementation. In particular, we observed a marked reversal of Pb-mediated alterations in synaptic bioenergetics with the rescue of defects in the mitochondrial membrane potential (MMP) and activities of the enzymes of the electron transport chain (ETC) [294,295]. Improvements in endogenous antioxidant pathways (particularly glutathione signaling) concomitantly with reduced oxidative damage to proteins and lipids were also observed in our model of AA supplementation in rats developmentally exposed to Pb [294,295]. An independent, recently published study also observed synergistic actions of AA and Schisandra chinensis extracts, but not AA alone, in the positive modulation of mitochondrial respiration in normal, wild-type mice [296]. Of note, we also provided evidence for altered de novo hippocampal protein translation in rats with early-life Pb exposure, mediated by deficits in signaling through the protein kinase Akt pathway [293]. These effects were also shown to be rescued by treatment with AA [293], which is consistent with other studies that have proposed positive modulatory actions of AA on hippocampal Akt signaling. For example, Fraga et al. found that AA supplementation in unchallenged rats resulted in the upregulation of hippocampal synaptic proteins such as synapsin, as well as increased dendritic spine density, maturation, and performance, in a novelty suppressed feeding test through the activation of the Akt pathway and its downstream effector 70S6K [297], which phosphorylates and regulates the function of ribosomal protein S6, a component of the translational machinery. Moreover, anti-depressant-like effects elicited by the oral delivery of AA have been shown to be mediated by the activation of phosphoinositide 3-kinase (PI3K) and Akt signaling, as well as the consequent activation of 70S6K that occurs simultaneously with a sharp elevation in postsynaptic density protein 95 (PSD-95) levels in the hippocampi of two-month-old young adult mice [298]. AA has also been shown to partially reverse the detrimental effects of maternal Pb exposure in rat pups on the morphology of hippocampal CA1 neurons, as assessed by Golgi staining and Sholl’s analysis [299].
In a series of studies, an independent contemporary research group based in South Korea also provided further evidence in favor of using AA as a therapeutic agent in developmental Pb neurotoxicity, particularly its effects on cellular apoptosis, endogenous antioxidant enzymes, synaptic dysfunction, and axonal myelination. Their data suggest that AA induces marked stimulation of the endogenous antioxidant enzymes Mn and Cu/Zn SODs and catalase in the hippocampi of rat pups that were gestationally and postnatally exposed to Pb, in parallel with the rescue of degenerating neurons and attenuation of the induction of the pro-apoptotic protein Bax [300,301]. In follow-up studies focusing on cerebellar cortices, the authors observed appreciable rescue of cellular degeneration, as assessed by TUNEL staining by AA administration, concomitantly with the amelioration of the Pb-induced induction of apoptotic protein Bax [302] and degenerative changes such as the reduction of glutamic acid decarboxylase 67 (GAD67) and receptor tyrosine kinase c-kit proteins [303]. The authors further provided evidence for AA-mediated attenuation of Pb-induced effects on glutamatergic signaling and oxidative stress, as assessed by the expression of the synaptic proteins synaptophysin, PSD95, NMDA receptor subunit 1 (NMDAR1), and brain-derived neurotrophic factor (BDNF), as well as antioxidant SODs, in a developmental Pb exposure model [304,305]. Furthermore, reversal of Pb-induced effects on Purkinje cells in parallel with rescue of Pb-induced alterations in calcium binding proteins such as calbindin, calretinin, and parvalbumin, as well as γ-aminobutyric acid transporter 1 (GABAT1) [306], osteopontin, Olig2-immunorecative oligodendrocytes, and axonal myelination (assessed by the expression of myelin-associated glycoprotein (MAG) and myelin basic protein (MBP)) upon AA supplementation was also reported [305,307]. A recent study corroborates the attenuation of toxicity of early-life (gestational and lactational) Pb exposure in the cerebellum of rat pups upon the supplementation of AA by oral gavage by using the assessments of oxidative stress markers and the antioxidant system, as well as histopathological examinations and tests for neuromotor functions (forelimb grip and negative geotaxis) [308]. Interestingly, ameliorative effects of AA in Pb poisoning in the cerebellum of adult rats have also been proposed [309].
8. Other Non-Neuronal Attenuative Effects of AA in Pb Poisoning
It should be noted that the effect of AA in the restoration of Pb-induced toxicity is not limited to the brain. Indeed, the non-neuronal amelioration of Pb-mediated effects in experimental animals by AA either alone or in combination with other proposed neuroprotective agents has been observed in blood biochemistry and hematological parameters [265,306,307,308,309,310,311,312,313,314,315,316], cardiac functions [317], hepatic physiology [277,285,312,318,319,320,321,322,323,324], renal functions [285,310,312,325,326], the colon [327], testicular functions and spermatogenesis [285,328,329,330,331,332], sperm morphology and physiology [333], thyroid hormone synthesis [334], the lungs [335], and clastogenicity in bone marrow cells [336,337,338]. Readers are suggested to refer to a recent review paper [339] for a brief overview of the non-neuronal effects of AA in Pb poisoning.
In vitro cell culture systems have also been employed to successfully access the ameliorative effects of AA on Pb uptake and release [340], Pb-induced genotoxicity and death [341,342], xenobiotics metabolism [343], and oxidative damage to cellular lipids, the nuclear factor (erythroid-derived 2) like 2–Kelch-like ECH-associated protein 1 (Nrf2–KEAP1) antioxidant pathway, and that pathway’s downstream enzyme effectors [344].
9. Conclusions
As a safe, readily available, and low-cost supplemental biomolecule, ascorbic acid might prove to be a useful ameliorative therapeutic agent against Pb toxicity, both in reducing BLLs and reversing the effects of Pb poisoning. This is particularly true in children, where prominent chelation therapeutic agents have largely failed. It should be noted that most of the recent studies delineating the effects of AA on Pb neurotoxicity have come from developing countries. This is expected because Pb toxicity is a major concern in these countries in particular because of their emerging industrial potential and issues such as a lack of proper legislation and compliance that aggravate the conditions. This can be related to recent outbreak of a ‘mystery’ illness that grasped several hundreds of individuals, including children, with symptoms such as nausea and seizures in the Eluru district of southern Indian state of Andhra Pradesh. A preliminary investigation of blood samples of the subjects indicated high level of contamination with heavy metals, particularly Pb and Ni. Our understanding of AA–Pb interactions is nevertheless still limited, and we are far from fully appreciating the usefulness of this vitamin as an effective ameliorative agent. Further studies are hence required (1) to confirm the prophylactic effects of AA in both animal models and human subjects with Pb exposure, as well as (2) to understand the mechanisms of protection offered by AA. With this review, we hope to rekindle research interest in this essential, water-soluble, and practically harmless vitamin as an effective neuroprotective agent, not only in developmental Pb neurotoxicity but also in other neuropathologies.
Funding
This research was supported by the Department of Anatomy, School of Biomedical Sciences, University of Otago, New Zealand.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
| 70S6K | S6 kinase 70 kDa |
| 8-OHdG | 8-hydroxy-2′-deoxyguanosine |
| AA | ascorbic acid; AD: Alzheimer’s disease |
| ADHD | attention deficit hyperactivity disorder |
| ALA | δ-aminolaevulinic acid |
| ALAD | δ-aminolaevulinic acid dehydratase |
| ASD | autism spectrum disorder |
| BBB | blood–brain barrier |
| BLL | blood lead level |
| CamKII | Ca2+/calmodulin-dependent protein kinase II |
| CaNa2EDTA | calcium disodium ethylenediaminetetraacetic acid |
| CREB | cAMP response element binding protein |
| CSF | cerebrospinal fluid |
| ECF | extracellular fluid |
| EECHO | environmental exposures and child health outcomes |
| ETC | electron transport chain |
| GABAT1 | γ-aminobutyric acid transporter 1 |
| GAD67 | glutamic acid decarboxylase 67 kDa |
| GD | gestational day |
| GLUT1 | glucose transporter 1 |
| GPx | glutathione peroxidase |
| GR | glutathione reductase |
| HPA | hypothalamic pituitary adrenal |
| IQ | intelligence quotient |
| KEAP1 | Kelch-like ECH-associated protein |
| LTP | long-term potentiation |
| MAG | myelin-associated glycoprotein |
| MBP | myelin basic protein |
| mDISC1 | mutated-in-schizophrenia 1 |
| MMP | mitochondrial membrane potential |
| NMDAR1 | N-methyl-D-aspartate receptor 1 |
| NOS | nitric oxide synthase |
| Nrf2 | nuclear factor (erythroid-derived 2)-like 2 |
| Pb | Lead (plumbum) |
| PI3K | phosphoinositide 3-kinase |
| PND | postnatal day |
| PSD95 | postsynaptic density protein 95 |
| SOD | superoxide dismutase |
| SVCT1 | sodium-dependent vitamin C transporter 1 |
| SVCT2 | sodium-dependent vitamin C transporter 1 |
| TBARS | thiobarbituric acid reactive substances |
| VDR | vitamin D receptor |
References
- Hsu, P.C.; Guo, Y.L. Antioxidant nutrients and lead toxicity. Toxicology 2002, 180, 33–44. [Google Scholar] [CrossRef]
- Boskabady, M.; Marefati, N.; Farkhondeh, T.; Shakeri, F.; Farshbaf, A.; Boskabady, M.H. The effect of environmental lead exposure on human health and the contribution of inflammatory mechanisms, a review. Environ. Int. 2018, 120, 404–420. [Google Scholar] [CrossRef] [PubMed]
- Assi, M.A.; Hezmee, M.N.M.; Haron, A.W.; Sabri, M.Y.M.; Rajion, M.A. The detrimental effects of lead on human and animal health. Vet. World 2016, 9, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Flora, G.; Gupta, D.; Tiwari, A. Toxicity of lead: A review with recent updates. Interdiscip. Toxicol. 2012, 5, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Wani, A.L.; Ara, A.; Usmani, J.A. Lead toxicity: A review. Interdiscip. Toxicol. 2015, 8, 55–64. [Google Scholar] [CrossRef] [Green Version]
- Winneke, G.; Lilienthal, H.; Krämer, U. The neurobehavioural toxicology and teratology of lead. Arch. Toxicol. Suppl. 1996, 18, 57–70. [Google Scholar] [CrossRef]
- Caito, S.; Aschner, M. Developmental neurotoxicity of lead. Adv. Neurobiol. 2017, 18, 3–12. [Google Scholar] [CrossRef]
- Rocha, A.; Trujillo, K.A. Neurotoxicity of low-level lead exposure: History, mechanisms of action, and behavioral effects in humans and preclinical models. Neurotoxicology 2019, 73, 58–80. [Google Scholar] [CrossRef]
- Shahid, M.; Pourrut, B.; Dumat, C.; Nadeem, M.; Aslam, M.; Pinelli, E. Heavy-metal-induced reactive oxygen species: Phytotoxicity and physicochemical changes in plants. Rev. Environ. Contam. Toxicol. 2014, 232, 1–44. [Google Scholar] [CrossRef]
- Green, A.J.; Planchart, A. The neurological toxicity of heavy metals: A fish perspective. Comp. Biochem. Physiol. Part—C Toxicol. Pharmacol. 2018, 208, 12–19. [Google Scholar] [CrossRef]
- Horzmann, K.A.; Freeman, J.L. Zebrafish get connected: Investigating neurotransmission targets and alterations in chemical toxicity. Toxics 2016, 4, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.W.; Choi, H.; Hwang, U.K.; Kang, J.C.; Kang, Y.J.; Kim, K.; Kim, J.H. Toxic effects of lead exposure on bioaccumulation, oxidative stress, neurotoxicity, and immune responses in fish: A review. Environ. Toxicol. Pharmacol. 2019, 68, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Bellinger, D.C.; Matthews-Bellinger, J.A.; Kordas, K. A developmental perspective on early-life exposure to neurotoxicants. Environ. Int. 2016, 94, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Bellinger, D.C.; Bellinger, A.M. Childhood lead poisoning: The torturous path from science to policy. J. Clin. Investig. 2006, 116, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.A. Is Prenatal Lead Exposure a Concern in Infancy? What Is the Evidence? Adv. Neonatal Care 2015, 15, 416–420. [Google Scholar] [CrossRef]
- Lidsky, T.I.; Schneider, J.S. Lead neurotoxicity in children: Basic mechanisms and clinical correlates. Brain 2003, 126, 5–19. [Google Scholar] [CrossRef]
- Chiodo, L.M.; Jacobson, S.W.; Jacobson, J.L. Neurodevelopmental effects of postnatal lead exposure at very low levels. Neurotoxicol. Teratol. 2004, 26, 359–371. [Google Scholar] [CrossRef]
- Shefa, S.T.; Héroux, P. Both physiology and epidemiology support zero tolerable blood lead levels. Toxicol. Lett. 2017, 280, 232–237. [Google Scholar] [CrossRef]
- Jakubowski, M. Low-level environmental lead exposure and intellectual impairment in children—The current concepts of risk assessment. Int. J. Occup. Med. Environ. Health 2011, 24, 1–7. [Google Scholar] [CrossRef]
- Vorvolakos, T.; Arseniou, S.; Samakouri, M. There is no safe threshold for lead exposure: Α literature review. Psychiatry 2016, 27, 204–214. [Google Scholar] [CrossRef]
- Arnold, O.M.; Liu, J. Blood lead levels ≤10 micrograms/deciliter and executive functioning across childhood development: A systematic review. Neurotoxicol. Teratol. 2020, 106888. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J. Low-level lead exposure and children’s IQ: A metaanalysis and search for a threshold. Environ. Res. 1994, 65, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Winneke, G. Developmental aspects of environmental neurotoxicology: Lessons from lead and polychlorinated biphenyls. J. Neurol. Sci. 2011, 308, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Maloney, B.; Bayon, B.L.; Zawia, N.H.; Lahiri, D.K. Latent consequences of early-life lead (Pb) exposure and the future: Addressing the Pb crisis. Neurotoxicology 2018, 68, 126–132. [Google Scholar] [CrossRef]
- Leggett, R.W. Research advances: An age-specific kinetic model of lead metabolism in humans. Environ. Health Perspect. 1993, 101, 598–616. [Google Scholar] [CrossRef]
- Santa Maria, M.P.; Hill, B.D.; Kline, J. Lead (Pb) neurotoxicology and cognition. Appl. Neuropsychol. Child 2019, 8, 272–293. [Google Scholar] [CrossRef]
- Sioen, I.; Den Hond, E.; Nelen, V.; Van de Mieroop, E.; Croes, K.; Van Larebeke, N.; Nawrot, T.S.; Schoeters, G. Prenatal exposure to environmental contaminants and behavioural problems at age 7-8 years. Environ. Int. 2013, 59, 225–231. [Google Scholar] [CrossRef]
- Dórea, J.G. Environmental exposure to low-level lead (Pb) co-occurring with other neurotoxicants in early life and neurodevelopment of children. Environ. Res. 2019, 177, 108641. [Google Scholar] [CrossRef]
- Hammad, T.A.; Sexton, M.; Langenberg, P. Relationship between blood lead and dietary iron intake in preschool children: A cross-sectional study. Ann. Epidemiol. 1996, 6, 30–33. [Google Scholar] [CrossRef]
- Yip, R.; Dallman, P.R. Developmental changes in erythrocyte protoporphyrin: Roles of iron deficiency and lead toxicity. J. Pediatrics 1984, 104, 710–713. [Google Scholar] [CrossRef]
- Marques, R.C.; Bernardi, J.V.E.; Dórea, J.G.; De Fatima, R.; Moreira, M.; Malm, O. Perinatal multiple exposure to neurotoxic (lead, methylmercury, ethylmercury, and aluminum) substances and neurodevelopment at six and 24 months of age. Environ. Pollut. 2014, 187, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Tatsuta, N.; Nakai, K.; Murata, K.; Suzuki, K.; Iwai-Shimada, M.; Kurokawa, N.; Hosokawa, T.; Satoh, H. Impacts of prenatal exposures to polychlorinated biphenyls, methylmercury, and lead on intellectual ability of 42-month-old children in Japan. Environ. Res. 2014, 133, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Gulson, B.L.; Mizon, K.J.; Korsch, M.J.; Palmer, J.M.; Donnelly, J.B. Mobilization of lead from human bone tissue during pregnancy and lactation—A summary of long-term research. Sci. Total Environ. 2003, 303, 79–104. [Google Scholar] [CrossRef]
- Ettinger, A.S.; Téllez-Rojo, M.M.; Amarasiriwardena, C.; Peterson, K.E.; Schwartz, J.; Aro, A.; Hu, H.; Hernández-Avila, M. Influence of maternal bone lead burden and calcium intake on levels of lead in breast milk over the course of lactation. Am. J. Epidemiol. 2006, 163, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Téllez-Rojo, M.M.; Bellinger, D.; Smith, D.; Ettinger, A.S.; Lamadrid-Figueroa, H.; Schwartz, J.; Schnaas, L.; Mercado-García, A.; Hernández-Avila, M. Fetal lead exposure at each stage of pregnancy as a predictor of infant mental development. Environ. Health Perspect. 2006, 114, 1730–1735. [Google Scholar] [CrossRef]
- Gardella, C. Lead exposure in pregnancy: A review of the literature and argument for routine prenatal screening. Obstet. Gynecol. Surv. 2001, 56, 231–238. [Google Scholar] [CrossRef]
- Moodie, S.; Ialongo, N.; López, P.; Rosado, J.; García-Vargas, G.; Ronquillo, D.; Kordas, K. The conjoint influence of home enriched environment and lead exposure on children’s cognition and behaviour in a Mexican lead smelter community. Neurotoxicology 2013, 34, 33–41. [Google Scholar] [CrossRef]
- Yabe, J.; Nakayama, S.M.M.; Ikenaka, Y.; Yohannes, Y.B.; Bortey-Sam, N.; Oroszlany, B.; Muzandu, K.; Choongo, K.; Kabalo, A.N.; Ntapisha, J.; et al. Lead poisoning in children from townships in the vicinity of a lead-zinc mine in Kabwe, Zambia. Chemosphere 2015, 119, 941–947. [Google Scholar] [CrossRef]
- Alegría-Torres, J.A.; Pérez-Rodríguez, R.Y.; García-Torres, L.; Costilla-Salazar, R.; Rocha-Amador, D. Exposure to arsenic and lead in children from Salamanca México, effects on telomeric lengthening and mitochondrial DNA. Environ. Sci. Pollut. Res. 2020, 27, 6420–6428. [Google Scholar] [CrossRef]
- Mathee, A.; Khan, T.; Naicker, N.; Kootbodien, T.; Naidoo, S.; Becker, P. Lead exposure in young school children in South African subsistence fishing communities. Environ. Res. 2013, 126, 179–183. [Google Scholar] [CrossRef]
- Liu, J.; Xu, X.; Wu, K.; Piao, Z.; Huang, J.; Guo, Y.; Li, W.; Zhang, Y.; Chen, A.; Huo, X. Association between lead exposure from electronic waste recycling and child temperament alterations. Neurotoxicology 2011, 32, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Bellinger, D.C. Lead neurotoxicity and socioeconomic status: Conceptual and analytical issues. Neurotoxicology 2008, 29, 828–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahamed, M.; Verma, S.; Kumar, A.; Siddiqui, M.K.J. Environmental exposure to lead and its correlation with biochemical indices in children. Sci. Total Environ. 2005, 346, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Kwon, H.J.; Ha, M.; Lim, J.A.; Lim, M.H.; Yoo, S.J.; Paik, K.C. How does low socioeconomic status increase blood lead levels in Korean children? Int. J. Environ. Res. Public Health 2018, 15, 1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordas, K.; Burganowski, R.; Roy, A.; Peregalli, F.; Baccino, V.; Barcia, E.; Mangieri, S.; Ocampo, V.; Mañay, N.; Martínez, G.; et al. Nutritional status and diet as predictors of children’s lead concentrations in blood and urine. Environ. Int. 2018, 111, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ai, Y.; McCauley, L.; Pinto-Martin, J.; Yan, C.; Shen, X.; Needleman, H. Blood lead levels and associated sociodemographic factors among preschool children in the South Eastern region of China. Paediatr. Perinat. Epidemiol. 2012, 26, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Hernanz, Á.; González-Estecha, M.; Blanco, M.; Fuentes, M.; Ordóñez-Iriarte, J.M.; Palazón-Bru, I.; Calvo-Manuel, E.; Bodas-Pinedo, A. Blood lead in children and associations with trace elements and sociodemographic factors. J. Trace Elem. Med. Biol. 2020, 58, 126424. [Google Scholar] [CrossRef]
- Jain, N.B.; Hu, H. Childhood correlates of blood lead levels in Mumbai and Delhi. Environ. Health Perspect. 2006, 114, 466–470. [Google Scholar] [CrossRef] [Green Version]
- Laidlaw, M.A.S.; Filippelli, G.; Mielke, H.; Gulson, B.; Ball, A.S. Lead exposure at firing ranges—A review. Environ. Health 2017, 16, 34. [Google Scholar] [CrossRef] [Green Version]
- Green, R.E.; Pain, D.J. Potential health risks to adults and children in the UK from exposure to dietary lead in gamebirds shot with lead ammunition. Food Chem. Toxicol. 2012, 50, 4180–4190. [Google Scholar] [CrossRef]
- Manduca, P.; Al Baraquni, N.; Al Baraquni, L.; Abu Abadi, D.; Abdallah, H.; Hamad, G.A.; Mosa, T.A.; Balousha, S.; Miqdad, H.; Mohammed, W.; et al. Hospital centered surveillance of births in Gaza, Palestine, 2011–2017 and heavy metal contamination of the mothers reveals long-term impact of wars. Reprod. Toxicol. 2019, 86, 23–32. [Google Scholar] [CrossRef]
- Al Baraquoni, N.; Qouta, S.R.; Vänskä, M.; Diab, S.Y.; Punamäki, R.L.; Manduca, P. It takes time to unravel the ecology of war in Gaza, Palestine: Long-term changes in maternal, newborn and toddlers’ heavy metal loads, and infant and toddler developmental milestones in the aftermath of the 2014 military attacks. Int. J. Environ. Res. Public Health 2020, 17, 6698. [Google Scholar] [CrossRef] [PubMed]
- Manduca, P.; Al Baraquni, N.; Parodi, S. Long term risks to neonatal health from exposure to war-9 years long survey of reproductive health and contamination by weapon-delivered heavy metals in Gaza, Palestine. Int. J. Environ. Res. Public Health 2020, 17, 2538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manduca, P.; Naim, A.; Signoriello, S. Specific association of teratogen and toxicant metals in hair of newborns with congenital birth defects or developmentally premature birth in a cohort of couples with documented parental exposure to military attacks: Observational study at Al Shifa Hospit. Int. J. Environ. Res. Public Health 2014, 11, 5208–5223. [Google Scholar] [CrossRef] [PubMed]
- Pawlas, N.; Broberg, K.; Olewińska, E.; Prokopowicz, A.; Skerfving, S.; Pawlas, K. Modification by the genes ALAD and VDR of lead-induced cognitive effects in children. Neurotoxicology 2012, 33, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Jedrychowski, W.; Perera, F.; Jankowski, J.; Mrozek-Budzyn, D.; Mroz, E.; Flak, E.; Edwards, S.; Skarupa, A.; Lisowska-Miszczyk, I. Gender specific differences in neurodevelopmental effects of prenatal exposure to very low-lead levels: The prospective cohort study in three-year olds. Early Hum. Dev. 2009, 85, 503–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llop, S.; Lopez-Espinosa, M.J.; Rebagliato, M.; Ballester, F. Gender differences in the neurotoxicity of metals in children. Toxicology 2013, 311, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Hauptman, M.; Stierman, B.; Woolf, A.D. Children with autism spectrum disorder and lead poisoning: Diagnostic challenges and management complexities. Clin. Pediatrics (Phila.) 2019, 58, 605–612. [Google Scholar] [CrossRef]
- Meyer, P.A.; McGeehin, M.A.; Falk, H. A global approach to childhood lead poisoning prevention. Int. J. Hyg. Environ. Health 2003, 206, 363–369. [Google Scholar] [CrossRef] [Green Version]
- Nichani, V.; Li, W.I.; Smith, M.A.; Noonan, G.; Kulkarni, M.; Kodavor, M.; Naeher, L.P. Blood lead levels in children after phase-out of leaded gasoline in Bombay, India. Sci. Total Environ. 2006, 363, 95–106. [Google Scholar] [CrossRef]
- Hwang, Y.H.; Ko, Y.; Chiang, C.D.; Hsu, S.P.; Lee, Y.H.; Yu, C.H.; Chiou, C.H.; Der Wang, J.; Chuang, H.Y. Transition of cord blood lead level, 1985-2002, in the Taipei area and its determinants after the cease of leaded gasoline use. Environ. Res. 2004, 96, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Mathee, A.; Röllin, H.; Von Schirnding, Y.; Levin, J.; Naik, I. Reductions in blood lead levels among school children following the introduction of unleaded petrol in South Africa. Environ. Res. 2006, 100, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Wood, S.K.; Sperling, R. Pediatric screening: Development, anemia, and lead. Prim. Care—Clin. Off. Pract. 2019, 46, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Lanphear, B.P.; Hornung, R.; Khoury, J.; Yolton, K.; Baghurst, P.; Bellinger, D.C.; Canfield, R.L.; Dietrich, K.N.; Bornschein, R.; Greene, T.; et al. Low-level environmental lead exposure and children’s intellectual function: An international pooled analysis. Environ. Health Perspect. 2005, 113, 894–899. [Google Scholar] [CrossRef]
- Chen, A.; Dietrich, K.N.; Ware, J.H.; Radcliffe, J.; Rogan, W.J. IQ and blood lead from 2 to 7 years of age: Are the effects in older children the residual of high blood lead concentrations in 2-year-olds? Environ. Health Perspect. 2005, 113, 597–601. [Google Scholar] [CrossRef] [Green Version]
- Baghurst, P.A.; McMichael, A.J.; Wigg, N.R.; Vimpani, G.V.; Robertson, E.F.; Roberts, R.J.; Tong, S.-L. Environmental exposure to lead and children’s intelligence at the age of seven years. N. Engl. J. Med. 1992, 327, 1279–1284. [Google Scholar] [CrossRef]
- Tong, S.; Baghurst, P.; McMichael, A.; Sawyer, M.; Mudge, J. Lifetime exposure to environmental lead and children’s intelligence at 11–13 years: The Port Pirie cohort study. Br. Med. J. 1996, 312, 1569–1575. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.C.; Su, P.H.; Chen, H.Y.; Bin Huang, H.; Tsai, J.L.; Huang, H.I.; Wang, S.L. Childhood blood lead levels and intellectual development after ban of leaded gasoline in Taiwan: A 9-year prospective study. Environ. Int. 2012, 40, 88–96. [Google Scholar] [CrossRef]
- Lucchini, R.G.; Zoni, S.; Guazzetti, S.; Bontempi, E.; Micheletti, S.; Broberg, K.; Parrinello, G.; Smith, D.R. Inverse association of intellectual function with very low blood lead but not with manganese exposure in Italian adolescents. Environ. Res. 2012, 118, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Bellinger, D.; Hu, H.; Schwartz, J.; Ettinger, A.S.; Wright, R.O.; Bouchard, M.; Palaniappan, K.; Balakrishnan, K. Lead exposure and behavior among young children in Chennai, India. Environ. Health Perspect. 2009, 117, 1607–1611. [Google Scholar] [CrossRef]
- Frndak, S.; Barg, G.; Canfield, R.L.; Quierolo, E.I.; Mañay, N.; Kordas, K. Latent subgroups of cognitive performance in lead- and manganese-exposed Uruguayan children: Examining behavioral signatures. Neurotoxicology 2019, 73, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Jedrychowski, W.; Perera, F.P.; Jankowski, J.; Mrozek-Budzyn, D.; Mroz, E.; Flak, E.; Edwards, S.; Skarupa, A.; Lisowska-Miszczyk, I. Very low prenatal exposure to lead and mental development of children in infancy and early childhood. Neuroepidemiology 2009, 32, 270–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaine, P.; Navas-Acien, A.; Lee, R.; Simon, P.; Diener-West, M.; Agnew, J. Elevated blood lead levels and reading readiness at the start of kindergarten. Pediatrics 2013, 131, 1081–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polanska, K.; Hanke, W.; Pawlas, N.; Wesolowska, E.; Jankowska, A.; Jagodic, M.; Mazej, D.; Dominowska, J.; Grzesiak, M.; Mirabella, F.; et al. Sex-dependent impact of low-level lead exposure during prenatal period on child psychomotor functions. Int. J. Environ. Res. Public Health 2018, 15, 2263. [Google Scholar] [CrossRef] [Green Version]
- Hou, S.; Yuan, L.; Jin, P.; Ding, B.; Qin, N.; Li, L.; Liu, X.; Wu, Z.; Zhao, G.; Deng, Y. A clinical study of the effects of lead poisoning on the intelligence and neurobehavioral abilities of children. Theor. Biol. Med. Model. 2013, 10, 13. [Google Scholar] [CrossRef] [Green Version]
- Reuben, A.; Caspi, A.; Belsky, D.W.; Broadbent, J.; Harrington, H.; Sugden, K.; Houts, R.M.; Ramrakha, S.; Poulton, R.; Moffitt, T.E. Association of childhood blood lead levels with cognitive function and socioeconomic status at age 38 years and with IQ change and socioeconomic mobility between childhood and adulthood. JAMA 2017, 317, 1244–1251. [Google Scholar] [CrossRef]
- Polańska, K.; Jurewicz, J.; Hanke, W. Review of current evidence on the impact of pesticides, polychlorinated biphenyls and selected metals on attention deficit / hyperactivity disorder in children. Int. J. Occup. Med. Environ. Health 2013, 26, 16–38. [Google Scholar] [CrossRef]
- Bellinger, D.; Hu, H.; Titlebaum, L.; Needleman, H.L. Attentional correlates of dentin and bone lead levels in adolescents. Arch. Environ. Health 1994, 49, 98–105. [Google Scholar] [CrossRef]
- Boucher, O.; Jacobson, S.W.; Plusquellec, P.; Dewailly, É.; Ayotte, P.; Forget-Dubois, N.; Jacobson, J.L.; Muckle, G. Prenatal methylmercury, postnatal lead exposure, and evidence of attention deficit/hyperactivity disorder among Inuit children in Arctic Québec. Environ. Health Perspect. 2012, 120, 1456–1461. [Google Scholar] [CrossRef] [Green Version]
- Braun, J.M.; Kahn, R.S.; Froehlich, T.; Auinger, P.; Lanphear, B.P. Exposures to environmental toxicants and attention deficit hyperactivity disorder in U.S. children. Environ. Health Perspect. 2006, 114, 1904–1909. [Google Scholar] [CrossRef] [Green Version]
- Nigg, J.T.; Nikolas, M.; Mark Knottnerus, G.; Cavanagh, K.; Friderici, K. Confirmation and extension of association of blood lead with attention-deficit/hyperactivity disorder (ADHD) and ADHD symptom domains at population-typical exposure levels. J. Child Psychol. Psychiatry 2010, 5, 58–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Arora, M.; Fernandez, C.; Landero, J.; Caruso, J.; Chen, A. Lead, mercury, and cadmium exposure and attention deficit hyperactivity disorder in children. Environ. Res. 2013, 126, 105–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolescu, R.; Petcu, C.; Cordeanu, A.; Fabritius, K.; Schlumpf, M.; Krebs, R.; Krämer, U.; Winneke, G. Environmental exposure to lead, but not other neurotoxic metals, relates to core elements of ADHD in Romanian children: Performance and questionnaire data. Environ. Res. 2010, 110, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Xu, Y.; Cai, R.; Tang, Y.; Ge, M.M.; Liu, Z.H.; Xu, L.; Hu, F.; Ruan, D.Y.; Wang, H.L. Epigenetic histone modification regulates developmental lead exposure induced hyperactivity in rats. Toxicol. Lett. 2014, 225, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martín, F.J.; Fan, Y.; Lindquist, D.M.; Xia, Y.; Puga, A. Lead induces similar gene expression changes in brains of gestationally exposed adult mice and in neurons differentiated from mouse embryonic stem cells. PLoS ONE 2013, 8, e80558. [Google Scholar] [CrossRef] [Green Version]
- Ruocco, L.A.; Treno, C.; Gironi Carnevale, U.A.; Arra, C.; Boatto, G.; Pagano, C.; Tino, A.; Nieddu, M.; Michel, M.; Prikulis, I.; et al. Immunization with DISC1 protein in an animal model of ADHD influences behavior and excitatory amino acids in prefrontal cortex and striatum. Amino Acids 2015, 47, 637–650. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, J.; Xu, Y. Epigenetic basis of lead-induced neurological disorders. Int. J. Environ. Res. Public Health 2020, 17, 4878. [Google Scholar] [CrossRef]
- Desrochers-Couture, M.; Courtemanche, Y.; Forget-Dubois, N.; Bélanger, R.E.; Boucher, O.; Ayotte, P.; Cordier, S.; Jacobson, J.L.; Jacobson, S.W.; Muckle, G. Association between early lead exposure and externalizing behaviors in adolescence: A developmental cascade. Environ. Res. 2019, 2019, 108679. [Google Scholar] [CrossRef]
- Dietrich, K.N.; Douglas, R.M.; Succop, P.A.; Berger, O.G.; Bornschein, R.L. Early exposure to lead and juvenile delinquency. Neurotoxicol. Teratol. 2001, 23, 511–518. [Google Scholar] [CrossRef]
- Needleman, H.L.; McFarland, C.; Ness, R.B.; Fienberg, S.E.; Tobin, M.J. Bone lead levels in adjudicated delinquents. Neurotoxicol. Teratol. 2002, 24, 711–717. [Google Scholar] [CrossRef]
- Gump, B.B.; Dykas, M.J.; MacKenzie, J.A.; Dumas, A.K.; Hruska, B.; Ewart, C.K.; Parsons, P.J.; Palmer, C.D.; Bendinskas, K. Background lead and mercury exposures: Psychological and behavioral problems in children. Environ. Res. 2017, 158, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Fruh, V.; Rifas-Shiman, S.L.; Amarasiriwardena, C.; Cardenas, A.; Bellinger, D.C.; Wise, L.A.; White, R.F.; Wright, R.O.; Oken, E.; Claus Henn, B. Prenatal lead exposure and childhood executive function and behavioral difficulties in project viva. Neurotoxicology 2019, 75, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Van Goozen, S.H.M.; Matthys, W.; Cohen-Kettenis, P.T.; Buitelaar, J.K.; Van Engeland, H. Hypothalamic-pituitary-adrenal axis and autonomic nervous system activity in disruptive children and matched controls. J. Am. Acad. Child Adolesc. Psychiatry 2000, 39, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- Popma, A.; Jansen, L.M.C.; Vermeiren, R.; Steiner, H.; Raine, A.; Van Goozen, S.H.M.; van Engeland, H.; Doreleijers, T.A.H. Hypothalamus pituitary adrenal axis and autonomic activity during stress in delinquent male adolescents and controls. Psychoneuroendocrinology 2006, 31, 948–957. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Lazcano, J.C.; López-Quiroz, A.; Alcantar-Almaraz, R.; Montes, S.; Sánchez-Mendoza, A.; Alcaraz-Zubeldia, M.; Tristán-López, L.A.; Sánchez-Hernández, B.E.; Morales-Martínez, A.; Ríos, C.; et al. A hypothesis of the interaction of the nitrergic and serotonergic systems in aggressive behavior induced by exposure to lead. Front. Behav. Neurosci. 2018, 12, 202. [Google Scholar] [CrossRef]
- White, L.D.; Cory-Slechta, D.A.; Gilbert, M.E.; Tiffany-Castiglioni, E.; Zawia, N.H.; Virgolini, M.; Rossi-George, A.; Lasley, S.M.; Qian, Y.C.; Basha, M.R. New and evolving concepts in the neurotoxicology of lead. Toxicol. Appl. Pharmacol. 2007, 225, 1–27. [Google Scholar] [CrossRef]
- Cory-Slechta, D.A.; Virgolini, M.B.; Rossi-George, A.; Thiruchelvam, M.; Lisek, R.; Weston, D. Lifetime consequences of combined maternal lead and stress. Basic Clin. Pharmacol. Toxicol. 2008, 102, 218–227. [Google Scholar] [CrossRef]
- Mason, L.H.; Harp, J.P.; Han, D.Y. Pb neurotoxicity: Neuropsychological effects of lead toxicity. Biomed Res. Int. 2014, 2014, 840547. [Google Scholar] [CrossRef] [Green Version]
- Fiłon, J.; Ustymowicz-Farbiszewska, J.; Krajewska-Kułak, E. Analysis of lead, arsenic and calcium content in the hair of children with autism spectrum disorder. BMC Public Health 2020, 20, 383. [Google Scholar] [CrossRef]
- Lakshmi Priya, M.D.; Geetha, A. Level of trace elements (copper, zinc, magnesium and selenium) and toxic elements (lead and mercury) in the hair and nail of children with autism. Biol. Trace Elem. Res. 2011, 142, 148–158. [Google Scholar] [CrossRef]
- Guilarte, T.R.; Opler, M.; Pletnikov, M. Is lead exposure in early life an environmental risk factor for Schizophrenia? Neurobiological connections and testable hypotheses. Neurotoxicology 2012, 33, 560–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opler, M.G.A.; Buka, S.L.; Groeger, J.; McKeague, I.; Wei, C.; Factor-Litvak, P.; Bresnahan, M.; Graziano, J.; Goldstein, J.M.; Seidman, L.J.; et al. Prenatal exposure to lead, δ-aminolevulinic acid, and schizophrenia: Further evidence. Environ. Health Perspect. 2008, 116, 1586–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opler, M.G.A.; Brown, A.S.; Graziano, J.; Desai, M.; Zheng, W.; Schaefer, C.; Factor-Litvak, P.; Susser, E.S. Prenatal lead exposure, δ-aminolevulinic acid, and schizophrenia. Environ. Health Perspect. 2004, 112, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Guilarte, T. 94. Early life lead exposure and schizophrenia: An environmental model of NMDA receptor hypofunction. Biol. Psychiatry 2017, 81, S39–S40. [Google Scholar] [CrossRef]
- Stansfield, K.H.; Ruby, K.N.; Soares, B.D.; McGlothan, J.L.; Liu, X.; Guilarte, T.R. Early-life lead exposure recapitulates the selective loss of parvalbumin-positive GABAergic interneurons and subcortical dopamine system hyperactivity present in schizophrenia. Transl. Psychiatry 2015, 5, e522. [Google Scholar] [CrossRef] [Green Version]
- Abazyan, B.; Dziedzic, J.; Hua, K.; Abazyan, S.; Yang, C.; Mori, S.; Pletnikov, M.V.; Guilarte, T.R. Chronic exposure of mutant DISC1 mice to lead produces sex-dependent abnormalities consistent with schizophrenia and related mental disorders: A gene-environment interaction study. Schizophr. Bull. 2014, 40, 575–584. [Google Scholar] [CrossRef] [Green Version]
- Bakulski, K.M.; Rozek, L.S.; Dolinoy, D.C.; Paulson, H.L.; Hu, H. Alzheimer’s disease and environmental exposure to lead: The epidemiologic evidence and potential role of epigenetics. Curr. Alzheimer Res. 2012, 9, 563–573. [Google Scholar] [CrossRef]
- Bakulski, K.M.; Seo, Y.A.; Hickman, R.C.; Brandt, D.; Vadari, H.S.; Hu, H.; Park, S.K. Heavy metals exposure and Alzheimer’s disease and related dementias. J. Alzheimer’s Dis. 2020, 76, 1215–1242. [Google Scholar] [CrossRef]
- Reuben, A. Childhood lead exposure and adult neurodegenerative disease. J. Alzheimer’s Dis. 2018, 64, 17–42. [Google Scholar] [CrossRef]
- Huat, T.J.; Camats-Perna, J.; Newcombe, E.A.; Valmas, N.; Kitazawa, M.; Medeiros, R. Metal toxicity links to Alzheimer’s disease and neuroinflammation. J. Mol. Biol. 2019, 431, 1843–1868. [Google Scholar] [CrossRef]
- Dosunmu, R.; Wu, J.; Basha, M.R.; Zawia, N.H. Environmental and dietary risk factors in Alzheimer’s disease. Expert Rev. Neurother. 2007, 7, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Zawia, N.H.; Lahiri, D.K.; Cardozo-Pelaez, F. Epigenetics, oxidative stress, and Alzheimer disease. Free Radic. Biol. Med. 2009, 46, 1241–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, A.; Khan, K.; Al-Khaledi, G.; Khan, I.; Attur, S. Early postnatal lead exposure induces tau phosphorylation in the brain of young rats. Acta Biol. Hung. 2012, 63, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Gassowska, M.; Baranowska-Bosiacka, I.; Moczydłowska, J.; Tarnowski, M.; Pilutin, A.; Gutowska, I.; Struzyńska, L.; Chlubek, D.; Adamczyk, A. Perinatal exposure to lead (Pb) promotes Tau phosphorylation in the rat brain in a GSK-3β and CDK5 dependent manner: Relevance to neurological disorders. Toxicology 2016, 347–349, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Yu, Z.L.; WaNg, L.; ZheNg, Y.T.; Jia, J.X.; WaNg, Q.; Zhu, M.J.; Liu, X.H.; Xia, X.; Li, W.J. Increased tau phosphorylation and beta amyloid in the hipocampus of mouse pups by early life lead exposure. Acta Biol. Hung. 2010, 61, 123–134. [Google Scholar] [CrossRef]
- Lee, J.; Freeman, J.L. Zebrafish as a model for investigating developmental lead (Pb) neurotoxicity as a risk factor in adult neurodegenerative disease: A mini-review. Neurotoxicology 2014, 43, 57–64. [Google Scholar] [CrossRef]
- Basha, R.; Rajarami, G.R. Developmental exposure to lead and late life abnormalities of nervous system. Indian J. Exp. Biol. 2010, 48, 636–641. [Google Scholar]
- Bihaqi, S.W. Early life exposure to lead (Pb) and changes in DNA methylation: Relevance to Alzheimer’s disease. Rev. Environ. Health 2019, 34, 187–195. [Google Scholar] [CrossRef]
- Chin-Chan, M.; Cobos-Puc, L.; Alvarado-Cruz, I.; Bayar, M.; Ermolaeva, M. Early-life Pb exposure as a potential risk factor for Alzheimer’s disease: Are there hazards for the Mexican population? J. Biol. Inorg. Chem. 2019, 24, 1285–1303. [Google Scholar] [CrossRef]
- Eid, A.; Zawia, N. Consequences of lead exposure, and it’s emerging role as an epigenetic modifier in the aging brain. Neurotoxicology 2016, 56, 254–261. [Google Scholar] [CrossRef]
- Feng, C.; Liu, S.; Zhou, F.; Gao, Y.; Li, Y.; Du, G.; Chen, Y.; Jiao, H.; Feng, J.; Zhang, Y.; et al. Oxidative stress in the neurodegenerative brain following lifetime exposure to lead in rats: Changes in lifespan profiles. Toxicology 2019, 411, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Zawia, N.H.; Basha, M.R. Environmental risk factors and the developmental basis for Alzheimer’s disease. Rev. Neurosci. 2005, 16, 325–337. [Google Scholar] [CrossRef] [PubMed]
- García-Lestón Julia, J.; Méndez, J.; Pásaro, E.; Laffon, B. Genotoxic effects of lead: An updated review. Environ. Int. 2010, 36, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Garza, A.; Vega, R.; Soto, E. Cellular mechanisms of lead neurotoxicity. Med. Sci. Monit. 2006, 12, RA57–RA65. [Google Scholar]
- Luo, W.; Ruan, D.; Yan, C.; Yin, S.; Chen, J. Effects of chronic lead exposure on functions of nervous system in Chinese children and developmental rats. Neurotoxicology 2012, 33, 862–871. [Google Scholar] [CrossRef]
- Verstraeten, S.V.; Aimo, L.; Oteiza, P.I. Aluminium and lead: Molecular mechanisms of brain toxicity. Arch. Toxicol. 2008, 82, 789–802. [Google Scholar] [CrossRef]
- Marchetti, C. Molecular targets of lead in brain neurotoxicity. Neurotox. Res. 2003, 5, 221–236. [Google Scholar] [CrossRef]
- Metryka, E.; Chibowska, K.; Gutowska, I.; Falkowska, A.; Kupnicka, P.; Barczak, K.; Chlubek, D.; Baranowska-Bosiacka, I. Lead (Pb) exposure enhances expression of factors associated with inflammation. Int. J. Mol. Sci. 2018, 19, 1813. [Google Scholar] [CrossRef] [Green Version]
- Neal, A.P.; Guilarte, T.R. Molecular neurobiology of lead (Pb2+): Effects on synaptic function. Mol. Neurobiol. 2010, 42, 151–160. [Google Scholar] [CrossRef] [Green Version]
- Chibowska, K.; Baranowska-Bosiacka, I.; Falkowska, A.; Gutowska, I.; Goschorska, M.; Chlubek, D. Effect of lead (Pb) on inflammatory processes in the brain. Int. J. Mol. Sci. 2016, 17, 2140. [Google Scholar] [CrossRef] [Green Version]
- Baranowska-Bosiacka, I.; Gutowska, I.; Rybicka, M.; Nowacki, P.; Chlubek, D. Neurotoxicity of lead. Hypothetical molecular mechanisms of synaptic function disorders. Neurol. Neurochir. Pol. 2012, 46, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Toscano, C.D.; Guilarte, T.R. Lead neurotoxicity: From exposure to molecular effects. Brain Res. Rev. 2005, 49, 529–554. [Google Scholar] [CrossRef] [PubMed]
- Kasten-Jolly, J.; Lawrence, D.A. Sex-specific effects of developmental lead exposure on the immune-neuroendocrine network. Toxicol. Appl. Pharmacol. 2017, 334, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Zhao, X.; Yu, J.; Xie, J.; Li, C.; Liu, D.; Tang, C.; Wang, C. Lead-induced oxidative damage in rats/mice: A meta-analysis. J. Trace Elem. Med. Biol. 2020, 58, 126443. [Google Scholar] [CrossRef]
- Karri, V.; Schuhmacher, M.; Kumar, V. Heavy metals (Pb, Cd, As and MeHg) as risk factors for cognitive dysfunction: A general review of metal mixture mechanism in brain. Environ. Toxicol. Pharmacol. 2016, 48, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Andrade, V.M.; Aschner, M.; Marreilha dos Santos, A.P. Neurotoxicity of metal mixtures. Adv. Neurobiol. 2017, 18, 227–265. [Google Scholar] [CrossRef]
- Wu, X.; Cobbina, S.J.; Mao, G.; Xu, H.; Zhang, Z.; Yang, L. A review of toxicity and mechanisms of individual and mixtures of heavy metals in the environment. Environ. Sci. Pollut. Res. 2016, 23, 8244–8259. [Google Scholar] [CrossRef]
- May, J.M. Vitamin C transport and its role in the central nervous system. Subcell. Biochem. 2012, 56, 85–103. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, U. The sour side of vitamin C might mediate neuroprotective, anticonvulsive and antidepressant-like effects. Med. Hypotheses 2019, 131, 109320. [Google Scholar] [CrossRef]
- Covarrubias-Pinto, A.; Acuña, A.I.; Beltrán, F.A.; Torres-Díaz, L.; Castro, M.A. Old things new view: Ascorbic acid protects the brain in neurodegenerative disorders. Int. J. Mol. Sci. 2015, 16, 28194–28217. [Google Scholar] [CrossRef]
- Carpenter, K.J. The discovery of vitamin C. Ann. Nutr. Metab. 2012, 61, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Nishikimi, M. Oxidation of ascorbic acid with superoxide anion generated by the xanthine-xanthine oxidase system. Biochem. Biophys. Res. Commun. 1975, 63, 463–468. [Google Scholar] [CrossRef]
- Bodannes, R.S.; Chan, P.C. Ascorbic acid as a scavenger of singlet oxygen. FEBS Lett. 1979, 105, 195–196. [Google Scholar] [CrossRef] [Green Version]
- Machlin, L.J.; Bendich, A. Free radical tissue damage: Protective role of antioxidant nutrients 1. FASEB J. 1987, 1, 441–445. [Google Scholar] [CrossRef]
- Niki, E. Action of ascorbic acid as a scavenger of active and stable oxygen radicals. Am. J. Clin. Nutr. 1991, 54, 1119S–1124S. [Google Scholar] [CrossRef] [Green Version]
- Winkler, B.S.; Orselli, S.M.; Rex, T.S. The redox couple between glutathione and ascorbic acid: A chemical and physiological perspective. Free Radic. Biol. Med. 1994, 17, 333–349. [Google Scholar] [CrossRef]
- Li, X.; Cobb, C.E.; Hill, K.E.; Burk, R.F.; May, J.M. Mitochondrial uptake and recycling of ascorbic acid. Arch. Biochem. Biophys. 2001, 387, 143–153. [Google Scholar] [CrossRef]
- Rose, R.C. Cerebral metabolism of oxidized ascorbate. Brain Res. 1993, 628, 49–55. [Google Scholar] [CrossRef]
- Fornai, F.; Piaggi, S.; Gesi, M.; Saviozzi, M.; Lenzi, P.; Paparelli, A.; Casini, A.F. Subcellular localization of a glutathione-dependent dehydroascorbate reductase within specific rat brain regions. Neuroscience 2001, 104, 15–31. [Google Scholar] [CrossRef]
- Getoff, N. Vitamin C: Electron emission, free radicals and biological versatility. In Vivo (Brooklyn) 2013, 27, 565–570. [Google Scholar]
- May, J.M. Is ascorbic acid an antioxidant for the plasma membrane? FASEB J. 1999, 13, 995–1006. [Google Scholar] [CrossRef] [PubMed]
- Barnes, M.J. Function of ascorbic acid in collagen metabolism. Ann. N. Y. Acad. Sci. 1975, 258, 264–277. [Google Scholar] [CrossRef] [PubMed]
- Murad, S.; Grove, D.; Lindberg, K.A.; Reynolds, G.; Sivarajah, A.; Pinnell, S.R. Regulation of collagen synthesis by ascorbic acid. Proc. Natl. Acad. Sci. USA 1981, 78, 2879–2882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diliberto, E.J.; Menniti, F.S.; Knoth, J.; Daniels, A.J.; Kizer, J.S.; Viveros, O.H. Adrenomedullary chromaffin cells as a model to study the neurobiology of ascorbic acid: From monooxygenation to neuromodulation. Ann. N. Y. Acad. Sci. 1987, 498, 28–53. [Google Scholar] [CrossRef] [PubMed]
- Glembotski, C.C. The role of ascorbic acid in the biosynthesis of the neuroendocrine peptides α-MSH and TRH. Ann. N. Y. Acad. Sci. 1987, 498, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Vitamin C: Antioxidant or pro-oxidant in vivo? Free Radic. Res. 1996, 25, 439–454. [Google Scholar] [CrossRef]
- Carr, A.; Frei, B. Does vitamin C act as a pro-oxidant under physiological conditions? FASEB J. 1999, 13, 1007–1024. [Google Scholar] [CrossRef] [Green Version]
- Podmore, I.D.; Griffiths, H.R.; Herbert, K.E.; Mistry, N.; Mistry, P.; Lunec, J. Vitamin C exhibits pro-oxidant properties. Nature 1998, 393, 559. [Google Scholar] [CrossRef]
- Poulsen, H.E.; Weimann, A.; Salonen, J.T.; Nyyssönen, K.; Loft, S.; Cadet, J.; T Douki, J.; Ravanat, L. Does vitamin C have a pro-oxidant effect? Nature 1998, 395, 231–232. [Google Scholar] [CrossRef]
- Levine, M.; Daruwala, R.C.; Park, J.B.; Rumsey, S.C.; Wang, Y. Does vitamin C have a pro-oxidant effect? Nature 1998, 395, 231. [Google Scholar] [CrossRef]
- Rowley, D.A.; Halliwell, B. Superoxide-dependent formation of hydroxyl radicals in the presence of thiol compounds. FEBS Lett. 1982, 138, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Laughton, M.J.; Halliwell, B.; Evans, P.J.; Robin, J.; Hoult, S. Antioxidant and pro-oxidant actions of the plant phenolics quercetin, gossypol and myricetin. Effects on lipid peroxidation, hydroxyl radical generation and bleomycin-dependent damage to DNA. Biochem. Pharmacol. 1989, 38, 2859–2865. [Google Scholar] [CrossRef]
- Ohno, S.; Ohno, Y.; Suzuki, N.; Soma, G.I.; Inoue, M. High-dose vitamin C (ascorbic acid) therapy in the treatment of patients with advanced cancer. Anticancer Res. 2009, 29, 809–815. [Google Scholar] [PubMed]
- Blanchard, J.; Tozer, T.N.; Rowland, M. Pharmacokinetic perspectives on mega doses of ascorbic acid. Am. J. Clin. Nutr. 1997, 66, 1165–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, G.J.; Sherry, S.; Ralli, E.P. The mechanisms of the excretion of vitamin C by the human kidney at low and normal plasma levels of ascorbic acid. J. Clin. Investig. 1940, 19, 685–689. [Google Scholar] [CrossRef] [Green Version]
- Hathcock, J.N.; Azzi, A.; Blumberg, J.; Bray, T.; Dickinson, A.; Frei, B.; Jialal, I.; Johnston, C.S.; Kelly, F.J.; Kraemer, K.; et al. Vitamins E and C are safe across a broad range of intakes. Am. J. Clin. Nutr. 2005, 81, 736–745. [Google Scholar] [CrossRef]
- Johnston, C.S. Biomarkers for establishing a tolerable upper intake level for vitamin C. Nutr. Rev. 1999, 57, 71–77. [Google Scholar] [CrossRef]
- Frei, B.; Birlouez-Aragon, I.; Lykkesfeldt, J. Authors’ perspective: What is the optimum intake of vitamin C in humans? Crit. Rev. Food Sci. Nutr. 2012, 52, 815–829. [Google Scholar] [CrossRef]
- Fowler, A.A.; Syed, A.A.; Knowlson, S.; Sculthorpe, R.; Farthing, D.; DeWilde, C.; Farthing, C.A.; Larus, T.L.; Martin, E.; Brophy, D.F.; et al. Phase I safety trial of intravenous ascorbic acid in patients with severe sepsis. J. Transl. Med. 2014, 12, 32. [Google Scholar] [CrossRef] [Green Version]
- Ou, J.; Zhu, X.; Lu, Y.; Zhao, C.; Zhang, H.; Wang, X.; Gui, X.; Wang, J.; Zhang, X.; Zhang, T.; et al. The safety and pharmacokinetics of high dose intravenous ascorbic acid synergy with modulated electrohyperthermia in Chinese patients with stage III-IV non-small cell lung cancer. Eur. J. Pharm. Sci. 2017, 109, 412–418. [Google Scholar] [CrossRef]
- Stephenson, C.M.; Levin, R.D.; Spector, T.; Lis, C.G. Phase I clinical trial to evaluate the safety, tolerability, and pharmacokinetics of high-dose intravenous ascorbic acid in patients with advanced cancer. Cancer Chemother. Pharmacol. 2013, 72, 139–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendich, A.; Langseth, L. The health effects of vitamin C supplementation: A review. J. Am. Coll. Nutr. 1995, 14, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Lykkesfeldt, J.; Tveden-Nyborg, P. The pharmacokinetics of vitamin C. Nutrients 2019, 11, 2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickey, S.; Roberts, H.J.; Miller, N.J. Pharmacokinetics of oral vitamin C. J. Nutr. Environ. Med. 2008, 17, 169–177. [Google Scholar] [CrossRef]
- Parhizkar, E.; Rashedinia, M.; Karimi, M.; Alipour, S. Design and development of vitamin C-encapsulated proliposome with improved in-vitro and ex-vivo antioxidant efficacy. J. Microencapsul. 2018, 35, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Łukawski, M.; Dałek, P.; Borowik, T.; Foryś, A.; Langner, M.; Witkiewicz, W.; Przybyło, M. New oral liposomal vitamin C formulation: Properties and bioavailability. J. Liposome Res. 2020, 30, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Hornig, D. Distribution of ascorbic acid, metabolites and alanogues in man and animals. Ann. N. Y. Acad. Sci. 1975, 258, 103–117. [Google Scholar] [CrossRef]
- Rice, M.E. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000, 23, 209–216. [Google Scholar] [CrossRef]
- Smythies, J.R. The role of ascorbate in brain: Therapeutic implications. J. R. Soc. Med. 1996, 89, 241. [Google Scholar] [CrossRef]
- Ballaz, S.J.; Rebec, G.V. Neurobiology of vitamin C: Expanding the focus from antioxidant to endogenous neuromodulator. Pharmacol. Res. 2019, 146, 104321. [Google Scholar] [CrossRef]
- Kratzing, C.C.; Kelly, J.D.; Kratzing, J.E. Ascorbic acid in fetal rat brain. J. Neurochem. 1985, 44, 1623–1624. [Google Scholar] [CrossRef] [PubMed]
- Spector, R.; Lorenzo, A.V. Ascorbic acid homeostasis in the central nervous system. Am. J. Physiol. 1973, 225, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Tsukaguchi, H.; Tokui, T.; Mackenzle, B.; Berger, U.V.; Chen, X.Z.; Wang, Y.; Brubaker, R.F.; Hediger, M.A. A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature 1999, 399, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; May, J.M. Vitamin C function in the brain: Vital role of the ascorbate transporter SVCT2. Free Radic. Biol. Med. 2009, 46, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Lam, D.K.C.; Daniel, P.M. The influx of ascorbic acid into the rat’s brain. Q. J. Exp. Physiol. 1986, 71, 483–489. [Google Scholar] [CrossRef] [Green Version]
- Agus, D.B.; Gambhir, S.S.; Pardridge, W.M.; Spielholz, C.; Baselga, J.; Vera, J.C.; Golde, D.W. Vitamin C crosses the blood-brain barrier in the oxidized form through the glucose transporters. J. Clin. Investig. 1997, 100, 2842–2848. [Google Scholar] [CrossRef] [Green Version]
- Nualart, F.J.; Rivas, C.I.; Montecinos, V.P.; Godoy, A.S.; Guaiquil, V.H.; Golde, D.W.; Vera, J.C. Recycling of vitamin C by a bystander effect. J. Biol. Chem. 2003, 278, 10128–10133. [Google Scholar] [CrossRef] [Green Version]
- Rice, M.E.; Russo-Menna, I. Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience 1997, 82, 1213–1223. [Google Scholar] [CrossRef]
- Nualart, F. Vitamin C Transporters, Recycling and the Bystander Effect in the Nervous System: SVCT2 versus Gluts. J. Stem Cell Res. Ther. 2014, 4, 209. [Google Scholar] [CrossRef] [Green Version]
- Miele, M.; Boutelle, M.G.; Fillenz, M. The physiologically induced release of ascorbate in rat brain is dependent on impulse traffic, calcium influx and glutamate uptake. Neuroscience 1994, 62, 87–91. [Google Scholar] [CrossRef]
- Cammack, J.; Ghasemzadeh, B.; Adams, R.N. The pharmacological profile of glutamate-evoked ascorbic acid efflux measured by in vivo electrochemistry. Brain Res. 1991, 565, 17–22. [Google Scholar] [CrossRef]
- Siushansian, R.; Tao, L.; Dixon, S.J.; Wilson, J.X. Cerebral astrocytes transport ascorbic acid and dehydroascorbic acid through distinct mechanisms regulated by cyclic AMP. J. Neurochem. 1997, 68, 2378–2385. [Google Scholar] [CrossRef] [PubMed]
- Vera, J.C.; Rivas, C.I.; Fischbarg, J.; Golde, D.W. Mammalian facilitative hexose transporters mediate the transport of dehydroascorbic acid. Nature 1993, 364, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Ghasemzedah, B.; Cammack, J.; Adams, R.N. Dynamic changes in extracellular fluid ascorbic acid monitored by in vivo electrochemistry. Brain Res. 1991, 547, 162–166. [Google Scholar] [CrossRef]
- O’Neill, R.D.; Fillenz, M.; Sundstrom, L.; Rawlins, J.N.P. Voltammetrically monitored brain ascorbate as an index of excitatory amino acid release in the unrestrained rat. Neurosci. Lett. 1984, 52, 227–233. [Google Scholar] [CrossRef]
- Acuña, A.I.; Esparza, M.; Kramm, C.; Beltrán, F.A.; Parra, A.V.; Cepeda, C.; Toro, C.A.; Vidal, R.L.; Hetz, C.; Concha, I.I.; et al. A failure in energy metabolism and antioxidant uptake precede symptoms of Huntington’s disease in mice. Nat. Commun. 2013, 4, 2917. [Google Scholar] [CrossRef] [Green Version]
- Rumsey, S.C.; Kwon, O.; Xu, G.W.; Burant, C.F.; Simpson, I.; Levine, M. Glucose transporter isoforms GLUT1 and GLUT3 transport dehydroascorbic acid. J. Biol. Chem. 1997, 272, 18982–18989. [Google Scholar] [CrossRef] [Green Version]
- Astuya, A.; Caprile, T.; Castro, M.; Salazar, K.; De Los Angeles García, M.; Reinicke, K.; Rodríguez, F.; Vera, J.C.; Millán, C.; Ulloa, V.; et al. Vitamin C uptake and recycling among normal and tumor cells from the central nervous system. J. Neurosci. Res. 2005, 79, 146–156. [Google Scholar] [CrossRef]
- Figueroa-Méndez, R.; Rivas-Arancibia, S. Vitamin C in health and disease: Its role in the metabolism of cells and redox state in the brain. Front. Physiol. 2015, 6, 397. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.X. Antioxidant defense of the brain: A role for astrocytes. Can. J. Physiol. Pharmacol. 1997, 75, 1149–1163. [Google Scholar] [CrossRef]
- Subramanian, N. On the brain ascorbic acid and its importance in metabolism of biogenic amines. Life Sci. 1977, 20, 1479–1484. [Google Scholar] [CrossRef]
- Grünewald, R.A. Ascorbic acid in the brain. Brain Res. Rev. 1993, 18, 123–133. [Google Scholar] [CrossRef]
- Rebec, G.V.; Christopher Pierce, R. A vitamin as neuromodulator: Ascorbate release into the extracellular fluid of the brain regulates dopaminergic and glutamatergic transmission. Prog. Neurobiol. 1994, 43, 537–565. [Google Scholar] [CrossRef]
- Han, Q.; Shen, T.; Wang, F.; Wu, P.; Chen, J. Preventive and therapeutic potential of vitamin C in mental disorders. Curr. Med. Sci. 2018, 38, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Padayatty, S.J.; Katz, A.; Wang, Y.; Eck, P.; Kwon, O.; Lee, J.H.; Chen, S.; Corpe, C.; Levine, M.; Dutta, A.; et al. Vitamin C as an antioxidant: Evaluation of its role in disease prevention. J. Am. Coll. Nutr. 2003, 22, 18–35. [Google Scholar] [CrossRef]
- Camarena, V.; Wang, G. The epigenetic role of vitamin C in health and disease. Cell. Mol. Life Sci. 2016, 73, 1645–1658. [Google Scholar] [CrossRef] [Green Version]
- Duarte, T.L.; Lunec, J. Review part of the series: From dietary antioxidants to regulators in cellular signalling and gene expression Review: When is an antioxidant not an antioxidant? A review of novel actions and reactions of vitamin C. Free Radic. Res. 2005, 39, 671–686. [Google Scholar] [CrossRef]
- Tveden-Nyborg, P.; Vogt, L.; Schjoldager, J.G.; Jeannet, N.; Hasselholt, S.; Paidi, M.D.; Christen, S.; Lykkesfeldt, J. Maternal vitamin C deficiency during pregnancy persistently impairs hippocampal neurogenesis in offspring of guinea pigs. PLoS ONE 2012, 7, e48488. [Google Scholar] [CrossRef] [Green Version]
- Harrison, F.E.; Meredith, M.E.; Dawes, S.M.; Saskowski, J.L.; May, J.M. Low ascorbic acid and increased oxidative stress in gulo (−/−) mice during development. Brain Res. 2010, 1349, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Eldridge, C.F.; Bunge, M.B.; Bunge, R.P.; Wood, P.M. Differentiation of axon-related Schwann cells in vitro. I. Ascorbic acid regulates basal lamina assembly and myelin formation. J. Cell Biol. 1987, 105, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Chang, M.Y.; Park, C.H.; Kim, H.Y.; Kim, J.H.; Son, H.; Lee, Y.S.; Lee, S.H. Ascorbate-induced differentiation of embryonic cortical precursors into neurons and astrocytes. J. Neurosci. Res. 2003, 73, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Oyarce, K.; Silva-Alvarez, C.; Ferrada, L.; Martínez, F.; Salazar, K.; Nualart, F. SVCT2 Is expressed by cerebellar precursor cells, which differentiate into neurons in response to ascorbic acid. Mol. Neurobiol. 2018, 55, 1136–1149. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Curran, C.P.; Nebert, D.W.; Patel, K.V.; Williams, M.T.; Vorhees, C.V. Effect of vitamin C deficiency during postnatal development on adult behavior: Functional phenotype of Gulo(−/−) knockout mice. Genes Brain Behav. 2012, 11, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.M.; Ahn, J.I.; Lee, K.H.; Lee, Y.S.; Lee, Y.S. Ascorbic acid responsive genes during neuronal differentiation of embryonic stem cells. Neuroreport 2004, 15, 1959–1963. [Google Scholar] [CrossRef]
- Nualart, F.; Salazar, K.; Oyarce, K.; Cisternas, P.; Jara, N.; Silva-Álvarez, C.; Pastor, P.; Martínez, F.; García, A.; de los Ángeles García-Robles, M.; et al. Typical and atypical stem cells in the brain, vitamin C effect and neuropathology. Biol. Res. 2012, 45, 243–256. [Google Scholar] [CrossRef]
- Bowman, G.L. Ascorbic acid, cognitive function, and Alzheimer’s disease: A current review and future direction. BioFactors 2012, 38, 114–122. [Google Scholar] [CrossRef] [Green Version]
- Siqueira, I.R.; Elsner, V.R.; Leite, M.C.; Vanzella, C.; Moysés, F.D.S.; Spindler, C.; Godinho, G.; Battú, C.; Wofchuk, S.; Souza, D.O.; et al. Ascorbate uptake is decreased in the hippocampus of ageing rats. Neurochem. Int. 2011, 58, 527–532. [Google Scholar] [CrossRef]
- Warner, T.A.; Kang, J.Q.; Kennard, J.A.; Harrison, F.E. Low brain ascorbic acid increases susceptibility to seizures in mouse models of decreased brain ascorbic acid transport and Alzheimer’s disease. Epilepsy Res. 2015, 110, 20–25. [Google Scholar] [CrossRef] [Green Version]
- Nam, S.M.; Seo, M.; Seo, J.S.; Rhim, H.; Nahm, S.S.; Cho, I.H.; Chang, B.J.; Kim, H.J.; Choi, S.H.; Nah, S.Y. Ascorbic acid mitigates D-galactose-induced brain aging by increasing hippocampal neurogenesis and improving memory function. Nutrients 2019, 11, 176. [Google Scholar] [CrossRef] [Green Version]
- Charlton, K.E.; Rabinowitz, T.L.; Geffen, L.N.; Dhansay, M.A. Lowered plasma vitamin C, but not vitamin E, concentrations in dementia patients. J. Nutr. Health Aging 2004, 8, 99–107. [Google Scholar]
- Rivière, S.; Birlouez-Aragon, I.; Nourhashémi, F.; Vellas, B. Low plasma vitamin C in Alzheimer patients despite an adequate diet. Int. J. Geriatr. Psychiatry 1998, 13, 749–754. [Google Scholar] [CrossRef]
- Zandi, P.P.; Anthony, J.C.; Khachaturian, A.S.; Stone, S.V.; Gustafson, D.; Tschanz, J.A.T.; Norton, M.C.; Welsh-Bohmer, K.A.; Breitner, J.C.S. Reduced risk of Alzheimer disease in users of antioxidant vitamin supplements: The Cache County Study. Arch. Neurol. 2004, 61, 82–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gariballa, S. Poor vitamin C status is associated with increased depression symptoms following acute illness in older people. Int. J. Vitam. Nutr. Res. 2014, 84, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Tandon, S.K.; Flora, S.J.S. Therapeutic efficacy of dimercaptosuccinic acid and thiamine/ascorbic acid on lead intoxication in rats. Bull. Environ. Contam. Toxicol. 1989, 43, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Niazi, S.; Lim, J.; Bederka, J.P. 1982. Effect of ascorbic acid on the renal excretion of lead in rats. J. Pharm. Sci. 1982, 71, 1189–1190. [Google Scholar] [CrossRef] [PubMed]
- Dalley, J.W.; Gupta, P.K.; Lam, F.C.; Hung, C.T. Interaction of L-ascorbic acid on the disposition of lead in rats. Pharmacol. Toxicol. 1989, 50, 337–348. [Google Scholar] [CrossRef]
- Morton, A.P.; Partridge, S.; Blair, J.A. The intestinal uptake of lead. Chem. Br. 1985, 15, 923–927. [Google Scholar]
- Lihm, H.; Kim, H.; Chang, H.; Yoon, M.; Lee, K.; Choi, J. Vitamin C modulates lead excretion in rats. Anat. Cell Biol. 2013, 46, 239–245. [Google Scholar] [CrossRef] [Green Version]
- Antonio-García, M.T.; Massó-Gonzalez, E.L. Toxic effects of perinatal lead exposure on the brain of rats: Involvement of oxidative stress and the beneficial role of antioxidants. Food Chem. Toxicol. 2008, 46, 2089–2095. [Google Scholar] [CrossRef]
- Flora, S.J.S.; Pande, M.; Mehta, A. Beneficial effect of combined administration of some naturally occurring antioxidants (vitamins) and thiol chelators in the treatment of chronic lead intoxication. Chem. Biol. Interact. 2003, 145, 267–280. [Google Scholar] [CrossRef]
- Tariq, S.A. Role of ascorbic acid in scavenging free radicals and lead toxicity from biosystems. Mol. Biotechnol. 2007, 37, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Gurer, H.; Ercal, N. Can antioxidants be beneficial in the treatment of lead poisoning? Free Radic. Biol. Med. 2000, 29, 927–945. [Google Scholar] [CrossRef]
- Zhai, Q.; Yang, L.; Zhao, J.; Zhang, H.; Tian, F.; Chen, W. Protective effects of dietary supplements containing probiotics, micronutrients, and plant extracts against lead toxicity in mice. Front. Microbiol. 2018, 9, 2134. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.C.; Jang, T.W.; Chae, H.J.; Choi, W.J.; Ha, M.N.; Ye, B.J.; Kim, B.G.; Jeon, M.J.; Kim, S.Y.; Hong, Y.S. Evaluation and management of lead exposure. Ann. Occup. Environ. Med. 2015, 27, 30. [Google Scholar] [CrossRef] [Green Version]
- Flora, S.J.S.; Pachauri, V. Chelation in metal intoxication. Int. J. Environ. Res. Public Health 2010, 7, 2745–2788. [Google Scholar] [CrossRef] [Green Version]
- Lowry, J. Oral chelation therapy for patients with lead poisoning. Am. Acad. Pediatr. 2010, 116, 1036–1046. [Google Scholar]
- Kalia, K.; Flora, S.J.S. Strategies for safe and effective therapeutic measures for chronic arsenic and lead poisoning. J. Occup. Health 2005, 47, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, K.N.; Ware, J.H.; Salganik, M.; Radcliffe, J.; Rogan, W.J.; Rhoads, G.G.; Fay, M.E.; Davoli, C.T.; Denckla, M.B.; Bornschein, R.L.; et al. Effect of chelation therapy on the neuropsychological and behavioral development of lead-exposed children after school entry. Pediatrics 2004, 114, 19–26. [Google Scholar] [CrossRef] [Green Version]
- McKay, C.A. Role of chelation in the treatment of lead poisoning: Discussion of the treatment of lead-exposed children trial (TLC). J. Med. Toxicol. 2013, 9, 339–343. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Liao, Y.; Jin, Y.; Zhao, Y.; Ren, Y.; Lu, C.; Li, G.; Li, Y.; Yang, J. Effects of in utero meso-2,3-dimercaptosuccinic acid with calcium and ascorbic acid on lead-induced fetal development. Arch. Toxicol. 2008, 82, 453–459. [Google Scholar] [CrossRef]
- Holmes, H.; Campbell, K.; Amberg, E. The effect of vitamin C on lead poisoning. J. Lab. Clin. Med. 1939, 24, 1119–1127. [Google Scholar]
- Marchmont-Robinson, S.W. Effect of vitamin C on workers exposed to lead dust. J. Lab. Clin. Med. 1941, 26, 1478–1481. [Google Scholar]
- Papaioannou, R.; Sohler, A.; Pfeiffer, C.C. Reduction of blood lead levels in battery workers by zinc and vitamin C. J. Orthomol. Psychiatry 1978, 7, 94–106. [Google Scholar]
- Ghanwat, G.; Patil, A.; Patil, J.; Kshirsagar, M.; Sontakke, A.; Ayachit, R.K. Effect of vitamin C supplementation on blood lead level, oxidative stress and antioxidant status of battery manufacturing workers of western Maharashtra, India. J. Clin. Diagn. Res. 2016, 10, BC08–BC11. [Google Scholar] [CrossRef]
- Abam, E.; Okediran, B.S.; Odukoya, O.O.; Adamson, I.; Ademuyiwa, O. Reversal of ionoregulatory disruptions in occupational lead exposure by vitamin C. Environ. Toxicol. Pharmacol. 2008, 26, 297–304. [Google Scholar] [CrossRef]
- Akinhanmi, O.; Ademuyiwa, O. Amelioration of lead toxicity in an occupationally exposed population with ascorbic acid. J. Chem. Soc. Niger. 2016, 41, 69–75. [Google Scholar]
- Onunkwor, B.; Dosumu, O.; Odukoya, O.O.; Arowolo, T.; Ademuyiwa, O. Biomarkers of lead exposure in petrol station attendants and auto-mechanics in Abeokuta, Nigeria: Effect of 2-week ascorbic acid supplementation. Environ. Toxicol. Pharmacol. 2004, 17, 169–176. [Google Scholar] [CrossRef]
- Ademuyiwa, O.; Ugbaja, R.N.; Ojo, D.A.; Owoigbe, A.O.; Adeokun, S.E. Reversal of aminolevulinic acid dehydratase (ALAD) inhibition and reduction of erythrocyte protoporphyrin levels by Vitamin C in occupational lead exposure in Abeokuta, Nigeria. Environ. Toxicol. Pharmacol. 2005. [Google Scholar] [CrossRef]
- Rendón-Ramírez, A.L.; Maldonado-Vega, M.; Quintanar-Escorza, M.A.; Hernández, G.; Arévalo-Rivas, B.I.; Zentella-Dehesa, A.; Calderón-Salinas, J.V. Effect of vitamin E and C supplementation on oxidative damage and total antioxidant capacity in lead-exposed workers. Environ. Toxicol. Pharmacol. 2014, 37, 45–54. [Google Scholar] [CrossRef]
- Vani, K.; Kurakula, M.; Syed, R.; Alharbi, K. Clinical relevance of vitamin C among lead-exposed infertile men. Genet. Test. Mol. Biomarkers 2012, 16, 1001–1006. [Google Scholar] [CrossRef]
- Tandon, S.K.; Chatterjee, M.; Bhargava, A.; Shukla, V.; Bihari, V. Lead poisoning in Indian silver refiners. Sci. Total Environ. 2001, 281, 177–182. [Google Scholar] [CrossRef]
- Sohler, A.; Kruesi, M.; Pfeiffer, C.C. Blood levels in psychiatric outpatients reduced by zinc and vitamin C. J. Orthomol. Psychiatry 1977, 6, 272–276. [Google Scholar]
- Flanagan, P.R.; Chamberlain, M.J.; Valberg, L.S. The relationship between iron and lead absorption in humans. Am. J. Clin. Nutr. 1982, 36, 823–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, E.B.; Evans, D.R.; Harris, W.A.; Teter, M.C.; McGanity, W.J. The effect of ascorbic acid supplementation on the blood lead levels of smokers. J. Am. Coll. Nutr. 1999, 18, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Lauwerys, R.; Roels, H.; Buchet, J.P.; Bernard, A.A.; Verhoeven, L.; Konings, J. The influence of orally-administered vitamin C or zinc on the absorption of and the biological response to lead. J. Occup. Med. 1983, 25, 668–678. [Google Scholar] [CrossRef]
- Calabrese, E.J.; Stoddard, A.; Leonard, D.A.; Dinardi, S.R. The effects of vitamin C supplementation on blood and hair levels of cadmium, lead, and mercury. Ann. N. Y. Acad. Sci. 1987, 498, 347–353. [Google Scholar] [CrossRef]
- Cheng, Y.; Willett, W.C.; Schwartz, J.; Sparrow, D.; Weiss, S.; Hu, H. Relation of nutrition to bone lead and blood lead levels in middle-aged to elderly men: The normative aging study. Am. J. Epidemiol. 1998, 147, 1162–1174. [Google Scholar] [CrossRef] [Green Version]
- Simon, J.A.; Hudes, E.S. Relationship of ascorbic acid to blood lead levels. J. Am. Med. Assoc. 1999, 281, 2289–2293. [Google Scholar] [CrossRef] [Green Version]
- Houston, D.K.; Johnson, M.A. Does vitamin C intake protect against lead toxicity? Nutr. Rev. 2000, 58, 73–79. [Google Scholar] [CrossRef]
- West, W.L.; Knight, E.M.; Edwards, C.H.; Manning, M.; Spurlock, B.; James, H.; Johnson, A.A.; Oyemade, U.J.; Cole, O.J.; Westney, O.E.; et al. Maternal low level lead and pregnancy outcomes. J. Nutr. 1994, 124, 981S–986S. [Google Scholar] [CrossRef]
- Altmann, P.; Maruna, R.F.L.; Maruna, H.; Michalica, W.; Wagner, G. Lead detoxication effect of a combined calcium phosphate and ascorbic acid therapy in pregnant women with increased lead burden. Wien. Med. Wochenschr. 1981, 131, 311–314. [Google Scholar]
- Hong, Y.C.; Oh, S.Y.; Kwon, S.O.; Park, M.S.; Kim, H.; Leem, J.H.; Ha, E.H. Blood lead level modifies the association between dietary antioxidants and oxidative stress in an urban adult population. Br. J. Nutr. 2013, 109, 148–154. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Yu, F.; Liao, Y.; Liu, S.; Liu, M.; Xu, J.; Yang, J. Therapeutic efficiency of succimer used with calcium and ascorbic acid in the treatment of mild lead-poisoning. Environ. Toxicol. Pharmacol. 2011, 31, 137–142. [Google Scholar] [CrossRef]
- Rudra Pal, D.; Chatterjee, J.; Chatterjee, G.C. Influence of lead administration on L-ascorbic acid metabolism in rats: Effect of L-ascorbic acid supplementation. Int. J. Vitam. Nutr. Res. 1975, 45, 429–437. [Google Scholar]
- Suzuki, T.; Yoshida, A. Effect of dietary supplementation of iron and ascorbic acid on lead toxicity in rats. J. Nutr. 1979, 109, 982–988. [Google Scholar] [CrossRef]
- Suzuki, T.; Yoshida, A. Effectiveness of dietary iron and ascorbic acid in the prevention and cure of moderately long-term lead toxicity in rats. J. Nutr. 1979, 109, 1974–1978. [Google Scholar] [CrossRef]
- Hill, C.H. Interactions of vitamin C with lead and mercury. Ann. N. Y. Acad. Sci. 1980, 355, 262–266. [Google Scholar] [CrossRef]
- Goyer, R.A.; Cherian, M.G. Ascorbic acid and EDTA treatment of lead toxicity in rats. Life Sci. 1979, 24, 433–438. [Google Scholar] [CrossRef]
- Flora, S.J.S.; Tandon, S.K. Preventive and therapeutic effects of thiamine, ascorbic acid and their combination in lead intoxication. Acta Pharmacol. Toxicol. 1986, 58, 374–378. [Google Scholar] [CrossRef]
- Dhawan, M.; Kachru, D.N.; Tandon, S.K. Influence of thiamine and ascorbic acid supplementation on the antidotal efficacy of thiol chelators in experimental lead intoxication. Arch. Toxicol. 1988, 62, 301–304. [Google Scholar] [CrossRef]
- Liao, Y.; Zhang, J.; Jin, Y.; Lu, C.; Li, G.; Yu, F.; Zhi, X.; An, L.; Yang, J. Therapeutic potentials of combined use of DMSA with calcium and ascorbic acid in the treatment of mild to moderately lead intoxicated mice. BioMetals 2008, 21, 1–8. [Google Scholar] [CrossRef]
- Hasan, M.Y.; Alshuaib, W.B.; Singh, S.; Fahim, M.A. Effects of ascorbic acid on lead induced alterations of synaptic transmission and contractile features in murine dorsiflexor muscle. Life Sci. 2003, 73, 1017–1025. [Google Scholar] [CrossRef]
- Moosavirad, S.A.; Rabbani, M.; Sharifzadeh, M.; Hosseini-Sharifabad, A. Protective effect of Vitamin C, vitamin B12 and omega-3 on lead induced memory impairment in rat. Res. Pharm. Sci. 2016, 11, 390–396. [Google Scholar] [CrossRef] [Green Version]
- Ebuehi, O.A.T.; Ayinde, O.C. Neurobehavioural and neurotoxic effects of L-ascorbic acid and L-tryptophan in lead exposed rats. Niger. Q. J. Hosp. Med. 2012, 22, 240–244. [Google Scholar] [CrossRef]
- Jiao, J.; Lü, G.; Liu, X.; Zhu, H.; Zhang, Y. Reduction of blood lead levels in lead-exposed mice by dietary supplements and natural antioxidants. J. Sci. Food Agric. 2011, 91, 485–491. [Google Scholar] [CrossRef]
- Das, K.K.; Saha, S. L-ascorbic acid and alpha tocopherol supplementation and antioxidant status in nickel- or lead-exposed rat brain tissue. J. Basic Clin. Physiol. Pharmacol. 2010, 21, 325–346. [Google Scholar] [CrossRef]
- Ebuehi, O.A.T.; Ogedegbe, R.A.; Ebuehi, O.M. Oral administration of vitamin C and vitamin E ameliorates lead-induced hepatotoxicity and oxidative stress in the rat brain. Niger. Q. J. Hosp. Med. 2012, 22, 85–90. [Google Scholar]
- Patra, R.C.; Swarup, D.; Dwivedi, S.K. Antioxidant effects of α tocopherol, ascorbic acid and L-methionine on lead induced oxidative stress to the liver, kidney and brain in rats. Toxicology 2001, 162, 81–88. [Google Scholar] [CrossRef]
- Ambali, S.; Angani, M.; Adole, A.; Kawu, M. Protective effect of vitamin C on biochemical alterations induced by subchronic co-administration of chlorpyrifos and lead in Wistar rats. J. Environ. Anal. Toxicol. 2011, 3, 234–244. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, Q.; Liu, X.; Zhu, H.; Song, A.; Jiao, J. Antioxidant and micronutrient-rich milk formula reduces lead poisoning and related oxidative damage in lead-exposed mice. Food Chem. Toxicol. 2013, 57, 201–208. [Google Scholar] [CrossRef]
- Massó-González, E.L.; Antonio-García, M.T. Natural antioxidants protect against lead-induced damage during pregnancy and lactation in rat’s pups. Ecotoxicol. Environ. Saf. 2009, 72, 2137–2142. [Google Scholar] [CrossRef]
- Ghasemi, S.; Hosseini, M.; Feizpour, A.; Alipour, F.; Sadeghi, A.; Vafaee, F.; Mohammadpour, T.; Soukhtanloo, M.; Ebrahimzadeh Bideskan, A.; Beheshti, F. Beneficial effects of garlic on learning and memory deficits and brain tissue damages induced by lead exposure during juvenile rat growth is comparable to the effect of ascorbic acid. Drug Chem. Toxicol. 2017, 40, 206–214. [Google Scholar] [CrossRef]
- Salehi, I.; Soleimani, M.S.; Pourjafar, M.; Moravej, G.F.; Asl, S.S. Vitamin C can alter lead-induced passive avoidance learning impairment in rats. J. Iran. Anat. Sci. 2015, 12, 137–140. [Google Scholar]
- Ghayoor, N.; Rasouli, B.; Afsharian, M.; Tehranipour, M.; Ghayoor, M. The protective effects of Melissa officinalis leaves usage on learning disorder induced by lead acetate administration during pre and postnatal periods in rats. Arak Med. Univ. J. 2010, 13, 97–104. [Google Scholar]
- Shaban El-Neweshy, M.; Said El-Sayed, Y. Influence of vitamin C supplementation on lead-induced histopathological alterations in male rats. Exp. Toxicol. Pathol. 2011, 63, 221–227. [Google Scholar] [CrossRef]
- Alipour, F.; Bideskan, A.E.; Fazel, A.; Sadeghi, A.; Hami, J.; Kheradmand, H.; Haghir, H. Protective effects of ascorbic acid and garlic extract against neurogenesis inhibition caused by developmental lead exposure in the dentate gyrus of rat. Comp. Clin. Pathol. 2014, 23, 1681–1687. [Google Scholar] [CrossRef]
- Khordad, E.; Fazel, A.; Bideskan, A.E. The effect of ascorbic acid and garlic administration on lead-induced apoptosis in rat offspring’s eye retina. Iran. Biomed. J. 2013, 17, 206–213. [Google Scholar] [CrossRef]
- Karamian, R.; Komaki, A.; Salehi, I.; Tahmasebi, L.; Komaki, H.; Shahidi, S.; Sarihi, A. Vitamin C reverses lead-induced deficits in hippocampal synaptic plasticity in rats. Brain Res. Bull. 2015, 116, 7–15. [Google Scholar] [CrossRef]
- Li, X.R.; Long, Y.H.; Fang, X.; Liu, X.G. Effect of vitamin C and E on antioxidative enzyme, NOS activity and NO contents in hippocampus of rats with lead poisoning. Zhejiang Da Xue Xue Bao Yi Xue Ban 2008, 37, 189–192. [Google Scholar]
- Fan, G.; Feng, C.; Li, Y.; Wang, C.; Yan, J.; Li, W.; Feng, J.; Shi, X.; Bi, Y. Selection of nutrients for prevention or amelioration of lead-induced learning and memory impairment in rats. Ann. Occup. Hyg. 2009, 53, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimzadeh-Bideskan, A.R.; Hami, J.; Alipour, F.; Haghir, H.; Fazel, A.R.; Sadeghi, A. Protective effects of ascorbic acid and garlic extract against lead-induced apoptosis in developing rat hippocampus. Metab. Brain Dis. 2016, 31, 1123–1132. [Google Scholar] [CrossRef]
- Sadeghi, A.; Bideskan, A.E.; Alipour, F.; Fazel, A.; Haghir, H. The effect of ascorbic acid and garlic administration on lead-induced neural damage in rat offspring’s hippocampus. Iran. J. Basic Med. Sci. 2013, 16, 157–164. [Google Scholar] [CrossRef]
- Ahmad, F.; Salahuddin, M.; Alsamman, K.; Almulla, A.A.; Salama, K.F. Developmental lead (Pb)-induced deficits in hippocampal protein translation at the synapses are ameliorated by ascorbate supplementation. Neuropsychiatr. Dis. Treat. 2018, 14, 3289–3298. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, F.; Haque, S.; Ravinayagam, V.; Ahmad, A.; Kamli, M.R.; Barreto, G.E.; Ashraf, G.M. Developmental lead (Pb)-induced deficits in redox and bioenergetic status of cerebellar synapses are ameliorated by ascorbate supplementation. Toxicology 2020, 440, 152492. [Google Scholar] [CrossRef]
- Ahmad, F.; Salahuddin, M.; Alamoudi, W.; Acharya, S. Dysfunction of cortical synapse-specific mitochondria in developing rats exposed to lead and its amelioration by ascorbate supplementation. Neuropsychiatr. Dis. Treat. 2018, 14, 813–824. [Google Scholar] [CrossRef] [Green Version]
- Jang, Y.; Lee, J.H.; Lee, M.J.; Kim, S.J.; Ju, X.; Cui, J.; Zhu, J.; Lee, Y.L.; Namgung, E.; Sung, H.W.J.; et al. Schisandra extract and ascorbic acid synergistically enhance cognition in mice through modulation of mitochondrial respiration. Nutrients 2020, 12, 897. [Google Scholar] [CrossRef] [Green Version]
- Fraga, D.B.; Costa, A.P.; Olescowicz, G.; Camargo, A.; Pazini, F.L.; Freitas, A.E.; Moretti, M.; Brocardo, P.S.; Ana, A.L. Ascorbic acid presents rapid behavioral and hippocampal synaptic plasticity effects. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2020, 96, 109757. [Google Scholar] [CrossRef]
- Moretti, M.; Budni, J.; Freitas, A.E.; Rosa, P.B.; Rodrigues, A.L.S. Antidepressant-like effect of ascorbic acid is associated with the modulation of mammalian target of rapamycin pathway. J. Psychiatr. Res. 2014, 48, 16–24. [Google Scholar] [CrossRef]
- Sepehri, H.; Ganji, F. The protective role of ascorbic acid on hippocampal CA1 pyramidal neurons in a rat model of maternal lead exposure. J. Chem. Neuroanat. 2016, 74, 5–10. [Google Scholar] [CrossRef]
- Han, J.M.; Chang, B.J.; Li, T.Z.; Choe, N.H.; Quan, F.S.; Jang, B.J.; Cho, I.H.; Hong, H.N.; Lee, J.H. Protective effects of ascorbic acid against lead-induced apoptotic neurodegeneration in the developing rat hippocampus in vivo. Brain Res. 2007, 1185, 68–74. [Google Scholar] [CrossRef]
- Chang, B.J.; Jang, B.J.; Son, T.G.; Cho, I.H.; Quan, F.S.; Choe, N.H.; Nahm, S.S.; Lee, J.H. Ascorbic acid ameliorates oxidative damage induced by maternal low-level lead exposure in the hippocampus of rat pups during gestation and lactation. Food Chem. Toxicol. 2012, 50, 104–108. [Google Scholar] [CrossRef]
- Nam, S.M.; Chang, B.J.; Kim, J.H.; Nahm, S.S.; Lee, J.H. Ascorbic acid ameliorates lead-induced apoptosis in the cerebellar cortex of developing rats. Brain Res. 2018, 1686, 10–18. [Google Scholar] [CrossRef]
- Nam, S.M.; Ahn, S.C.; Go, T.H.; Seo, J.S.; Nahm, S.S.; Chang, B.J.; Lee, J.H. Ascorbic acid ameliorates gestational lead exposure-induced developmental alteration in GAD67 and c-Kit expression in the rat cerebellar cortex. Biol. Trace Elem. Res. 2018, 182, 278–286. [Google Scholar] [CrossRef]
- Nam, S.M.; Seo, J.S.; Go, T.H.; Nahm, S.S.; Chang, B.J. Ascorbic acid supplementation prevents the detrimental effects of prenatal and postnatal lead exposure on the Purkinje cell and related proteins in the cerebellum of developing rats. Biol. Trace Elem. Res. 2019, 190, 446–456. [Google Scholar] [CrossRef]
- Nam, S.M.; Cho, I.S.; Seo, J.S.; Go, T.H.; Kim, J.H.; Nahm, S.S.; Chang, B.J.; Lee, J.H. Ascorbic acid attenuates lead-induced alterations in the synapses in the developing rat cerebellum. Biol. Trace Elem. Res. 2019, 187, 142–150. [Google Scholar] [CrossRef]
- Nam, S.M.; Seo, J.S.; Nahm, S.S.; Chang, B.J. Effects of ascorbic acid treatment on developmental alterations in calcium-binding proteins and gamma-aminobutyric acid transporter 1 in the cerebellum of lead-exposed rats during pregnancy and lactation. J. Toxicol. Sci. 2019, 44, 799–809. [Google Scholar] [CrossRef]
- Nam, S.M.; Seo, J.S.; Nahm, S.S.; Chang, B.J. Effects of ascorbic acid on osteopontin expression and axonal myelination in the developing cerebellum of lead-exposed rat pups. Int. J. Environ. Res. Public Health 2019, 16, 983. [Google Scholar] [CrossRef] [Green Version]
- Imosemi, I.O.; Owoeye, O.; Ao, M. Lead-induced oxidative stress in postnatal developing cerebellum of Wistar rats: Role of aqueous extract of Cucumis sativus Linn and vitamin. MOJ Anat. Physiol. 2020, 7, 104–113. [Google Scholar] [CrossRef]
- Abraham Musa, S.; Musa Omoniye, I.; Oliver Hamman, W.; Oseloka Ibegbu, A.; Emmanuel Umana, U. Preventive activity of ascorbic acid on lead acetate induced cerebellar damaged in adult Wistar rats. Med. Health Sci. J. 2012, 13, 99–104. [Google Scholar] [CrossRef]
- Vij, A.G.; Satija, N.K.; Flora, S.J.S. Lead induced disorders in hematopoietic and drug metabolizing enzyme system and their protection by ascorbic acid supplementation. Biomed. Environ. Sci. 1998, 11, 7–14. [Google Scholar]
- Farooq, Y.; Farooq, M.A.; Hussain, A. Role of ascorbic acid supplement in amelioration of anaemia in lead intoxication. J. Pak. Med. Assoc. 2016, 66, 1073–1076. [Google Scholar]
- Upadhyay, A.K.; Mathur, R.; Bhadauria, M.; Nirala, S.K. Therapeutic influence of zinc and ascorbic acid against lead induced biochemical alterations. Therapie 2009, 64, 383–388. [Google Scholar] [CrossRef]
- Bhattacharjee, C.R.; Dey, S.; Goswami, P. Protective role of ascorbic acid against lead toxicity in blood of albino mice as revealed by metal uptake, lipid profiles, and ultrastructural features of erythrocytes. Bull. Environ. Contam. Toxicol. 2003, 70, 1189–1196. [Google Scholar] [CrossRef]
- Kumar, A.; Rahal, A.; Zoheb, S.M.; Prakash, A.; Mandil, R. Antioxidant role of ascorbic acid on oxidative stress induced by sub-acute exposure of lead and cypermethrin in erythrocytes of Wistar rats. Toxicol. Environ. Chem. 2014, 96, 1248–1259. [Google Scholar] [CrossRef]
- Hafez, L.M.; El-Sebeay, A.S.; Ibrahim, A.F.M.; Kishk, A.M. Protective role of different doses of ascorbic acid against harmful effects of lead in liver, kidney and brain in male rats. J. Agric. Chem. Biotechnol. 2017. [Google Scholar]
- Hafez, L.; Kishk, A. Antioxidant effect of escorbic acid (Vit.C) in different concentration against harmful effect of toxic heavy metal (lead acetate) in rats. J. Agric. Chem. Biotechnol. 2017, 8, 191–196. [Google Scholar] [CrossRef]
- Ramadan, A.S.; El-Sayed, M.H. Hemodynamic and cardiac functions in rats exposed to lead toxicity, the possible effect of vitamin C (ascorbic acid). Life Sci. J. 2014, 11, 167–179. [Google Scholar]
- Shalan, M.G.; Mostafa, M.S.; Hassouna, M.M.; Hassab El-Nabi, S.E.; El-Refaie, A. Amelioration of lead toxicity with vitamin C and silymarin supplementation. J. Toxicol. Clin. Toxicol. 2004, 42, 798. [Google Scholar]
- Shalan, M.G.; Mostafa, M.S.; Hassouna, M.M.; El-Nabi, S.E.H.; El-Refaie, A. Amelioration of lead toxicity on rat liver with Vitamin C and silymarin supplements. Toxicology 2005, 206, 1–15. [Google Scholar] [CrossRef]
- Wang, C.; Liang, J.; Zhang, C.; Bi, Y.; Shi, X.; Shi, Q. Effect of ascorbic acid and thiamine supplementation at different concentrations on lead toxicity in liver. Ann. Occup. Hyg. 2007, 51, 563–569. [Google Scholar] [CrossRef]
- Osifo, J.E.; Idemili, N.G.; Osubor, C.C. Ameliorative effect of vitamin C on lead induced hepatotoxicity in rats. JMBR 2015, 14, 123–131. [Google Scholar]
- Alhusaini, A.M.; Faddah, L.M.; Hasan, I.H.; Jarallah, S.J.; Alghamdi, S.H.; Alhadab, N.M.; Badr, A.; Elorabi, N.; Zakaria, E.; Al-anazi, A. Vitamin C and turmeric attenuate Bax and Bcl-2 proteins’ expressions and DNA damage in lead acetate-induced liver injury. Dose-Response 2019, 17, 1559325819885782. [Google Scholar] [CrossRef]
- Alhusaini, A.; Fadda, L.; Hasan, I.H.; Zakaria, E.; Alenazi, A.M.; Mahmoud, A.M. Curcumin ameliorates lead-induced hepatotoxicity by suppressing oxidative stress and inflammation, and modulating Akt/GSK-3β signaling pathway. Biomolecules 2019, 9, 703. [Google Scholar] [CrossRef] [Green Version]
- El Shafai, A.; Zohdy, N.; El Mulla, K.; Hassan, M.; Morad, N. Light and electron microscopic study of the toxic effect of prolonged lead exposure on the seminiferous tubules of albino rats and the possible protective effect of ascorbic acid. Food Chem. Toxicol. 2011, 49, 734–743. [Google Scholar] [CrossRef]
- Ali, S.; Hussain, S.; Khan, R.; Mumtaz, S.; Ashraf, N.; Andleeb, S.; Shakir, H.A.; Tahir, H.M.; Khan, M.K.A.; Ulhaq, M. Renal toxicity of heavy metals (cadmium and mercury) and their amelioration with ascorbic acid in rabbits. Environ. Sci. Pollut. Res. 2019, 26, 3909–3920. [Google Scholar] [CrossRef]
- Kosik-Bogacka, D.I.; Baranowska-Bosiacka, I.; Marchlewicz, M.; Kolasa, A.; Jakubowska, K.; Olszewska, M.; Łanocha, N.; Wiernicki, I.; Millo, B.; Wiszniewska, B.; et al. The effect of L-ascorbic acid and/or tocopherol supplementation on electrophysiological parameters of the colon of rats chronically exposed to lead. Case Rep. Clin. Pract. Rev. 2011, 17, BR16–BR26. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, Y.; Liang, J.; Shan, G.; Wang, Y.; Shi, Q. Impacts of ascorbic acid and thiamine supplementation at different concentrations on lead toxicity in testis. Clin. Chim. Acta 2006, 370, 82–88. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, C.; Liang, J.; Zhang, C.; Wang, C. qiang Impacts of combined supplementation with ascorbic acid and thiamine on certain biochemical and morphologic indexes of testes in mice treated by lead. Wei Sheng Yan Jiu 2006, 35, 731–734. [Google Scholar]
- Mishra, M.; Acharya, U.R. Protective action of vitamins on the spermatogenesis in lead-treated Swiss mice. J. Trace Elem. Med. Biol. 2004, 18, 173–178. [Google Scholar] [CrossRef]
- Shan, G.; Tang, T.; Zhang, X. The protective effect of ascorbic acid and thiamine supplementation against damage caused by lead in the testes of mice. J. Huazhong Univ. Sci. Technol. Med. Sci. 2009, 29, 68–72. [Google Scholar] [CrossRef]
- Ayinde, O.C.; Ogunnowo, S.; Ogedegbe, R.A. Influence of vitamin C and vitamin E on testicular zinc content and testicular toxicity in lead exposed albino rats. BMC Pharmacol. Toxicol. 2012, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.C.; Liu, M.Y.; Hsu, C.C.; Chen, L.Y.; Leon Guo, Y. Effects of vitamin E and/or C on reactive oxygen species-related lead toxicity in the rat sperm. Toxicology 1998, 128, 169–179. [Google Scholar] [CrossRef]
- Ambali, S.F.; Orieji, C.; Abubakar, W.O.; Shittu, M.; Kawu, M.U. Ameliorative effect of vitamin C on alterations in thyroid hormones concentrations induced by subchronic coadministration of chlorpyrifos and lead in Wistar rats. J. Thyroid Res. 2011, 2011, 214924. [Google Scholar] [CrossRef] [Green Version]
- Upasani, C.D.; Balaraman, R. Effect of vitamin E, vitamin C and spirulina on the levels of membrane bound enzymes and lipids in some organs of rats exposed to lead. Indian J. Pharmacol. 2001, 33, 185–191. [Google Scholar]
- Roy, A.K.; Dhir, H.; Sharma, A. Modification of metal-induced micronuclei formation in mouse bone marrow erythrocytes by Phyllanthus fruit extract and ascorbic acid. Toxicol. Lett. 1992, 62, 9–17. [Google Scholar] [CrossRef]
- Dhir, H.; Roy, A.K.; Sharma, A.; Talukder, G. Modification of clastogenicity of lead and aluminium in mouse bone marrow cells by dietary ingestion of Phyllanthus emblica fruit extract. Mutat. Res. Toxicol. 1990, 241, 305–312. [Google Scholar] [CrossRef]
- Dhir, H.; Roy, A.K.; Sharma, A. Relative efficiency of Phyllanthus emblica fruit extract and ascorbic acid in modifying lead and aluminium-induced sister-chromatid exchanges in mouse bone marrow. Environ. Mol. Mutagenesis 1993, 21, 229–236. [Google Scholar] [CrossRef]
- Mumtaz, S.; Ali, S.; Khan, R.; Shakir, H.A.; Tahir, H.M.; Mumtaz, S.; Andleeb, S. Therapeutic role of garlic and vitamins C and E against toxicity induced by lead on various organs. Environ. Sci. Pollut. Res. 2020, 27, 8953–8964. [Google Scholar] [CrossRef]
- Fischer, A.B.; Hess, C.; Neubauer, T.; Eikmann, T. Testing of chelating agents and vitamins against lead toxicity using mammalian cell cultures. Analyst 1998, 123, 55–58. [Google Scholar] [CrossRef]
- Blankenship, L.J.; Carlisle, D.L.; Wise, J.P.; Orenstein, J.M.; Dye, L.E.; Patierno, S.R. Induction of apoptotic cell death by particulate lead chromate: Differential effects of vitamins C and E on genotoxicity and survival. Toxicol. Appl. Pharmacol. 1997, 146, 270–280. [Google Scholar] [CrossRef]
- Wise, J.P.; Orenstein, J.M.; Patierno, S.R. Inhibition of lead chromate clastogenesis by ascorbate: Relationship to particle dissolution and uptake. Carcinogenesis 1993, 14, 429–434. [Google Scholar] [CrossRef]
- Darwish, W.S.; Ikenaka, Y.; Nakayama, S.M.M.; Mizukawa, H.; Ishizuka, M. Constitutive effects of lead on aryl hydrocarbon receptor gene battery and protection by β-carotene and ascorbic acid in human HepG2 cells. J. Food Sci. 2016, 81, T275–T281. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Darwish, W.S.; Chen, Z.; Hui, T.; Wu, Y.; Hirotaka, S.; Chiba, H.; Hui, S.P. Identification of lead-produced lipid hydroperoxides in human HepG2 cells and protection using rosmarinic and ascorbic acids with a reference to their regulatory roles on Nrf2-Keap1 antioxidant pathway. Chem. Biol. Interact. 2019, 314, 108847. [Google Scholar] [CrossRef]
- Wang, J.; Ren, T.; Han, Y.; Zhao, Y.; Liao, M.; Wang, F.; Jiang, Z. Effects of dietary vitamin C supplementation on lead-treated sea cucumbers, Apostichopus japonicus. Ecotoxicol. Environ. Saf. 2015, 118, 21–26. [Google Scholar] [CrossRef]
- Kim, J.H.; Kang, J.C. Effects of sub-chronic exposure to lead (Pb) and ascorbic acid in juvenile rockfish: Antioxidant responses, MT gene expression, and neurotransmitters. Chemosphere 2017, 171, 520–527. [Google Scholar] [CrossRef]
- Shawahna, R.; Zyoud, A.; Yahia, E.H.; Sulieman, R.; Haddad, A.; Makhlof, M.; Abu-Hilal, B.; Murtaza, G.; Hilal, H. Sub-chronic treatment with high doses of ascorbic acid reduces lead levels in hen eggs intentionally exposed to a concentrated source of lead: A pilot study. BMC Pharmacol. Toxicol. 2020, 21, 17. [Google Scholar] [CrossRef] [Green Version]
- Mumtaz, S.; Ali, S.; Khan, R.; Andleeb, S.; Ulhaq, M.; Khan, M.A.; Shakir, H.A. The protective role of ascorbic acid in the hepatotoxicity of cadmium and mercury in rabbits. Environ. Sci. Pollut. Res. 2019, 26, 14087–14096. [Google Scholar] [CrossRef]
- Khan, R.; Ali, S.; Mumtaz, S.; Andleeb, S.; Ulhaq, M.; Tahir, H.M.; Khan, M.K.A.; Khan, M.A.; Shakir, H.A. Toxicological effects of toxic metals (cadmium and mercury) on blood and the thyroid gland and pharmacological intervention by vitamin C in rabbits. Environ. Sci. Pollut. Res. 2019, 26, 16727–16741. [Google Scholar] [CrossRef]
- BBC News. Andhra Pradesh’s Eluru: India Experts Investigate “Mystery” Illness. 2020. Available online: https://www.bbc.com/news/world-asia-india-55227388 (accessed on 7 December 2020).
Figure 1.
Ascorbate oxidation and recycling. Ascorbate is converted to an ascorbyl radical or semi-dehydroascorbate upon the loss of an e−, which is further oxidized to dehydroascorbate. Both the ascorbyl radical and dehydroascorbate can be recycled back to a monovalent, negatively-charged ascorbate ion by the action of enzyme coupled reactions utilizing thiol-based antioxidants such as glutathione and thioredoxin.
Figure 1.
Ascorbate oxidation and recycling. Ascorbate is converted to an ascorbyl radical or semi-dehydroascorbate upon the loss of an e−, which is further oxidized to dehydroascorbate. Both the ascorbyl radical and dehydroascorbate can be recycled back to a monovalent, negatively-charged ascorbate ion by the action of enzyme coupled reactions utilizing thiol-based antioxidants such as glutathione and thioredoxin.

Figure 2.
Ascorbate uptake and metabolism. Asc: Ascorbic acid in its anionic ascorbate form; GLUT: glucose transporter; SVCT: sodium-dependent ascorbate transporter; CSF: cerebrospinal fluid; DHA: dehydroascorbate; X•: free radical species; XH: reduced/scavenged radicals. Ascorbic acid enters the CSF in its anionic ascorbate form (Asc) through the choroid plexus via SVCT2 in the epithelial cells and as dehydroascorbate (DHA) through the GLUTs across the BBB. The entry of Asc in neurons is also mediated by SVCT2, wherein it scavenges various free radicals (X•) upon its oxidation to DHA. Neuronal DHA is released into the extracellular fluid for uptake by astrocytes via the GLUT-mediated transport. DHA is then recycled back to Asc for its own need or release into the extracellular space for uptake by neurons.
Figure 2.
Ascorbate uptake and metabolism. Asc: Ascorbic acid in its anionic ascorbate form; GLUT: glucose transporter; SVCT: sodium-dependent ascorbate transporter; CSF: cerebrospinal fluid; DHA: dehydroascorbate; X•: free radical species; XH: reduced/scavenged radicals. Ascorbic acid enters the CSF in its anionic ascorbate form (Asc) through the choroid plexus via SVCT2 in the epithelial cells and as dehydroascorbate (DHA) through the GLUTs across the BBB. The entry of Asc in neurons is also mediated by SVCT2, wherein it scavenges various free radicals (X•) upon its oxidation to DHA. Neuronal DHA is released into the extracellular fluid for uptake by astrocytes via the GLUT-mediated transport. DHA is then recycled back to Asc for its own need or release into the extracellular space for uptake by neurons.

Figure 3.
Mechanisms of ascorbic acid-mediated neuroprotection in Pb neurotoxicity. Stemming from its ability for metal ion chelation, antioxidant potential, and neuromodulation function, ascorbic acid attenuates Pb neurotoxicity in a multimodal manner, ranging from decreasing the Pb burden to alleviating biochemical and neurobehavioral deficits.
Figure 3.
Mechanisms of ascorbic acid-mediated neuroprotection in Pb neurotoxicity. Stemming from its ability for metal ion chelation, antioxidant potential, and neuromodulation function, ascorbic acid attenuates Pb neurotoxicity in a multimodal manner, ranging from decreasing the Pb burden to alleviating biochemical and neurobehavioral deficits.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ahmad, F.; Liu, P. (Ascorb)ing Pb Neurotoxicity in the Developing Brain. Antioxidants 2020, 9, 1311. https://doi.org/10.3390/antiox9121311
AMA Style
Ahmad F, Liu P. (Ascorb)ing Pb Neurotoxicity in the Developing Brain. Antioxidants. 2020; 9(12):1311. https://doi.org/10.3390/antiox9121311
Chicago/Turabian StyleAhmad, Faraz, and Ping Liu. 2020. "(Ascorb)ing Pb Neurotoxicity in the Developing Brain" Antioxidants 9, no. 12: 1311. https://doi.org/10.3390/antiox9121311
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.