Protein Carbonylation and Lipid Peroxidation in Hematological Malignancies

 , , , ,
, , , ,  and
and
Abstract
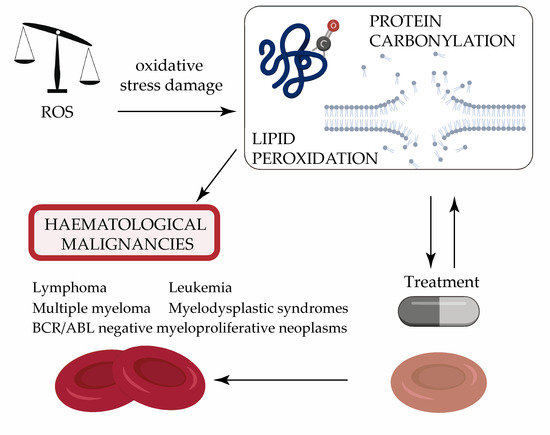
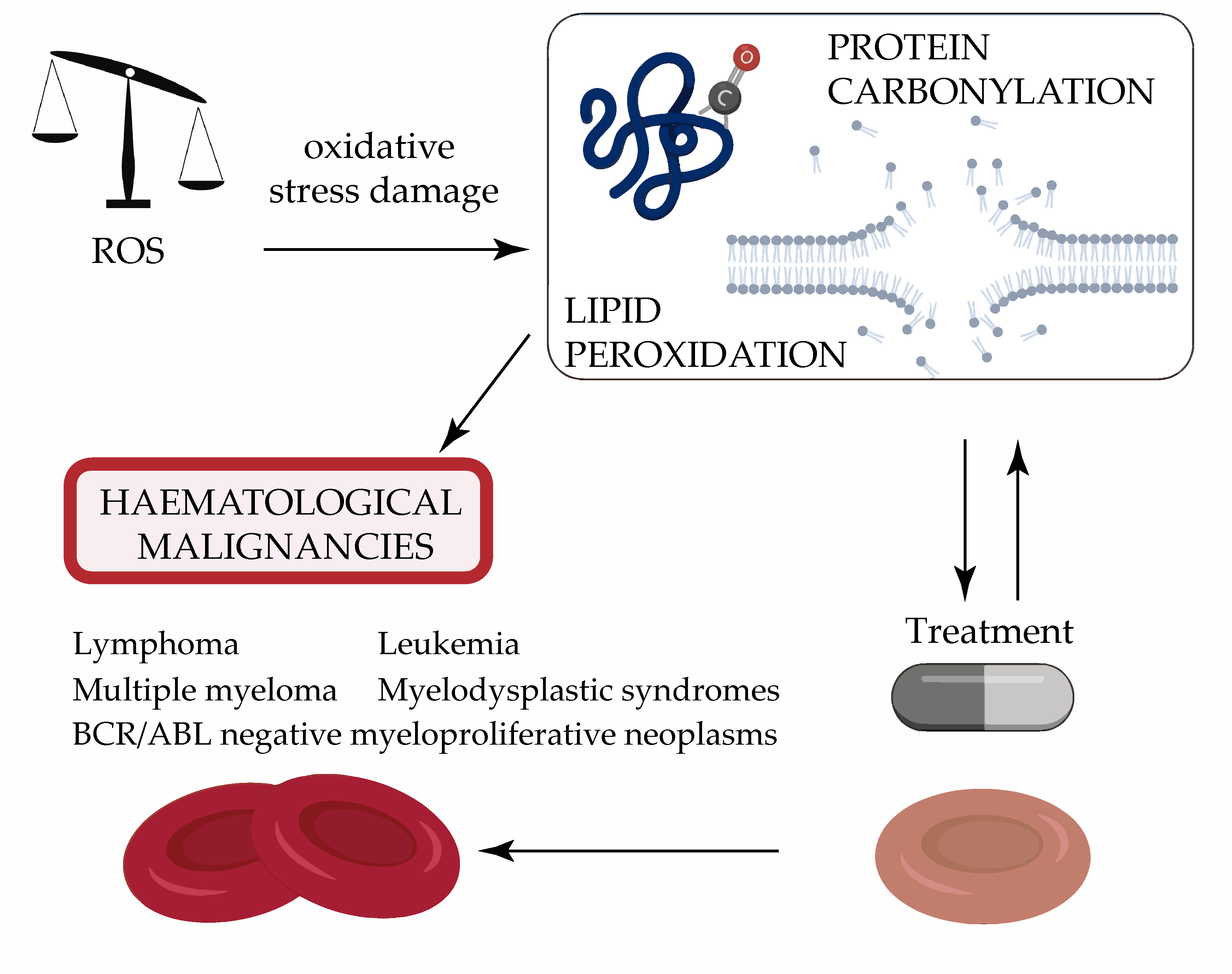
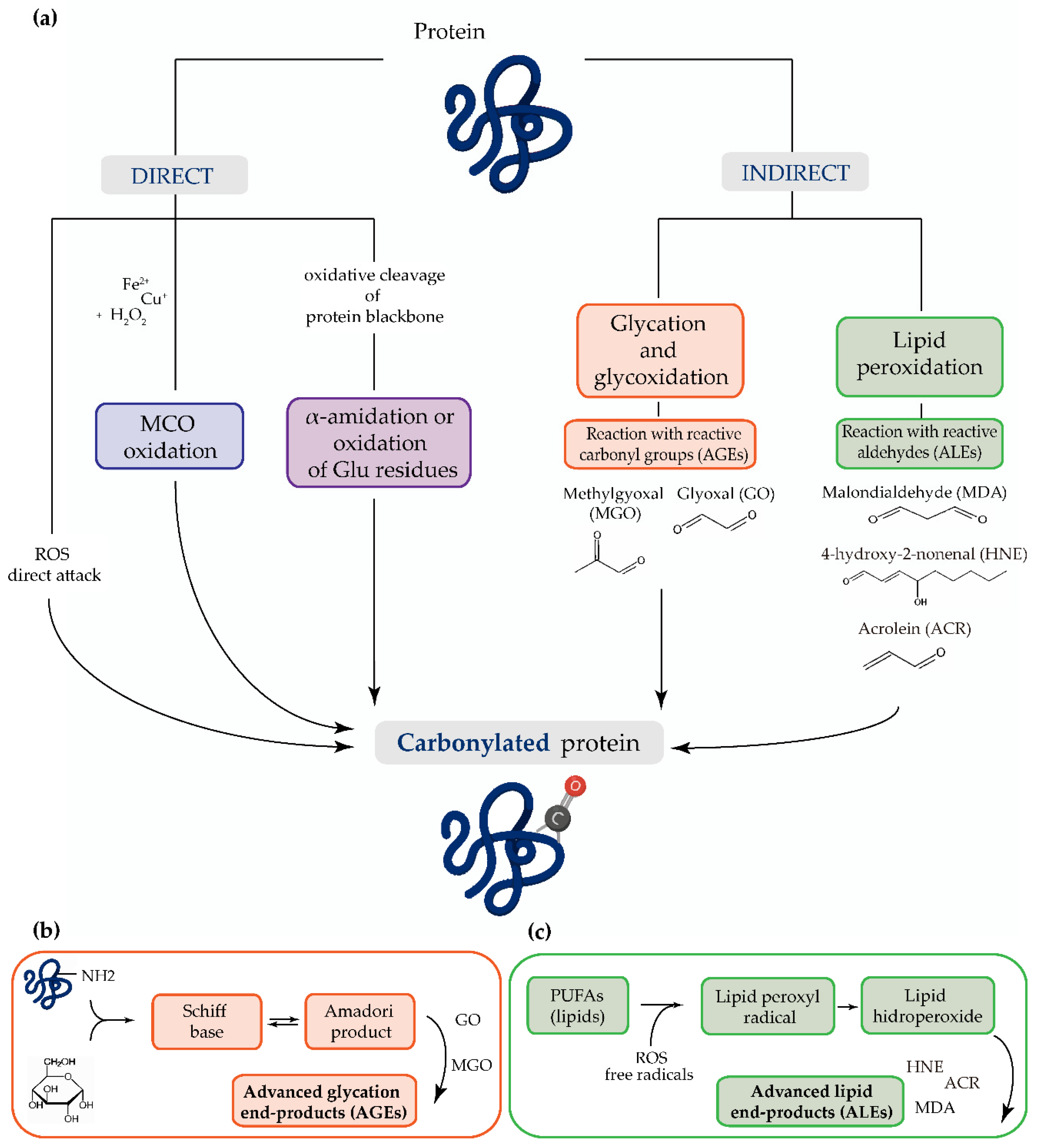
:
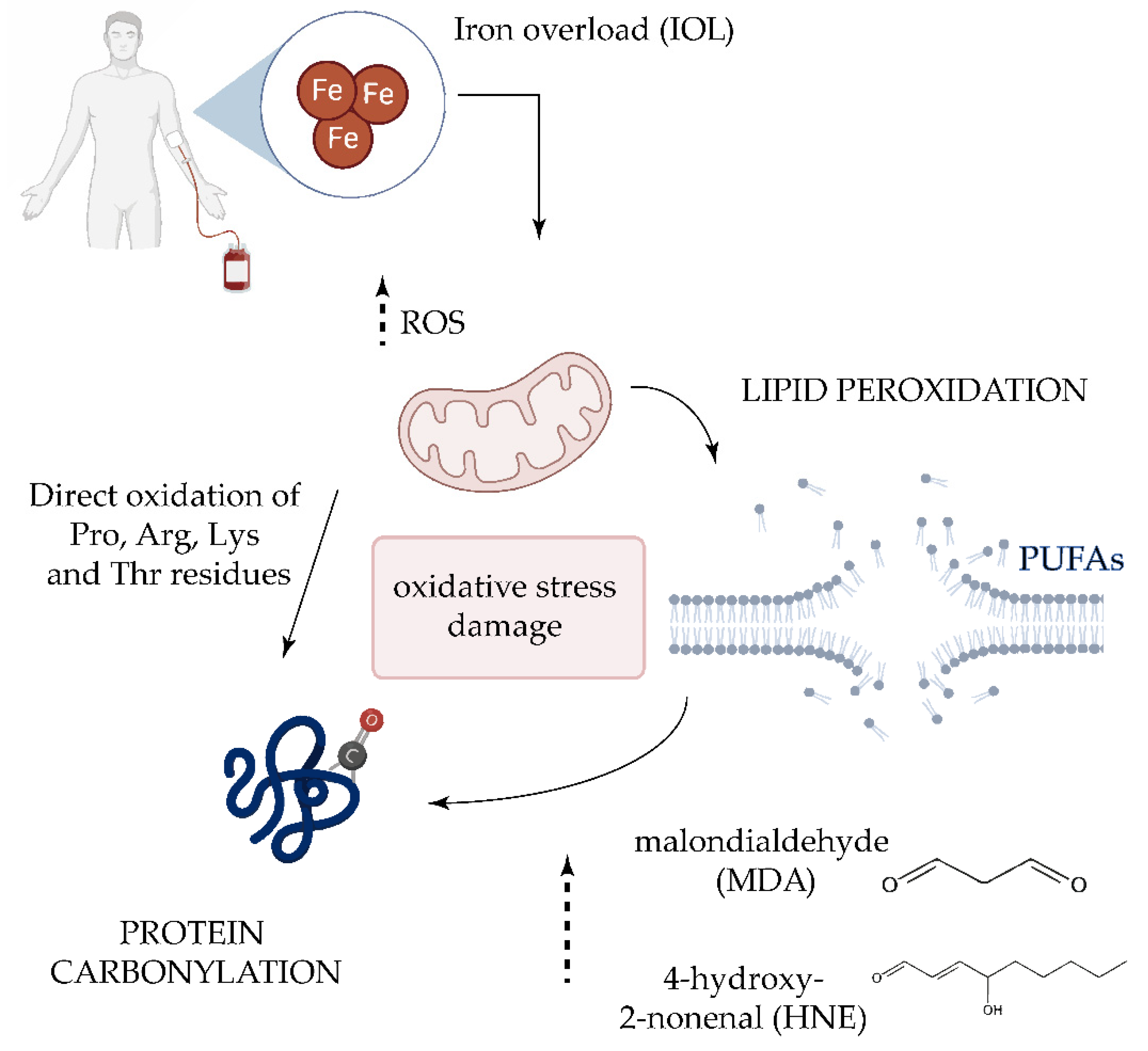
1. Introduction
2. Lymphoma
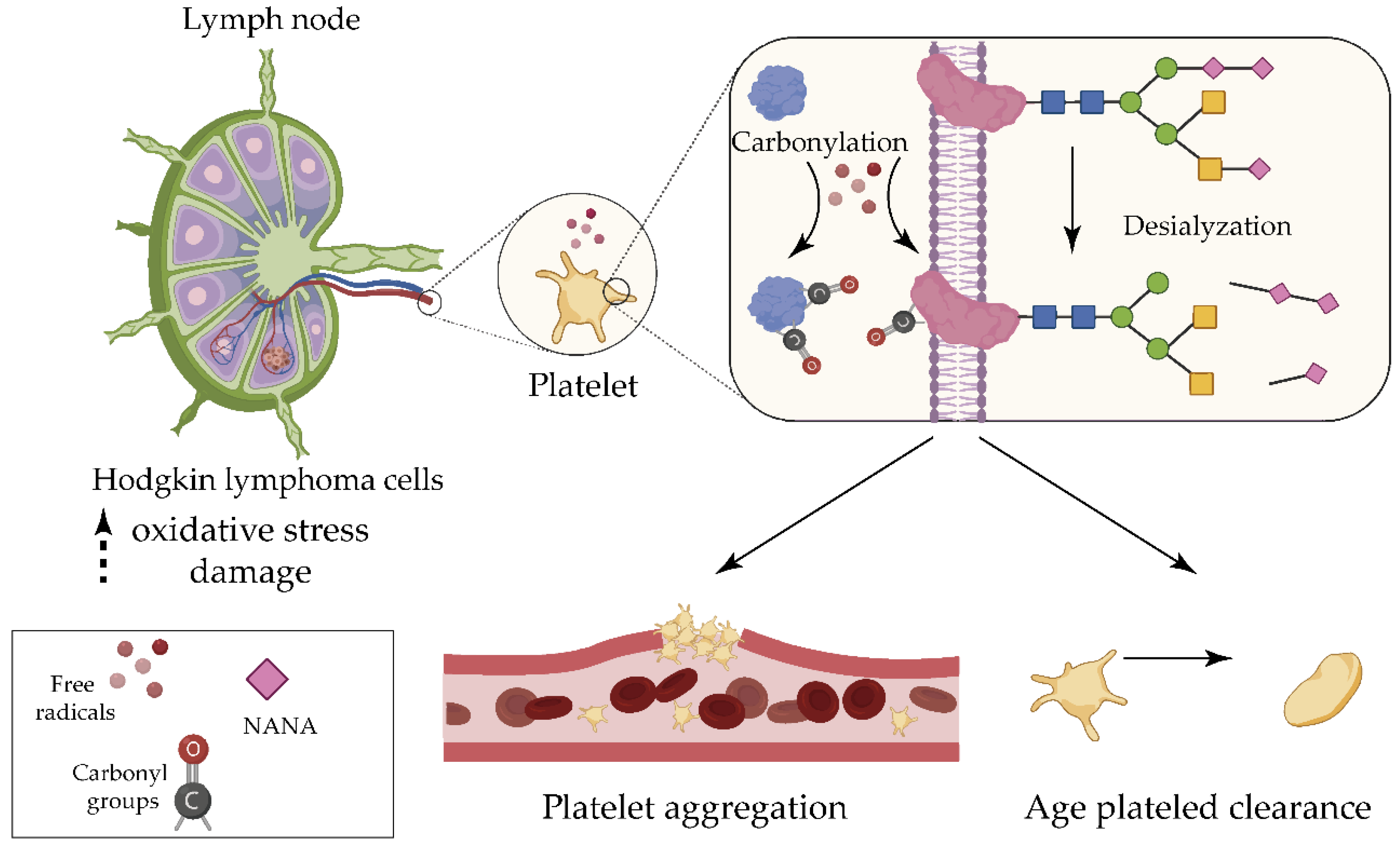
2.1. Hodgkin Lymphoma
2.2. Non-Hodgkin Lymphoma
3. Multiple Myeloma
4. Leukemia
4.1. Acute Lymphoblastic Leukemia
4.2. Chronic Lymphoblastic Leukemia
4.3. Acute Myeloid Leukemia
4.4. Chronic Myeloid Leukemia
5. Myelodysplastic Syndromes
6. BCR/ABL Negative Myeloproliferative Neoplasms
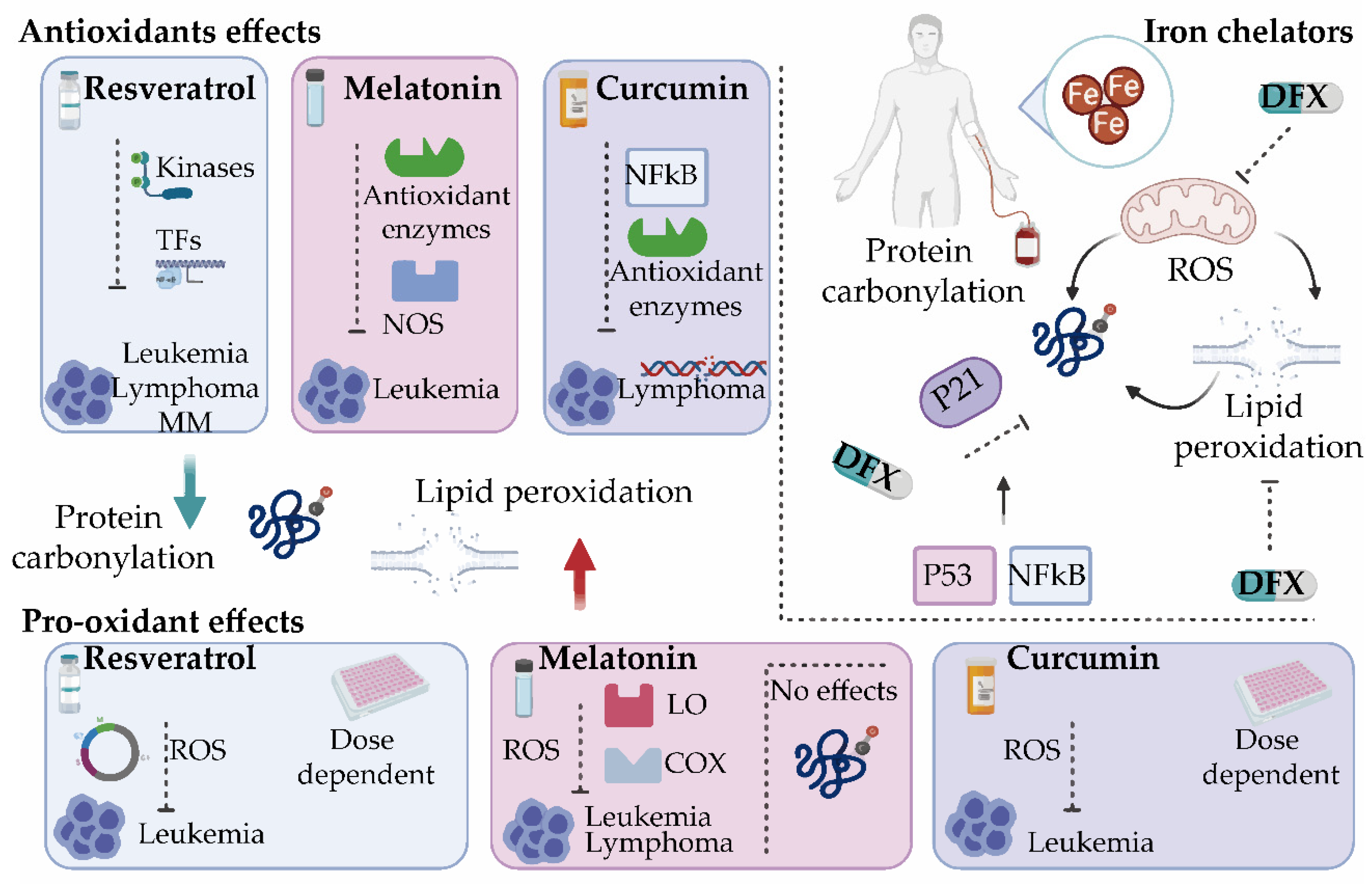
7. Oxidative Stress Modulators for the Treatment of Hematological Malignancies
7.1. Potential Antioxidant Drugs
7.2. Potential Pro-Oxidant Drugs
7.3. Iron Chelators
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 4-HNE | 4-hydroxy-2,3-nonenal |
| ABVD | Adriamycin, Bleomycin, Vincristine and Dexamethasone |
| ACR | Acrolein |
| AGEs | Advanced glycation end-products |
| ALEs | Advanced lipid peroxidation end-products |
| ALL | Acute lymphoblastic leukemia |
| AML | Acute myeloid leukemia |
| AOPPs | Advanced oxidation protein products |
| APL | Acute promyelocytic leukemia |
| B CLL | B-cell chronic lymphoblastic leukemia |
| BM | Bone marrow |
| CAT | Catalase |
| CHOP | Cyclophosphamide, Vincristine, Doxorubicin, and Prednisone |
| CML | Chronic myeloid leukemia |
| COX | Cyclooxygenase |
| DFX | Deferasirox |
| DLBCL | Diffuse large B-cell lymphoma |
| DNP | Dinitrophenylhydrazone |
| DNPH | Dinitrophenylhydrazine |
| DOX-TRF | Doxorubicin-transferrin |
| ET | Essential thrombocythemia |
| GO | Glyoxal |
| GPX | Glutathione peroxidase |
| HL | Hodgkin lymphoma |
| IOL | Iron overload |
| LO | Lipoxygenase |
| MAD | Malondialdehyde |
| MGO | Methyglyoxal |
| MGUS | Monoclonal gammapathy of undetermined significance |
| MM | Multiple myeloma |
| MPN | Myeloproliferative neoplasms |
| NANA | N-acetyl neuraminic acid |
| NHL | Non-Hodgkin lymphoma |
| NOS | Nitric oxide synthase |
| ns | Nonsignificant |
| ONE | 4-oxo-2-nonenal |
| PC | Protein carbonylation |
| PMF | Primary myelofibrosis |
| PN | Protein nitrosylated |
| PUFA | Polyunsaturated fatty acyl |
| PV | Polycythemia vera |
| ROS | Reactive oxygen species |
| SOD | Superoxide dismutase |
| TBARS | Thiobarbituric reactive substances |
| Trx | Thioredoxin |
| VAD | Vincristine–Adriamycin–Dexamethasone |
References
- Singh, R.K.; Tripathi, A.K.; Tripathi, P.; Singh, S.; Singh, R.; Ahmad, R. Studies on biomarkers for oxidative stress in patients with chronic myeloid leukemia. Hematol. Oncol. Stem. Cell 2009, 2, 285–288. [Google Scholar] [CrossRef] [Green Version]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Battisti, V.; Maders, L.D.K.; Bagatini, M.D.; Santos, K.F.; Spanevello, R.M.; Maldonado, P.A.; Brulé, A.O.; do Carmo Araújo, M.; Schetinger, M.R.C.; Morsch, V.M. Measurement of oxidative stress and antioxidant status in acute lymphoblastic leukemia patients. Clin. Biochem. 2008, 41, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Tripathi, A.K.; Tripathi, P.; Singh, S.; Singh, R.; Singh, R.K. Malondialdehyde and protein carbonyl as biomarkers for oxidative stress and disease progression in patients with chronic myeloid leukemia. In Vivo 2008, 22, 525–528. [Google Scholar] [PubMed]
- Ahmad, R.; Tripathi, A.K.; Tripathi, P.; Singh, R.; Singh, S.; Singh, R.K. Studies on lipid peroxidation and non-enzymatic antioxidant status as indices of oxidative stress in patients with chronic myeloid leukaemia. Singap. Med. J. 2010, 51, 110–115. [Google Scholar]
- Butterfield, D.A.; Reed, T.; Newman, S.F.; Sultana, R. Roles of Amyloid β-Peptide-Associated Oxidative Stress and Brain Protein Modifications in the Pathogenesis of Alzheimer’s Disease and Mild Cognitive Impairment. Free Radic. Biol. Med. 2007, 43, 658–677. [Google Scholar] [CrossRef] [Green Version]
- Cecarini, V.; Gee, J.; Fioretti, E.; Amici, M.; Angeletti, M.; Eleuteri, A.M.; Keller, J.N. Protein oxidation and cellular homeostasis: Emphasis on metabolism. Biochim. Biophys. Acta 2007, 1773, 93–104. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Giustarini, D.; Colombo, R.; Rossi, R.; Milzani, A. Protein carbonylation in human diseases. Trends Mol. Med. 2003, 9, 169–176. [Google Scholar] [CrossRef]
- Barrera, G.; Pizzimenti, S.; Daga, M.; Dianzani, C.; Arcaro, A.; Cetrangolo, G.P.; Giordano, G.; Cucci, M.A.; Graf, M.; Gentile, F. Lipid Peroxidation-Derived Aldehydes, 4-Hydroxynonenal and Malondialdehyde in Aging-Related Disorders. Antioxidants 2018, 7, 102. [Google Scholar] [CrossRef] [Green Version]
- Linares, M.; Marín-Garcíía, P.; Méndez, D.; Puyet, A.; Diez, A.; Bautista, J.M. Proteomic approaches to identifying carbonylated proteins in brain tissue. J. Proteome Res. 2011, 10, 1719–1727. [Google Scholar] [CrossRef]
- Levine, R.L.; Williams, J.A.; Stadtman, E.R.; Shacter, E. Carbonyl assays for determination of oxidatively modified proteins. Meth. Enzymol. 1994, 233, 346–357. [Google Scholar] [CrossRef]
- Fedorova, M.; Bollineni, R.C.; Hoffmann, R. Protein carbonylation as a major hallmark of oxidative damage: Update of analytical strategies. Mass Spec. Rev. 2014, 33, 79–97. [Google Scholar] [CrossRef] [PubMed]
- Rudzińska, M.; Parodi, A.; Balakireva, A.V.; Chepikova, O.E.; Venanzi, F.M.; Zamyatnin, A.A. Cellular Aging Characteristics and Their Association with Age-Related Disorders. Antioxidants 2020, 9, 94. [Google Scholar] [CrossRef] [Green Version]
- Morabito, F.; Cristani, M.; Saija, A.; Stelitano, C.; Callea, V.; Tomaino, A.; Minciullo, P.L.; Gangemi, S. Lipid peroxidation and protein oxidation in patients affected by Hodgkin’s lymphoma. Mediat. Inflamm. 2004, 13, 381–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda, C.L.; Reed, R.L.; Kuiper, H.C.; Alber, S.; Stevens, J.F. Ascorbic acid promotes detoxification and elimination of 4-hydroxy-2(E)-nonenal in human monocytic THP-1 cells. Chem. Res. Toxicol. 2009, 22, 863–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Previati, M.; Lanzoni, I.; Corbacella, E.; Magosso, S.; Guaran, V.; Martini, A.; Capitani, S. Cisplatin-induced apoptosis in human promyelocytic leukemia cells. Int. J. Mol. Med. 2006, 18, 511–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negre-Salvayre, A.; Coatrieux, C.; Ingueneau, C.; Salvayre, R. Advanced lipid peroxidation end products in oxidative damage to proteins. Potential role in diseases and therapeutic prospects for the inhibitors. Br. J. Pharmacol. 2008, 153, 6–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, S.; Green, P.; Claxton, R.; Simcox, S.; Williams, M.V.; Walsh, K.; Leeuwenburgh, C. Reactive carbonyl formation by oxidative and non-oxidative pathways. Front. Biosci. 2001, 6, A17–A24. [Google Scholar] [CrossRef] [Green Version]
- Gangemi, S.; Allegra, A.; Aguennouz, M.; Alonci, A.; Speciale, A.; Cannavò, A.; Cristani, M.; Russo, S.; Spatari, G.; Alibrandi, A.; et al. Relationship between advanced oxidation protein products, advanced glycation end products, and S-nitrosylated proteins with biological risk and MDR-1 polymorphisms in patients affected by B-chronic lymphocytic leukemia. Cancer Investig. 2012, 30, 20–26. [Google Scholar] [CrossRef]
- Sayre, L.M.; Lin, D.; Yuan, Q.; Zhu, X.; Tang, X. Protein adducts generated from products of lipid oxidation: Focus on HNE and one. Drug Metab. Rev. 2006, 38, 651–675. [Google Scholar] [CrossRef]
- Lee, S.H.; Oe, T.; Blair, I.A. Vitamin C-induced decomposition of lipid hydroperoxides to endogenous genotoxins. Science 2001, 292, 2083–2086. [Google Scholar] [CrossRef] [PubMed]
- De Souza, G.F.; Ribeiro, H.L.; De Sousa, J.C.; Heredia, F.F.; De Freitas, R.M.; Martins, M.R.A.; Gonçalves, R.P.; Pinheiro, R.F.; Magalhães, S.M.M. HFE gene mutation and oxidative damage biomarkers in patients with myelodysplastic syndromes and its relation to transfusional iron overload: An observational cross-sectional study. BMJ Open 2015, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Güven, M.; Oztürk, B.; Sayal, A.; Ozet, A. Lipid peroxidation and antioxidant system in the blood of patients with Hodgkin’s disease. Clin. Biochem. 2000, 33, 209–212. [Google Scholar] [CrossRef]
- Eissa, L.A.; Esmaeel, M.I. Relevance of some serum biomarkers (E cadherin, GAGs & MDA in patients with diffuse large B-cell lymphoma. Pak. J. Pharm. Sci. 2008, 21, 29–35. [Google Scholar] [PubMed]
- El-Mezayen, H.; Darwish, H.; Hasheim, M.; El-Baz, H.A.; Mohamed, M.A. Oxidant/antioxidant status and their relations to chemotherapy in non-Hodgkin’s lymphoma. Int. J. Pharm. Clin. Res. 2015, 7, 269–274. [Google Scholar]
- Haddouche, M.; Meziane, W.; Hadjidj, Z.; Mesli, N.; Aribi, M. Clinical association of baseline levels of conjugated dienes in low-density lipoprotein and nitric oxide with aggressive B-cell non-Hodgkin lymphoma and their relationship with immunoglobulins and Th1-to-Th2 ratio. J. Blood Med. 2016, 7, 111–119. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Tripathi, M.; Satyam, A.; Kumar, L. Study of antioxidant levels in patients with multiple myeloma. Leuk. Lymphoma 2009, 50, 809–815. [Google Scholar] [CrossRef]
- Musolino, C.; Alonci, A.; Allegra, A.; Saija, A.; Penna, G.; Cannavò, A.; Cristani, M.; Saitta, S.; Gangemi, S. Increase in serum protein carbonyl groups is associated with more advanced stage of disease in multiple myeloma patients. Biomarkers 2011, 16, 718–719. [Google Scholar] [CrossRef]
- Gangemi, S.; Allegra, A.; Alonci, A.; Cristani, M.; Russo, S.; Speciale, A.; Penna, G.; Spatari, G.; Cannavò, A.; Bellomo, G.; et al. Increase of novel biomarkers for oxidative stress in patients with plasma cell disorders and in multiple myeloma patients with bone lesions. Inflamm. Res. 2012, 61, 1063–1067. [Google Scholar] [CrossRef]
- Lodh, M.; Goswami, B.; Gupta, N.; Patra, S.K.; Saxena, A. Assessment of oxidative stress and inflammatory process in patients of multiple myeloma. Indian J. Clin. Biochem. 2012, 27, 410–413. [Google Scholar] [CrossRef] [Green Version]
- Katz, J.; Moreb, J.; Baitinger, C.; Singer, C.; Caudle, R.M. Advanced glycation endproducts (AGEs) in saliva of patients with multiple myeloma—A pilot study. Leuk. Lymphoma 2017, 58, 2934–2938. [Google Scholar] [CrossRef]
- Nowak, W.; Treliński, J.; Chojnowski, K.; Matczak, J.; Robak, M.; Misiewicz, M.; Nowak, P. Assessment of oxidative/nitrative modifications of plasma proteins, selected ROTEM parameters and kinetics of fibrinogen polymerization in patients with multiple myeloma at diagnosis. Med. Oncol. 2017, 34, 4. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Musolino, C.; Pace, E.; Innao, V.; Di Salvo, E.; Ferraro, M.; Casciaro, M.; Spatari, G.; Tartarisco, G.; Allegra, A.G.; et al. Evaluation of the AGE/sRAGE Axis in Patients with Multiple Myeloma. Antioxidants 2019, 8, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devi, G.S.; Prasad, M.H.; Saraswathi, I.; Raghu, D.; Rao, D.N.; Reddy, P.P. Free radicals antioxidant enzymes and lipid peroxidation in different types of leukemias. Clin. Chim. Acta 2000, 293, 53–62. [Google Scholar] [CrossRef]
- Zuo, X.L.; Chen, J.M.; Zhou, X.; Li, X.Z.; Mei, G.Y. Levels of selenium, zinc, copper, and antioxidant enzyme activity in patients with leukemia. Biol Trace Elem. Res. 2006, 114, 41–53. [Google Scholar] [CrossRef]
- Musolino, C.; Allegra, A.; Alonci, A.; Saija, A.; Russo, S.; Cannavò, A.; Cristani, M.; Centorrino, R.; Saitta, S.; Alibrandi, A.; et al. Carbonyl group serum levels are associated with CD38 expression in patients with B chronic lymphocytic leukemia. Clin. Biochem. 2011, 44, 1487–1490. [Google Scholar] [CrossRef] [PubMed]
- Zelen, I.; Djurdjevic, P.; Popovic, S.; Stojanovic, M.; Jakovljevic, V.; Radivojevic, S.; Baskic, D.; Arsenijevic, N. Antioxidant enzymes activities and plasma levels of oxidative stress markers in B-chronic lymphocytic leukemia patients. J. BUON 2010, 15, 330–336. [Google Scholar]
- Jabłońska, E.; Kiersnowska-Rogowska, B.; Ratajczak, W.; Rogowski, F.; Sawicka-Powierza, J. Reactive oxygen and nitrogen species in the course of B-CLL. Adv. Med. Sci. 2007, 52, 154–158. [Google Scholar]
- Ahmad, R.; Tripathi, A.K.; Tripathi, P.; Singh, R.; Singh, S.; Singh, R.K. Oxidative stress and antioxidant status in patients with chronic myeloid leukemia. Indian J. Clin. Biochem. 2008, 23, 328–333. [Google Scholar] [CrossRef] [Green Version]
- Hlaváčková, A.; Štikarová, J.; Pimková, K.; Chrastinová, L.; Májek, P.; Kotlín, R.; Čermák, J.; Suttnar, J.; Dyr, J.E. Enhanced plasma protein carbonylation in patients with myelodysplastic syndromes. Free Radic. Biol. Med. 2017, 108, 1–7. [Google Scholar] [CrossRef]
- De Souza, G.F.; Barbosa, M.C.; de Jesus Santos, T.E.; de Jesus Ponte Carvalho, T.M.; de Freitas, R.M.; Martins, M.R.A.; Gonçalves, R.P.; Pinheiro, R.F.; Magalhães, S.M.M. Increased parameters of oxidative stress and its relation to transfusion iron overload in patients with myelodysplastic syndromes. J. Clin. Pathol. 2013, 66, 996–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortelezzi, A.; Cattaneo, C.; Cristiani, S.; Duca, L.; Sarina, B.; Deliliers, G.L.; Fiorelli, G.; Cappellini, M.D. Non-transferrin-bound iron in myelodysplastic syndromes: A marker of ineffective erythropoiesis? Hematol. J. 2000, 1, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Pimková, K.; Chrastinová, L.; Suttnar, J.; Štikarová, J.; Kotlín, R.; Čermák, J.; Dyr, J.E. Plasma levels of aminothiols, nitrite, nitrate, and malondialdehyde in myelodysplastic syndromes in the context of clinical outcomes and as a consequence of iron overload. Oxid. Med. Cell Longev. 2014, 2014, 416028. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-García, A.; Morales, M.L.; Garrido-García, V.; García-Baquero, I.; Leivas, A.; Carreño-Tarragona, G.; Sánchez, R.; Arenas, A.; Cedena, T.; Ayala, R.M.; et al. Protein Carbonylation in Patients with Myelodysplastic Syndrome: An Opportunity for Deferasirox Therapy. Antioxidants 2019, 8, 508. [Google Scholar] [CrossRef] [Green Version]
- Djikic, D.; Markovic, D.; Bogdanovic, A.; Mitrovic-Ajtic, O.; Suboticki, T.; Diklic, M.; Beleslin-Cokic, B.; Bjelica, S.; Kovacic, M.; P Cokic, V. Oxidative and nitrosative stress in myeloproliferative neoplasms: The impact on the AKT/mTOR signaling pathway. J. BUON 2018, 23, 1481–1491. [Google Scholar]
- Kaya, E.; Keskin, L.; Aydogdu, I.; Kuku, I.; Bayraktar, N.; Erkut, M.A. Oxidant/antioxidant parameters and their relationship with chemotherapy in Hodgkin’s lymphoma. J. Int. Med. Res. 2005, 33, 687–692. [Google Scholar] [CrossRef] [Green Version]
- Abou-Seif, M.A.; Rabia, A.; Nasr, M. Antioxidant status, erythrocyte membrane lipid peroxidation and osmotic fragility in malignant lymphoma patients. Clin. Chem. Lab. Med. 2000, 38, 737–742. [Google Scholar] [CrossRef]
- Bottari, N.B.; Munhoz, T.D.; Torbitz, V.D.; Tonin, A.A.; Anai, L.A.; Semolin, L.M.S.; Jark, P.C.; Bollick, Y.S.; Moresco, R.N.; França, R.T.; et al. Oxidative stress in dogs with multicentric lymphoma: Effect of chemotherapy on oxidative and antioxidant biomarkers. Redox Rep. 2015, 20, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Kuku, I.; Aydogdu, I.; Bayraktar, N.; Kaya, E.; Akyol, O.; Erkurt, M.A. Oxidant/antioxidant parameters and their relationship with medical treatment in multiple myeloma. Cell Biochem. Funct. 2005, 23, 47–50. [Google Scholar] [CrossRef]
- Mehdi, W.A.; Zainulabdeen, J.A.; Mehde, A.A. Investigation of the Antioxidant Status in Multiple Myeloma Patients: Effects of Therapy. Asian Pac. J. Cancer Prev. 2013, 14, 3663–3667. [Google Scholar] [CrossRef] [Green Version]
- Szwed, M.; Kania, K.D.; Jozwiak, Z. Molecular damage caused by generation of reactive oxygen species in the redox cycle of doxorubicin-transferrin conjugate in human leukemia cell lines. Leuk. Lymphoma 2015, 56, 1475–1483. [Google Scholar] [CrossRef]
- Esfahani, A.; Ghoreishi, Z.; Nikanfar, A.; Sanaat, Z.; Ghorbanihaghjo, A. Influence of chemotherapy on the lipid peroxidation and antioxidant status in patients with acute myeloid leukemia. Acta Med. Iran. 2012, 50, 454–458. [Google Scholar]
- Kumar, S.; Tchounwou, P.B. Molecular mechanisms of cisplatin cytotoxicity in acute promyelocytic leukemia cells. Oncotarget 2015, 6, 40734–40746. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Yedjou, C.G.; Tchounwou, P.B. Arsenic trioxide induces oxidative stress, DNA damage, and mitochondrial pathway of apoptosis in human leukemia (HL-60) cells. J. Exp. Clin. Cancer Res. 2014, 33, 42. [Google Scholar] [CrossRef] [Green Version]
- Mathews, V.V.; Paul, M.V.S.; Abhilash, M.; Manju, A.; Abhilash, S.; Nair, R.H. Myocardial toxicity of acute promyelocytic leukaemia drug-arsenic trioxide. Eur. Rev. Med. Pharm. Sci. 2013, 17 (Suppl. 1), 34–38. [Google Scholar]
- Sun, R.; Medeiros, L.J.; Young, K.H. Diagnostic and predictive biomarkers for lymphoma diagnosis and treatment in the era of precision medicine. Mod. Pathol. 2016, 29, 1118–1142. [Google Scholar] [CrossRef] [Green Version]
- Shankland, K.R.; Armitage, J.O.; Hancock, B.W. Non-Hodgkin lymphoma. Lancet 2012, 380, 848–857. [Google Scholar] [CrossRef]
- Shanbhag, S.; Ambinder, R.F. Hodgkin lymphoma: A review and update on recent progress. CA Cancer J. Clin. 2018, 68, 116–132. [Google Scholar] [CrossRef]
- Intlekofer, A.M.; Younes, A. Precision therapy for lymphoma--current state and future directions. Nat. Rev. Clin. Oncol. 2014, 11, 585–596. [Google Scholar] [CrossRef]
- Bur, H.; Haapasaari, K.-M.; Turpeenniemi-Hujanen, T.; Kuittinen, O.; Auvinen, P.; Marin, K.; Koivunen, P.; Sormunen, R.; Soini, Y.; Karihtala, P. Oxidative stress markers and mitochondrial antioxidant enzyme expression are increased in aggressive Hodgkin lymphomas. Histopathology 2014, 65, 319–327. [Google Scholar] [CrossRef]
- Weinberg, F.; Ramnath, N.; Nagrath, D. Reactive Oxygen Species in the Tumor Microenvironment: An Overview. Cancers 2019, 11, 1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irigoyen, M.; García-Ruiz, J.C.; Berra, E. The hypoxia signalling pathway in haematological malignancies. Oncotarget 2017, 8, 36832–36844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matolay, O.; Méhes, G. Sustain, Adapt, and Overcome-Hypoxia Associated Changes in the Progression of Lymphatic Neoplasia. Front. Oncol. 2019, 9, 1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhalla, K.; Jaber, S.; Nahid, M.N.; Underwood, K.; Beheshti, A.; Landon, A.; Bhandary, B.; Bastian, P.; Evens, A.M.; Haley, J.; et al. Role of hypoxia in Diffuse Large B-cell Lymphoma: Metabolic repression and selective translation of HK2 facilitates development of DLBCL. Sci Rep. 2018, 8, 744. [Google Scholar] [CrossRef] [Green Version]
- Al-Gayyar, M.M.H.; Eissa, L.A.; Rabie, A.M.; El-Gayar, A.M. Measurements of oxidative stress status and antioxidant activity in chronic leukaemia patients. J. Pharm. Pharmacol. 2007, 59, 409–417. [Google Scholar] [CrossRef]
- Ansell, S.M. Hodgkin lymphoma: 2018 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2018, 93, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Imbesi, S.; Musolino, C.; Allegra, A.; Saija, A.; Morabito, F.; Calapai, G.; Gangemi, S. Oxidative stress in oncohematologic diseases: An update. Expert Rev. Hematol. 2013, 6, 317–325. [Google Scholar] [CrossRef]
- Gopas, J.; Stern, E.; Zurgil, U.; Ozer, J.; Ben-Ari, A.; Shubinsky, G.; Braiman, A.; Sinay, R.; Ezratty, J.; Dronov, V.; et al. Reed-Sternberg cells in Hodgkin’s lymphoma present features of cellular senescence. Cell Death Dis. 2016, 7, e2457. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Aziz, A.F.; El-Naggar, M.M. Superoxide dismutase activities in serum and white blood cells of patients with some malignancies. Cancer Lett. 1997, 113, 61–64. [Google Scholar] [CrossRef]
- Bewick, M.; Coutie, W.; Tudhope, G.R. Superoxide dismutase, glutathione peroxidase and catalase in the red cells of patients with malignant lymphoma. Br. J. Haematol. 1987, 65, 347–350. [Google Scholar] [CrossRef]
- Gonzales, R.; Auclair, C.; Voisin, E.; Gautero, H.; Dhermy, D.; Boivin, P. Superoxide dismutase, catalase, and glutathione peroxidase in red blood cells from patients with malignant diseases. Cancer Res. 1984, 44, 4137–4139. [Google Scholar]
- Zhang, J.; Li, X.; Han, X.; Liu, R.; Fang, J. Targeting the Thioredoxin System for Cancer Therapy. Trends Pharmacol. Sci. 2017, 38, 794–808. [Google Scholar] [CrossRef]
- Vassilakopoulos, T.P.; Levidou, G.; Milionis, V.; Hartmann, S.; Lakiotaki, E.; Sepsa, A.; Thymara, I.; Ntailiani, P.; Spanou, K.; K Angelopoulou, M.; et al. Thioredoxin-1, chemokine (C-X-C motif) ligand-9 and interferon-γ expression in the neoplastic cells and macrophages of Hodgkin lymphoma: Clinicopathologic correlations and potential prognostic implications. Leuk. Lymphoma 2017, 58, 1–13. [Google Scholar] [CrossRef]
- Pasanen, A.K.; Kuitunen, H.; Haapasaari, K.-M.; Karihtala, P.; Kyllönen, H.; Soini, Y.; Turpeenniemi-Hujanen, T.; Kuittinen, O. Expression and prognostic evaluation of oxidative stress markers in an immunohistochemical study of B-cell derived lymphomas. Leuk. Lymphoma 2012, 53, 624–631. [Google Scholar] [CrossRef]
- Pérez, V.I.; Cortez, L.A.; Lew, C.M.; Rodriguez, M.; Webb, C.R.; Van Remmen, H.; Chaudhuri, A.; Qi, W.; Lee, S.; Bokov, A.; et al. Thioredoxin 1 overexpression extends mainly the earlier part of life span in mice. J. Gerontol. A Biol. Sci. Med. Sci. 2011, 66, 1286–1299. [Google Scholar] [CrossRef] [Green Version]
- Slovak, M.L.; Bedell, V.; Hsu, Y.-H.; Estrine, D.B.; Nowak, N.J.; Delioukina, M.L.; Weiss, L.M.; Smith, D.D.; Forman, S.J. Molecular karyotypes of Hodgkin and Reed-Sternberg cells at disease onset reveal distinct copy number alterations in chemosensitive versus refractory Hodgkin lymphoma. Clin. Cancer Res. 2011, 17, 3443–3454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spindel, O.N.; World, C.; Berk, B.C. Thioredoxin interacting protein: Redox dependent and independent regulatory mechanisms. Antioxid. Redox Signal. 2012, 16, 587–596. [Google Scholar] [CrossRef]
- Goswami, K.; Koner, B.C. Level of sialic acid residues in platelet proteins in diabetes, aging, and Hodgkin’s lymphoma: A potential role of free radicals in desialylation. Biochem. Biophys. Res. Commun. 2002, 297, 502–505. [Google Scholar] [CrossRef]
- Quach, M.E.; Chen, W.; Li, R. Mechanisms of platelet clearance and translation to improve platelet storage. Blood 2018, 131, 1512–1521. [Google Scholar] [CrossRef]
- Prete, A.; Urtula, A.; Grozovsky, R. Sialic Acid Content on Platelet Surface Glycoproteins Modulates Thrombin-Induced Activation. Blood 2018, 132, 3730. [Google Scholar] [CrossRef]
- Läubli, H.; Borsig, L. Altered Cell Adhesion and Glycosylation Promote Cancer Immune Suppression and Metastasis. Front. Immunol. 2019, 10, 2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drillenburg, P.; Pals, S.T. Cell adhesion receptors in lymphoma dissemination. Blood 2000, 95, 1900–1910. [Google Scholar] [CrossRef] [PubMed]
- Armitage, J.O.; Gascoyne, R.D.; Lunning, M.A.; Cavalli, F. Non-Hodgkin lymphoma. Lancet 2017, 390, 298–310. [Google Scholar] [CrossRef]
- Alexander, D.D.; Mink, P.J.; Adami, H.-O.; Chang, E.T.; Cole, P.; Mandel, J.S.; Trichopoulos, D. The non-Hodgkin lymphomas: A review of the epidemiologic literature. Int. J. Cancer 2007, 120 (Suppl. 12), 1–39. [Google Scholar] [CrossRef]
- Lan, Q.; Zheng, T.; Shen, M.; Zhang, Y.; Wang, S.S.; Zahm, S.H.; Holford, T.R.; Leaderer, B.; Boyle, P.; Chanock, S. Genetic polymorphisms in the oxidative stress pathway and susceptibility to non-Hodgkin lymphoma. Hum. Genet. 2007, 121, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Lightfoot, T.J.; Skibola, C.F.; Smith, A.G.; Forrest, M.S.; Adamson, P.J.; Morgan, G.J.; Bracci, P.M.; Roman, E.; Smith, M.T.; Holly, E.A. Polymorphisms in the oxidative stress genes, superoxide dismutase, glutathione peroxidase and catalase and risk of non-Hodgkin’s lymphoma. Haematologica 2006, 91, 1222–1227. [Google Scholar]
- Wang, S.S.; Davis, S.; Cerhan, J.R.; Hartge, P.; Severson, R.K.; Cozen, W.; Lan, Q.; Welch, R.; Chanock, S.J.; Rothman, N. Polymorphisms in oxidative stress genes and risk for non-Hodgkin lymphoma. Carcinogenesis 2006, 27, 1828–1834. [Google Scholar] [CrossRef] [Green Version]
- Hirota, K.; Murata, M.; Sachi, Y.; Nakamura, H.; Takeuchi, J.; Mori, K.; Yodoi, J. Distinct roles of thioredoxin in the cytoplasm and in the nucleus. A two-step mechanism of redox regulation of transcription factor NF-kappaB. J. Biol. Chem. 1999, 274, 27891–27897. [Google Scholar] [CrossRef] [Green Version]
- Peroja, P.; Pasanen, A.K.; Haapasaari, K.-M.; Jantunen, E.; Soini, Y.; Turpeenniemi-Hujanen, T.; Bloigu, R.; Lilja, L.; Kuittinen, O.; Karihtala, P. Oxidative stress and redox state-regulating enzymes have prognostic relevance in diffuse large B-cell lymphoma. Exp. Hematol. Oncol. 2012, 1, 2. [Google Scholar] [CrossRef] [Green Version]
- Roschewski, M.; Staudt, L.M.; Wilson, W.H. Diffuse large B-cell lymphoma-treatment approaches in the molecular era. Nat. Rev. Clin. Oncol. 2014, 11, 12–23. [Google Scholar] [CrossRef]
- Tome, M.E.; Johnson, D.B.F.; Rimsza, L.M.; Roberts, R.A.; Grogan, T.M.; Miller, T.P.; Oberley, L.W.; Briehl, M.M. A redox signature score identifies diffuse large B-cell lymphoma patients with a poor prognosis. Blood 2005, 106, 3594–3601. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Yang, Z.; Jiang, S.; Di, W.; Ma, Z.; Hu, W.; Chen, F.; Reiter, R.J.; Yang, Y. Melatonin: Does it have utility in the treatment of haematological neoplasms? Br. J. Pharm. 2018, 175, 3251–3262. [Google Scholar] [CrossRef]
- Camiolo, G.; Barbato, A.; Giallongo, C.; Vicario, N.; Romano, A.; Parrinello, N.L.; Parenti, R.; Sandoval, J.C.; García-Moreno, D.; Lazzarino, G.; et al. Iron regulates myeloma cell/macrophage interaction and drives resistance to bortezomib. Redox Biol. 2020, 36. [Google Scholar] [CrossRef]
- Zima, T.; Spicka, I.; Stípek, S.; Crkovská, J.; Pláteník, J.; Merta, M.; Tesar, V. Antioxidant enzymes and lipid peroxidation in patients with multiple myeloma. Neoplasma 1996, 43, 69–73. [Google Scholar]
- Raninga, P.V.; Di Trapani, G.; Vuckovic, S.; Tonissen, K.F. TrxR1 inhibition overcomes both hypoxia-induced and acquired bortezomib resistance in multiple myeloma through NF-кβ inhibition. Cell Cycle 2016, 15, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Raninga, P.V.; Di Trapani, G.; Vuckovic, S.; Bhatia, M.; Tonissen, K.F. Inhibition of thioredoxin 1 leads to apoptosis in drug-resistant multiple myeloma. Oncotarget 2015, 6, 15410–15424. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; Fan, S.; Zheng, J.; Huang, W.; Gasparetto, C.; Chao, N.J.; Hu, J.; Kang, Y. Inhibition of thioredoxin activates mitophagy and overcomes adaptive bortezomib resistance in multiple myeloma. J. Hematol. Oncol. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Fink, E.E.; Mannava, S.; Bagati, A.; Bianchi-Smiraglia, A.; Nair, J.R.; Moparthy, K.; Lipchick, B.C.; Drokov, M.; Utley, A.; Ross, J.; et al. Mitochondrial thioredoxin reductase regulates major cytotoxicity pathways of proteasome inhibitors in multiple myeloma cells. Leukemia 2016, 30, 104–111. [Google Scholar] [CrossRef] [Green Version]
- Sze, J.H.; Raninga, P.V.; Nakamura, K.; Casey, M.; Khanna, K.K.; Berners-Price, S.J.; Di Trapani, G.; Tonissen, K.F. Anticancer activity of a Gold(I) phosphine thioredoxin reductase inhibitor in multiple myeloma. Redox Biol. 2019, 28. [Google Scholar] [CrossRef]
- Hurt, E.M.; Thomas, S.B.; Peng, B.; Farrar, W.L. Integrated molecular profiling of SOD2 expression in multiple myeloma. Blood 2007, 109, 3953–3962. [Google Scholar] [CrossRef] [Green Version]
- Allegra, A.; Pace, E.; Tartarisco, G.; Innao, V.; DI Salvo, E.; Allegra, A.G.; Ferraro, M.; Musolino, C.; Gangemi, S. Changes in Serum Interleukin-8 and sRAGE Levels in Multiple Myeloma Patients. Anticancer Res. 2020, 40, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Asadipooya, K.; Uy, E.M. Advanced Glycation End Products (AGEs), Receptor for AGEs, Diabetes, and Bone: Review of the Literature. J. Endocr. Soc. 2019, 3, 1799–1818. [Google Scholar] [CrossRef] [Green Version]
- Anchoori, R.K.; Tan, M.; Tseng, S.-H.; Peng, S.; Soong, R.-S.; Algethami, A.; Foran, P.; Das, S.; Wang, C.; Wang, T.-L.; et al. Structure-function analyses of candidate small molecule RPN13 inhibitors with antitumor properties. PLoS ONE 2020, 15, e0227727. [Google Scholar] [CrossRef] [Green Version]
- Glynn, S.J.; Gaffney, K.J.; Sainz, M.A.; Louie, S.G.; Petasis, N.A. Molecular characterization of the boron adducts of the proteasome inhibitor bortezomib with epigallocatechin-3-gallate and related polyphenols. Org. Biomol. Chem. 2015, 13, 3887–3899. [Google Scholar] [CrossRef] [Green Version]
- Nabissi, M.; Morelli, M.B.; Offidani, M.; Amantini, C.; Gentili, S.; Soriani, A.; Cardinali, C.; Leoni, P.; Santoni, G. Cannabinoids synergize with carfilzomib, reducing multiple myeloma cells viability and migration. Oncotarget 2016, 7, 77543–77557. [Google Scholar] [CrossRef] [Green Version]
- Szalat, R.; Samur, M.K.; Fulciniti, M.; Lopez, M.; Nanjappa, P.; Cleynen, A.; Wen, K.; Kumar, S.; Perini, T.; Calkins, A.S.; et al. Nucleotide excision repair is a potential therapeutic target in multiple myeloma. Leukemia 2018, 32, 111–119. [Google Scholar] [CrossRef]
- Karademir, B.; Sari, G.; Jannuzzi, A.T.; Musunuri, S.; Wicher, G.; Grune, T.; Mi, J.; Hacioglu-Bay, H.; Forsberg-Nilsson, K.; Bergquist, J.; et al. Proteomic approach for understanding milder neurotoxicity of Carfilzomib against Bortezomib. Sci. Rep. 2018, 8, 16318. [Google Scholar] [CrossRef]
- Wei, J.-Y.; Liu, C.-C.; Ouyang, H.-D.; Ma, C.; Xie, M.-X.; Liu, M.; Lei, W.-L.; Ding, H.-H.; Wu, S.-L.; Xin, W.-J. Activation of RAGE/STAT3 pathway by methylglyoxal contributes to spinal central sensitization and persistent pain induced by bortezomib. Exp. Neurol. 2017, 296, 74–82. [Google Scholar] [CrossRef]
- Imam, F.; Al-Harbi, N.O.; Al-Harbi, M.M.; Ansari, M.A.; Almutairi, M.M.; Alshammari, M.; Almukhlafi, T.S.; Ansari, M.N.; Aljerian, K.; Ahmad, S.F. Apremilast reversed carfilzomib-induced cardiotoxicity through inhibition of oxidative stress, NF-κB and MAPK signaling in rats. Toxicol. Mech. Methods 2016, 26, 700–708. [Google Scholar] [CrossRef]
- Aryal, B.; Jeong, J.; Rao, V.A. Doxorubicin-induced carbonylation and degradation of cardiac myosin binding protein C promote cardiotoxicity. Proc. Natl. Acad. Sci. USA 2014, 111, 2011–2016. [Google Scholar] [CrossRef] [Green Version]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malard, F.; Mohty, M. Acute lymphoblastic leukaemia. Lancet 2020, 395, 1146–1162. [Google Scholar] [CrossRef]
- Milne, K.; Sturrock, B.; Chevassut, T. Chronic Lymphocytic Leukaemia in 2020: The Future Has Arrived. Curr. Oncol. Rep. 2020, 22, 36. [Google Scholar] [CrossRef] [Green Version]
- Short, N.J.; Rytting, M.E.; Cortes, J.E. Acute myeloid leukaemia. Lancet 2018, 392, 593–606. [Google Scholar] [CrossRef]
- Gale, R.P.; Apperley, J. What Does Chronic Myeloid Leukaemia Tell Us About Other Leukaemias? Curr. Hematol. Malig. Rep. 2019, 14, 477–479. [Google Scholar] [CrossRef] [PubMed]
- Er, T.-K.; Tsai, S.-M.; Wu, S.-H.; Chiang, W.; Lin, H.-C.; Lin, S.-F.; Wu, S.-H.; Tsai, L.-Y.; Liu, T.-Z. Antioxidant status and superoxide anion radical generation in acute myeloid leukemia. Clin. Biochem. 2007, 40, 1015–1019. [Google Scholar] [CrossRef] [PubMed]
- Antoszewska-Smith, J.; Pawlowska, E.; Blasiak, J. Reactive oxygen species in BCR-ABL1-expressing cells—Relevance to chronic myeloid leukemia. Acta Biochim. Pol. 2017, 64, 1–10. [Google Scholar] [CrossRef]
- Oltra, A.M.; Carbonell, F.; Tormos, C.; Iradi, A.; Sáez, G.T. Antioxidant enzyme activities and the production of MDA and 8-oxo-dG in chronic lymphocytic leukemia. Free Radic. Biol. Med. 2001, 30, 1286–1292. [Google Scholar] [CrossRef]
- Collado, R.; Ivars, D.; Oliver, I.; Tormos, C.; Egea, M.; Miguel, A.; Sáez, G.T.; Carbonell, F. Increased oxidative damage associated with unfavorable cytogenetic subgroups in chronic lymphocytic leukemia. Biomed. Res. Int. 2014, 2014, 686392. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Zhang, W.; Wei, Y.; Zhou, D.; Su, Z.; Meng, X.; Hui, L.; Tian, W. The changes of oxidative stress and human 8-hydroxyguanine glycosylase1 gene expression in depressive patients with acute leukemia. Leuk. Res. 2007, 31, 387–393. [Google Scholar] [CrossRef]
- Papiez, M.A.; Dybala, M.; Sowa-Kucma, M.; Krzysciak, W.; Taha, H.; Jozkowicz, A.; Nowak, G. Evaluation of oxidative status and depression-like responses in Brown Norway rats with acute myeloid leukemia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2009, 33, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.; Yan, S.; Vestal, C. Oxidative Stress, Bone Marrow Failure, and Genome Instability in Hematopoietic Stem Cells. Int. J. Mol. Sci. 2015, 16, 2366–2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimer, S.D. Myelodysplastic syndromes. Blood 2008, 111, 4841–4851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cazzola, M.; Malcovati, L. Myelodysplastic syndromes—Coping with ineffective hematopoiesis. N. Engl. J. Med. 2005, 352, 536–538. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.J.; Robert, C.; Gough, S.M.; Rassool, F.V.; Aplan, P.D. Oxidative stress leads to increased mutation frequency in a murine model of myelodysplastic syndrome. Leuk. Res. 2014, 38, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Guglielmelli, P.; Larson, D.R.; Finke, C.; Wassie, E.A.; Pieri, L.; Gangat, N.; Fjerza, R.; Belachew, A.A.; Lasho, T.L.; et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014, 124, 2507–2513. [Google Scholar] [CrossRef]
- Farquhar, M.J.; Bowen, D.T. Oxidative stress and the myelodysplastic syndromes. Int. J. Hematol. 2003, 77, 342–350. [Google Scholar] [CrossRef]
- Ghoti, H.; Amer, J.; Winder, A.; Rachmilewitz, E.; Fibach, E. Oxidative stress in red blood cells, platelets and polymorphonuclear leukocytes from patients with myelodysplastic syndrome. Eur. J. Haematol. 2007, 79, 463–467. [Google Scholar] [CrossRef]
- Gonçalves, A.C.; Cortesão, E.; Oliveiros, B.; Alves, V.; Espadana, A.I.; Rito, L.; Magalhães, E.; Pereira, S.; Pereira, A.; Costa, J.M.N.; et al. Oxidative stress levels are correlated with P15 and P16 gene promoter methylation in myelodysplastic syndrome patients. Clin. Exp. Med. 2016, 16, 333–343. [Google Scholar] [CrossRef]
- Shenoy, N.; Vallumsetla, N.; Rachmilewitz, E.; Verma, A.; Ginzburg, Y. Impact of iron overload and potential benefit from iron chelation in low-risk myelodysplastic syndrome. Blood 2014, 124, 873–881. [Google Scholar] [CrossRef]
- Chai, X.; Li, D.; Cao, X.; Zhang, Y.; Mu, J.; Lu, W.; Xiao, X.; Li, C.; Meng, J.; Chen, J.; et al. ROS-mediated iron overload injures the hematopoiesis of bone marrow by damaging hematopoietic stem/progenitor cells in mice. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Britton, R.S.; Ramm, G.A.; Olynyk, J.; Singh, R.; O’Neill, R.; Bacon, B.R. Pathophysiology of Iron Toxicity. In Progress in Iron Research; Hershko, C., Konijn, A.M., Aisen, P., Eds.; Advances in Experimental Medicine and Biology; Springer: Boston, MA, USA, 1994; pp. 239–253. ISBN 978-1-4615-2554-7. [Google Scholar]
- Malcovati, L.; Porta, M.G.D.; Pascutto, C.; Invernizzi, R.; Boni, M.; Travaglino, E.; Passamonti, F.; Arcaini, L.; Maffioli, M.; Bernasconi, P.; et al. Prognostic factors and life expectancy in myelodysplastic syndromes classified according to WHO criteria: A basis for clinical decision making. J. Clin. Oncol. 2005, 23, 7594–7603. [Google Scholar] [CrossRef]
- Semlitsch, T.; Tillian, H.M.; Neven, Z.; Martin, P. Differential influence of the lipid peroxidation product 4-hydroxynonenal on the growth of human lymphatic leukaemia cells and human periopherial blood lymphocytes. Anticancer Res. 2002, 22, 1689–1697. [Google Scholar]
- Gattermann, N.; Rachmilewitz, E.A. Iron overload in MDS-pathophysiology, diagnosis, and complications. Ann. Hematol. 2011, 90, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Frank, J.; Pompella, A.; Biesalski, H.K. Immunohistochemical Detection of Protein Oxidation. In Oxidants and Antioxidants. Methods in Molecular BiologyTM; Armstrong, D., Ed.; Humana Press: Totowa, NJ, USA, 2002; Volume 196, pp. 35–40. ISBN 978-0-89603-851-6. [Google Scholar]
- Tefferi, A.; Vardiman, J.W. Myelodysplastic Syndromes. N. Engl. J. Med. 2009, 361, 1872–1885. [Google Scholar] [CrossRef] [PubMed]
- Spivak, J.L. Myeloproliferative Neoplasms. N. Engl. J. Med. 2017, 376, 2168–2181. [Google Scholar] [CrossRef] [Green Version]
- Iurlo, A.; Cattaneo, D.; Gianelli, U. Blast Transformation in Myeloproliferative Neoplasms: Risk Factors, Biological Findings, and Targeted Therapeutic Options. Int. J. Mol. Sci. 2019, 20, 1839. [Google Scholar] [CrossRef] [Green Version]
- Vener, C.; Novembrino, C.; Catena, F.B.; Fracchiolla, N.S.; Gianelli, U.; Savi, F.; Radaelli, F.; Fermo, E.; Cortelezzi, A.; Lonati, S.; et al. Oxidative stress is increased in primary and post-polycythemia vera myelofibrosis. Exp. Hematol. 2010, 38, 1058–1065. [Google Scholar] [CrossRef]
- Durmus, A.; Mentese, A.; Yilmaz, M.; Sumer, A.; Akalin, I.; Topal, C.; Alver, A. Increased oxidative stress in patients with essential thrombocythemia. Eur. Rev. Med. Pharm. Sci. 2013, 17, 2860–2866. [Google Scholar]
- Musolino, C.; Allegra, A.; Saija, A.; Alonci, A.; Russo, S.; Spatari, G.; Penna, G.; Gerace, D.; Cristani, M.; David, A.; et al. Changes in advanced oxidation protein products, advanced glycation end products, and s-nitrosylated proteins, in patients affected by polycythemia vera and essential thrombocythemia. Clin. Biochem. 2012, 45, 1439–1443. [Google Scholar] [CrossRef]
- Chandra, J. Oxidative Stress by Targeted Agents Promotes Cytotoxicity in Hematologic Malignancies. Antioxid. Redox Signal. 2009, 11, 1123–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelkel, M.; Jacob, C.; Dicato, M.; Diederich, M. Potential of the Dietary Antioxidants Resveratrol and Curcumin in Prevention and Treatment of Hematologic Malignancies. Molecules 2010, 15, 7035–7074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messa, E.; Carturan, S.; Maffè, C.; Pautasso, M.; Bracco, E.; Roetto, A.; Messa, F.; Arruga, F.; Defilippi, I.; Rosso, V.; et al. Deferasirox is a powerful NF-kappaB inhibitor in myelodysplastic cells and in leukemia cell lines acting independently from cell iron deprivation by chelation and reactive oxygen species scavenging. Haematologica 2010, 95, 1308–1316. [Google Scholar] [CrossRef] [Green Version]
- Hole, P.S.; Darley, R.L.; Tonks, A. Do reactive oxygen species play a role in myeloid leukemias? Blood 2011, 117, 5816–5826. [Google Scholar] [CrossRef] [Green Version]
- Gargouri, M.; Soussi, A.; Akrouti, A.; Magné, C.; El Feki, A. Potential protective effects of the edible alga Arthrospira platensis against lead-induced oxidative stress, anemia, kidney injury, and histopathological changes in adult rats. Appl. Physiol. Nutr. Metab. 2019, 44, 271–281. [Google Scholar] [CrossRef]
- Dua, T.K.; Dewanjee, S.; Khanra, R. Prophylactic role of Enhydra fluctuans against arsenic-induced hepatotoxicity via anti-apoptotic and antioxidant mechanisms. Redox Rep. 2016, 21, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Acker, C.I.; Souza, A.C.G.; Dos Santos, M.P.; Mazzanti, C.M.; Nogueira, C.W. Diphenyl diselenide attenuates hepatic and hematologic toxicity induced by chlorpyrifos acute exposure in rats. Env. Sci. Pollut Res. Int. 2012, 19, 3481–3490. [Google Scholar] [CrossRef]
- Soudani, N.; Ben Amara, I.; Troudi, A.; Hakim, A.; Bouaziz, H.; Ayadi Makni, F.; Zeghal, K.M.; Zeghal, N. Oxidative damage induced by chromium (VI) in rat erythrocytes: Protective effect of selenium. J. Physiol. Biochem. 2011, 67, 577–588. [Google Scholar] [CrossRef]
- Espinoza, J.L.; Kurokawa, Y.; Takami, A. Rationale for assessing the therapeutic potential of resveratrol in hematological malignancies. Blood Rev. 2019, 33, 43–52. [Google Scholar] [CrossRef]
- Quoc Trung, L.; Espinoza, J.L.; Takami, A.; Nakao, S. Resveratrol induces cell cycle arrest and apoptosis in malignant NK cells via JAK2/STAT3 pathway inhibition. PLoS ONE 2013, 8, e55183. [Google Scholar] [CrossRef] [Green Version]
- Roccaro, A.M.; Leleu, X.; Sacco, A.; Moreau, A.-S.; Hatjiharissi, E.; Jia, X.; Xu, L.; Ciccarelli, B.; Patterson, C.J.; Ngo, H.T.; et al. Resveratrol exerts antiproliferative activity and induces apoptosis in Waldenström’s macroglobulinemia. Clin. Cancer Res. 2008, 14, 1849–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salehi, B.; Mishra, A.P.; Nigam, M.; Sener, B.; Kilic, M.; Sharifi-Rad, M.; Fokou, P.V.T.; Martins, N.; Sharifi-Rad, J. Resveratrol: A Double-Edged Sword in Health Benefits. Biomedicines 2018, 6, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zykova, T.A.; Zhu, F.; Zhai, X.; Ma, W.-Y.; Ermakova, S.P.; Lee, K.W.; Bode, A.M.; Dong, Z. Resveratrol directly targets COX-2 to inhibit carcinogenesis. Mol. Carcinog. 2008, 47, 797–805. [Google Scholar] [CrossRef] [Green Version]
- Varoni, E.M.; Lo Faro, A.F.; Sharifi-Rad, J.; Iriti, M. Anticancer Molecular Mechanisms of Resveratrol. Front. Nutr. 2016, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Pezzuto, J.M. Resveratrol as an Inhibitor of Carcinogenesis. Pharm. Biol. 2008, 46, 443–573. [Google Scholar] [CrossRef]
- Kundu, J.K.; Surh, Y.-J. Cancer chemopreventive and therapeutic potential of resveratrol: Mechanistic perspectives. Cancer Lett. 2008, 269, 243–261. [Google Scholar] [CrossRef]
- Rimmelé, P.; Lofek-Czubek, S.; Ghaffari, S. Resveratrol increases the bone marrow hematopoietic stem and progenitor cell capacity. Am. J. Hematol. 2014, 89, E235–E238. [Google Scholar] [CrossRef] [Green Version]
- Ferry-Dumazet, H.; Garnier, O.; Mamani-Matsuda, M.; Vercauteren, J.; Belloc, F.; Billiard, C.; Dupouy, M.; Thiolat, D.; Kolb, J.P.; Marit, G.; et al. Resveratrol inhibits the growth and induces the apoptosis of both normal and leukemic hematopoietic cells. Carcinogenesis 2002, 23, 1327–1333. [Google Scholar] [CrossRef]
- Gautam, S.C.; Xu, Y.X.; Dumaguin, M.; Janakiraman, N.; Chapman, R.A. Resveratrol selectively inhibits leukemia cells: A prospective agent for ex vivo bone marrow purging. Bone Marrow Transplant. 2000, 25, 639–645. [Google Scholar] [CrossRef] [Green Version]
- Popat, R.; Plesner, T.; Davies, F.; Cook, G.; Cook, M.; Elliott, P.; Jacobson, E.; Gumbleton, T.; Oakervee, H.; Cavenagh, J. A phase 2 study of SRT501 (resveratrol) with bortezomib for patients with relapsed and or refractory multiple myeloma. Br. J. Haematol. 2013, 160, 714–717. [Google Scholar] [CrossRef]
- Srinivasan, V.; Spence, D.W.; Pandi-Perumal, S.R.; Brown, G.M.; Cardinali, D.P. Melatonin in Mitochondrial Dysfunction and Related Disorders. Int. J. Alzheimers Dis. 2011, 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín, M.; Macías, M.; Escames, G.; León, J.; Acuña-Castroviejo, D. Melatonin but not vitamins C and E maintains glutathione homeostasis in t-butyl hydroperoxide-induced mitochondrial oxidative stress. FASEB J. 2000, 14, 1677–1679. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.I.; Escames, G.; López, L.C.; García, J.A.; Ortiz, F.; López, A.; Acuña-Castroviejo, D. Melatonin administration prevents cardiac and diaphragmatic mitochondrial oxidative damage in senescence-accelerated mice. J. Endocrinol. 2007, 194, 637–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapias, V.; Escames, G.; López, L.C.; López, A.; Camacho, E.; Carrión, M.D.; Entrena, A.; Gallo, M.A.; Espinosa, A.; Acuña-Castroviejo, D. Melatonin and its brain metabolite N(1)-acetyl-5-methoxykynuramine prevent mitochondrial nitric oxide synthase induction in parkinsonian mice. J. Neurosci. Res. 2009, 87, 3002–3010. [Google Scholar] [CrossRef]
- Poeggeler, B.; Thuermann, S.; Dose, A.; Schoenke, M.; Burkhardt, S.; Hardeland, R. Melatonin’s unique radical scavenging properties—Roles of its functional substituents as revealed by a comparison with its structural analogs. J. Pineal Res. 2002, 33, 20–30. [Google Scholar] [CrossRef]
- León, J.; Acuña-Castroviejo, D.; Escames, G.; Tan, D.-X.; Reiter, R.J. Melatonin mitigates mitochondrial malfunction. J. Pineal Res. 2005, 38, 1–9. [Google Scholar] [CrossRef]
- Antolín, I.; Rodríguez, C.; Saínz, R.M.; Mayo, J.C.; Uría, H.; Kotler, M.L.; Rodríguez-Colunga, M.J.; Tolivia, D.; Menéndez-Peláez, A. Neurohormone melatonin prevents cell damage: Effect on gene expression for antioxidant enzymes. FASEB J. 1996, 10, 882–890. [Google Scholar] [CrossRef]
- Escames, G.; León, J.; Macías, M.; Khaldy, H.; Acuña-Castroviejo, D. Melatonin counteracts lipopolysaccharide-induced expression and activity of mitochondrial nitric oxide synthase in rats. FASEB J. 2003, 17, 932–934. [Google Scholar] [CrossRef]
- Rodriguez, C.; Mayo, J.C.; Sainz, R.M.; Antolín, I.; Herrera, F.; Martín, V.; Reiter, R.J. Regulation of antioxidant enzymes: A significant role for melatonin. J. Pineal Res. 2004, 36, 1–9. [Google Scholar] [CrossRef]
- Zhelev, Z.; Ivanova, D.; Bakalova, R.; Aoki, I.; Higashi, T. Synergistic Cytotoxicity of Melatonin and New-generation Anticancer Drugs against Leukemia Lymphocytes but Not Normal Lymphocytes. Anticancer Res. 2017, 37, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Strasser, A.; Carra, M.; Ghareeb, K.; Awad, W.; Böhm, J. Protective effects of antioxidants on deoxynivalenol-induced damage in murine lymphoma cells. Mycotoxin Res. 2013, 29, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.; Das, N.P.; Tan, C.H. Effects of gamma-linolenic acid, flavonoids, and vitamins on cytotoxicity and lipid peroxidation. Free Radic. Biol. Med. 1994, 16, 43–48. [Google Scholar] [CrossRef]
- Palozza, P.; Luberto, C.; Ricci, P.; Sgarlata, E.; Calviello, G.; Bartoli, G.M. Effect of beta-carotene and canthaxanthin on tert-butyl hydroperoxide-induced lipid peroxidation in murine normal and tumor thymocytes. Arch. Biochem. Biophys. 1996, 325, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, B.K.; Zaidi, A.H.; Gupta, P.; Mokhamatam, R.B.; Raviprakash, N.; Mahali, S.K.; Manna, S.K. A natural xanthone increases catalase activity but decreases NF-kappa B and lipid peroxidation in U-937 and HepG2 cell lines. Eur. J. Pharmacol. 2015, 764, 520–528. [Google Scholar] [CrossRef]
- Thavamani, B.S.; Mathew, M.; Palaniswamy, D.S. Anticancer activity of Cocculus hirsutus against Dalton’s lymphoma ascites (DLA) cells in mice. Pharm. Biol. 2014, 52, 867–872. [Google Scholar] [CrossRef]
- Basu, M.; Banerjee, A.; Bhattacharya, U.K.; Bishayee, A.; Chatterjee, M. Beta-carotene prolongs survival, decreases lipid peroxidation and enhances glutathione status in transplantable murine lymphoma. Phytomedicine 2000, 7, 151–159. [Google Scholar] [CrossRef]
- Sharma, R.; Vinayak, M. α-Tocopherol prevents lymphoma by improving antioxidant defence system of mice. Mol. Biol. Rep. 2013, 40, 839–849. [Google Scholar] [CrossRef]
- Priyadarsini, K.I.; Khopde, S.M.; Kumar, S.S.; Mohan, H. Free radical studies of ellagic acid, a natural phenolic antioxidant. J. Agric. Food Chem. 2002, 50, 2200–2206. [Google Scholar] [CrossRef]
- Mishra, S.; Vinayak, M. Anti-carcinogenic action of ellagic acid mediated via modulation of oxidative stress regulated genes in Dalton lymphoma bearing mice. Leuk. Lymphoma 2011, 52, 2155–2161. [Google Scholar] [CrossRef]
- Das, L.; Vinayak, M. Anti-carcinogenic action of curcumin by activation of antioxidant defence system and inhibition of NF-κB signalling in lymphoma-bearing mice. Biosci. Rep. 2012, 32, 161–170. [Google Scholar] [CrossRef]
- Bizzarri, M.; Proietti, S.; Cucina, A.; Reiter, R.J. Molecular mechanisms of the pro-apoptotic actions of melatonin in cancer: A review. Expert Opin. Ther. Targets 2013, 17, 1483–1496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-M.; Zhang, Y. Melatonin: A well-documented antioxidant with conditional pro-oxidant actions. J. Pineal Res. 2014, 57, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Pasciu, V.; Posadino, A.M.; Cossu, A.; Sanna, B.; Tadolini, B.; Gaspa, L.; Marchisio, A.; Dessole, S.; Capobianco, G.; Pintus, G. Akt downregulation by flavin oxidase-induced ROS generation mediates dose-dependent endothelial cell damage elicited by natural antioxidants. Toxicol. Sci. 2010, 114, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiss, E.H.; Schilder, Y.D.C.; Dirsch, V.M. Chronic treatment with resveratrol induces redox stress- and ataxia telangiectasia-mutated (ATM)-dependent senescence in p53-positive cancer cells. J. Biol. Chem. 2007, 282, 26759–26766. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.K.; Zhang, W.; Sanderson, B.J.S. Selective growth inhibition of human leukemia and human lymphoblastoid cells by resveratrol via cell cycle arrest and apoptosis induction. J. Agric. Food Chem. 2008, 56, 7572–7577. [Google Scholar] [CrossRef]
- Dörrie, J.; Gerauer, H.; Wachter, Y.; Zunino, S.J. Resveratrol induces extensive apoptosis by depolarizing mitochondrial membranes and activating caspase-9 in acute lymphoblastic leukemia cells. Cancer Res. 2001, 61, 4731–4739. [Google Scholar]
- Aggarwal, B.B.; Bhardwaj, A.; Aggarwal, R.S.; Seeram, N.P.; Shishodia, S.; Takada, Y. Role of resveratrol in prevention and therapy of cancer: Preclinical and clinical studies. Anticancer Res. 2004, 24, 2783–2840. [Google Scholar]
- Bejarano, I.; Espino, J.; Marchena, A.M.; Barriga, C.; Paredes, S.D.; Rodríguez, A.B.; Pariente, J.A. Melatonin enhances hydrogen peroxide-induced apoptosis in human promyelocytic leukaemia HL-60 cells. Mol. Cell. Biochem. 2011, 353, 167–176. [Google Scholar] [CrossRef]
- Paternoster, L.; Radogna, F.; Accorsi, A.; Cristina Albertini, M.; Gualandi, G.; Ghibelli, L. Melatonin as a modulator of apoptosis in B-lymphoma cells. Ann. N. Y. Acad. Sci. 2009, 1171, 345–349. [Google Scholar] [CrossRef]
- Jang, S.S.; Kim, W.D.; Park, W.-Y. Melatonin exerts differential actions on X-ray radiation-induced apoptosis in normal mice splenocytes and Jurkat leukemia cells. J. Pineal Res. 2009, 47, 147–155. [Google Scholar] [CrossRef]
- Radogna, F.; Paternoster, L.; De Nicola, M.; Cerella, C.; Ammendola, S.; Bedini, A.; Tarzia, G.; Aquilano, K.; Ciriolo, M.; Ghibelli, L. Rapid and transient stimulation of intracellular reactive oxygen species by melatonin in normal and tumor leukocytes. Toxicol. Appl. Pharmacol. 2009, 239, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kang, J.; Da, W.; Ou, Y. Combination with water-soluble antioxidants increases the anticancer activity of quercetin in human leukemia cells. Pharmazie 2004, 59, 859–863. [Google Scholar] [PubMed]
- Kong, Y.; Ma, W.; Liu, X.; Zu, Y.; Fu, Y.; Wu, N.; Liang, L.; Yao, L.; Efferth, T. Cytotoxic Activity of Curcumin towards CCRF-CEM Leukemia Cells and Its Effect on DNA Damage. Molecules 2009, 14, 5328–5338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.; Chen, J.; Shi, Y.; Jia, J.; Zhang, Y. Curcumin-induced histone hypoacetylation: The role of reactive oxygen species. Biochem. Pharmacol. 2005, 69, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Lu, J.; Holmgren, A. Thioredoxin reductase is irreversibly modified by curcumin: A novel molecular mechanism for its anticancer activity. J. Biol. Chem. 2005, 280, 25284–25290. [Google Scholar] [CrossRef] [Green Version]
- Piwocka, K.; Jaruga, E.; Skierski, J.; Gradzka, I.; Sikora, E. Effect of glutathione depletion on caspase-3 independent apoptosis pathway induced by curcumin in Jurkat cells. Free Radic. Biol. Med. 2001, 31, 670–678. [Google Scholar] [CrossRef]
- Talib, W. Melatonin and Cancer Hallmarks. Molecules 2018, 23, 518. [Google Scholar] [CrossRef] [Green Version]
- Nelson, K.M.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F.; Walters, M.A. The Essential Medicinal Chemistry of Curcumin: Miniperspective. J. Med. Chem. 2017, 60, 1620–1637. [Google Scholar] [CrossRef]
- Keylor, M.H.; Matsuura, B.S.; Stephenson, C.R.J. Chemistry and Biology of Resveratrol-Derived Natural Products. Chem. Rev. 2015, 115, 8976–9027. [Google Scholar] [CrossRef]
- Biaglow, J.E.; Miller, R.A. The thioredoxin reductase/thioredoxin system: Novel redox targets for cancer therapy. Cancer Biol. Ther. 2005, 4, 6–13. [Google Scholar] [CrossRef]
- Sardina, J.L.; López-Ruano, G.; Sánchez-Sánchez, B.; Llanillo, M.; Hernández-Hernández, A. Reactive oxygen species: Are they important for haematopoiesis? Crit. Rev. Oncol. Hematol. 2012, 81, 257–274. [Google Scholar] [CrossRef] [PubMed]
- Zafar, A.; Singh, S.; Naseem, I. Cu(II)-coumestrol interaction leads to ROS-mediated DNA damage and cell death: A putative mechanism for anticancer activity. J. Nutr. Biochem. 2016, 33, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Gasparetto, M.; Jordan, C.; Pollyea, D.A.; Vasiliou, V. The effects of alcohol and aldehyde dehydrogenases on disorders of hematopoiesis. Adv. Exp. Med. Biol. 2015, 815, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Rose, C.; Brechignac, S.; Vassilief, D.; Pascal, L.; Stamatoullas, A.; Guerci, A.; Larbaa, D.; Dreyfus, F.; Beyne-Rauzy, O.; Chaury, M.P.; et al. Does iron chelation therapy improve survival in regularly transfused lower risk MDS patients? A multicenter study by the GFM (Groupe Francophone des Myélodysplasies). Leuk. Res. 2010, 34, 864–870. [Google Scholar] [CrossRef]
- Rassool, F.V.; Gaymes, T.J.; Omidvar, N.; Brady, N.; Beurlet, S.; Pla, M.; Reboul, M.; Lea, N.; Chomienne, C.; Thomas, N.S.B.; et al. Reactive oxygen species, DNA damage, and error-prone repair: A model for genomic instability with progression in myeloid leukemia? Cancer Res. 2007, 67, 8762–8771. [Google Scholar] [CrossRef] [Green Version]
- Merkel, D.G.; Nagler, A. Toward resolving the unsettled role of iron chelation therapy in myelodysplastic syndromes. Expert Rev. Anticancer 2014, 14, 817–829. [Google Scholar] [CrossRef]
- Malcovati, L.; Hellström-Lindberg, E.; Bowen, D.; Adès, L.; Cermak, J.; Del Cañizo, C.; Della Porta, M.G.; Fenaux, P.; Gattermann, N.; Germing, U.; et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: Recommendations from the European LeukemiaNet. Blood 2013, 122, 2943–2964. [Google Scholar] [CrossRef] [Green Version]
- Zeidan, A.M.; Hendrick, F.; Friedmann, E.; Baer, M.R.; Gore, S.D.; Sasane, M.; Paley, C.; Davidoff, A.J. Deferasirox therapy is associated with reduced mortality risk in a medicare population with myelodysplastic syndromes. J. Comp. Eff. Res. 2015, 4, 327–340. [Google Scholar] [CrossRef] [Green Version]
- Angelucci, E.; Santini, V.; Di Tucci, A.A.; Quaresmini, G.; Finelli, C.; Volpe, A.; Quarta, G.; Rivellini, F.; Sanpaolo, G.; Cilloni, D.; et al. Deferasirox for transfusion-dependent patients with myelodysplastic syndromes: Safety, efficacy, and beyond (GIMEMA MDS0306 Trial). Eur. J. Haematol. 2014, 92, 527–536. [Google Scholar] [CrossRef]
- Ko, B.-S.; Chang, M.-C.; Chiou, T.-J.; Chang, T.-K.; Chen, Y.-C.; Lin, S.-F.; Chang, C.-S.; Lu, Y.-C.; Yeh, S.-P.; Chen, T.-Y.; et al. Long-term safety and efficacy of deferasirox in patients with myelodysplastic syndrome, aplastic anemia and other rare anemia in Taiwan. Hematology 2019, 24, 247–254. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Shi, P.; Liu, J.; Li, J.; Cao, Y. Efficacy and safety of iron chelator for transfusion-dependent patients with myelodysplastic syndrome: A meta-analysis. Hematology 2019, 24, 669–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tataranni, T.; Agriesti, F.; Mazzoccoli, C.; Ruggieri, V.; Scrima, R.; Laurenzana, I.; D’Auria, F.; Falzetti, F.; Di Ianni, M.; Musto, P.; et al. The iron chelator deferasirox affects redox signalling in haematopoietic stem/progenitor cells. Br. J. Haematol. 2015, 170, 236–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivars, D.; Orero, M.T.; Javier, K.; Díaz-Vico, L.; García-Giménez, J.L.; Mena, S.; Tormos, C.; Egea, M.; Pérez, P.L.; Arrizabalaga, B.; et al. Oxidative imbalance in low/intermediate-1-risk myelodysplastic syndrome patients: The influence of iron overload. Clin. Biochem. 2017, 50, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Ghoti, H.; Fibach, E.; Merkel, D.; Perez-Avraham, G.; Grisariu, S.; Rachmilewitz, E.A. Changes in parameters of oxidative stress and free iron biomarkers during treatment with deferasirox in iron-overloaded patients with myelodysplastic syndromes. Haematologica 2010, 95, 1433–1434. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, S.; Kobune, M.; Iyama, S.; Sato, T.; Murase, K.; Kawano, Y.; Takada, K.; Ono, K.; Kaneko, Y.; Miyanishi, K.; et al. Improvement of iron-mediated oxidative DNA damage in patients with transfusion-dependent myelodysplastic syndrome by treatment with deferasirox. Free Radic. Biol. Med. 2012, 53, 643–648. [Google Scholar] [CrossRef]
- Callens, C.; Coulon, S.; Naudin, J.; Radford-Weiss, I.; Boissel, N.; Raffoux, E.; Wang, P.H.M.; Agarwal, S.; Tamouza, H.; Paubelle, E.; et al. Targeting iron homeostasis induces cellular differentiation and synergizes with differentiating agents in acute myeloid leukemia. J. Exp. Med. 2010, 207, 731–750. [Google Scholar] [CrossRef]
- Fang, D.; Bao, Y.; Li, X.; Liu, F.; Cai, K.; Gao, J.; Liao, Q. Effects of iron deprivation on multidrug resistance of leukemic K562 cells. Chemotherapy 2010, 56, 9–16. [Google Scholar] [CrossRef]
- Kim, J.-L.; Kang, H.-N.; Kang, M.H.; Yoo, Y.A.; Kim, J.S.; Choi, C.W. The oral iron chelator deferasirox induces apoptosis in myeloid leukemia cells by targeting caspase. Acta Haematol. 2011, 126, 241–245. [Google Scholar] [CrossRef]
- Ohyashiki, J.H.; Kobayashi, C.; Hamamura, R.; Okabe, S.; Tauchi, T.; Ohyashiki, K. The oral iron chelator deferasirox represses signaling through the mTOR in myeloid leukemia cells by enhancing expression of REDD1. Cancer Sci. 2009, 100, 970–977. [Google Scholar] [CrossRef]
- Lee, D.-H.; Jang, P.S.; Chung, N.G.; Cho, B.; Jeong, D.C.; Kim, H.K. Deferasirox shows in vitro and in vivo antileukemic effects on murine leukemic cell lines regardless of iron status. Exp. Hematol. 2013, 41, 539–546. [Google Scholar] [CrossRef]
- Shapira, S.; Raanani, P.; Samara, A.; Nagler, A.; Lubin, I.; Arber, N.; Granot, G. Deferasirox selectively induces cell death in the clinically relevant population of leukemic CD34+CD38- cells through iron chelation, induction of ROS, and inhibition of HIF1α expression. Exp. Hematol. 2019, 70, 55–69.e4. [Google Scholar] [CrossRef]
- Rychtarcikova, Z.; Lettlova, S.; Tomkova, V.; Korenkova, V.; Langerova, L.; Simonova, E.; Zjablovskaja, P.; Alberich-Jorda, M.; Neuzil, J.; Truksa, J. Tumor-initiating cells of breast and prostate origin show alterations in the expression of genes related to iron metabolism. Oncotarget 2017, 8, 6376–6398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Chen, Q.; Gu, J.; Li, S.; Zhao, G.; Wang, W.; Wang, Z.; Wang, X. Synergistic inhibitory effects of deferasirox in combination with decitabine on leukemia cell lines SKM-1, THP-1, and K-562. Oncotarget 2017, 8, 36517–36530. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, S.; Takahashi, R.; Ohsuga, M.; Ohyashiki, K.; Ohyashiki, J.H. Effect of combined deferasirox and 5-azacytidine treatment on human leukemia cells in vitro. Ann. Hematol. 2015, 94, 1601–1602. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.-S.; Jeon, Y.-W.; Hahn, A.-R.; Lee, T.-H.; Park, S.-S.; Yoon, J.-H.; Lee, S.-E.; Eom, K.-S.; Kim, Y.-J.; Lee, S.; et al. Improved survival outcomes and restoration of graft-vs-leukemia effect by deferasirox after allogeneic stem cell transplantation in acute myeloid leukemia. Cancer Med. 2019, 8, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Sivgin, S.; Eser, B.; Bahcebasi, S.; Kaynar, L.; Kurnaz, F.; Uzer, E.; Pala, C.; Deniz, K.; Ozturk, A.; Cetin, M.; et al. Efficacy and safety of oral deferasirox treatment in the posttransplant period for patients who have undergone allogeneic hematopoietic stem cell transplantation (alloHSCT). Ann. Hematol. 2012, 91, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, T.; Kawabata, H.; Nakamura, T.; Iwao, H.; Nakajima, A.; Miki, M.; Sakai, T.; Sawaki, T.; Fujita, Y.; Tanaka, M.; et al. Iron chelation therapy with deferasirox induced complete remission in a patient with chemotherapy-resistant acute monocytic leukemia. Anticancer Res. 2011, 31, 1741–1744. [Google Scholar]
- Lui, G.Y.L.; Kovacevic, Z.; Richardson, V.; Merlot, A.M.; Kalinowski, D.S.; Richardson, D.R. Targeting cancer by binding iron: Dissecting cellular signaling pathways. Oncotarget 2015, 6, 18748–18779. [Google Scholar] [CrossRef] [Green Version]
- Babosova, O.; Kapralova, K.; Raskova Kafkova, L.; Korinek, V.; Divoky, V.; Prchal, J.T.; Lanikova, L. Iron chelation and 2-oxoglutarate-dependent dioxygenase inhibition suppress mantle cell lymphoma’s cyclin D1. J. Cell. Mol. Med. 2019, 23, 7785–7795. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, J.A.; Luria-Pérez, R.; López-Valdés, H.E.; Casero, D.; Daniels, T.R.; Patel, S.; Avila, D.; Leuchter, R.; So, S.; Ortiz-Sánchez, E.; et al. Lethal iron deprivation induced by non-neutralizing antibodies targeting transferrin receptor 1 in malignant B cells. Leuk. Lymphoma 2011, 52, 2169–2178. [Google Scholar] [CrossRef]
- Moura, I.C.; Lepelletier, Y.; Arnulf, B.; England, P.; Baude, C.; Beaumont, C.; Bazarbachi, A.; Benhamou, M.; Monteiro, R.C.; Hermine, O. A neutralizing monoclonal antibody (mAb A24) directed against the transferrin receptor induces apoptosis of tumor T lymphocytes from ATL patients. Blood 2004, 103, 1838–1845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benadiba, J.; Rosilio, C.; Nebout, M.; Heimeroth, V.; Neffati, Z.; Popa, A.; Mary, D.; Griessinger, E.; Imbert, V.; Sirvent, N.; et al. Iron chelation: An adjuvant therapy to target metabolism, growth and survival of murine PTEN-deficient T lymphoma and human T lymphoblastic leukemia/lymphoma. Leuk. Lymphoma 2017, 58, 1433–1445. [Google Scholar] [CrossRef]
- Eberhard, Y.; McDermott, S.P.; Wang, X.; Gronda, M.; Venugopal, A.; Wood, T.E.; Hurren, R.; Datti, A.; Batey, R.A.; Wrana, J.; et al. Chelation of intracellular iron with the antifungal agent ciclopirox olamine induces cell death in leukemia and myeloma cells. Blood 2009, 114, 3064–3073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamihara, Y.; Takada, K.; Sato, T.; Kawano, Y.; Murase, K.; Arihara, Y.; Kikuchi, S.; Hayasaka, N.; Usami, M.; Iyama, S.; et al. The iron chelator deferasirox induces apoptosis by targeting oncogenic Pyk2/β-catenin signaling in human multiple myeloma. Oncotarget 2016, 7, 64330–64341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordini, J.; Morisi, F.; Cerruti, F.; Cascio, P.; Camaschella, C.; Ghia, P.; Campanella, A. Iron Causes Lipid Oxidation and Inhibits Proteasome Function in Multiple Myeloma Cells: A Proof of Concept for Novel Combination Therapies. Cancers 2020, 12, 970. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Malignancy | Sample Tested | N Patient | N Control | Biomarkers | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MDA | PC | AOPPs | PN | AGEs | TBARS | 4-HNE | |||||
| HL | Plasma | 30 | 30 | ↑ | [23] | ||||||
| HL | Erythrocytes | 30 | 30 | ↑↑ | [23] | ||||||
| HL | Serum | 15 | 10 | ↑↑↑ | ↑↑↑ | [14] | |||||
| DLBCL | Serum | 40 | 15 | ↑↑↑ | [24] | ||||||
| NHL | Serum | 146 | 60 | ↑↑↑ | [25] | ||||||
| B-NHL | Serum | 32 | 25 | ↑↑ | ↑↑ | [26] | |||||
| MM | Serum | 50 | 50 | ↑↑↑ | [27] | ||||||
| MM | Serum | 21 | 30 | ↑↑↑ | [28] | ||||||
| MM | Serum | 20 | 23 | ↑↑↑ | ↑↑↑ | [29] | |||||
| MGUS | Serum | 8 | 23 | ns | ns | [29] | |||||
| MM | Serum | 20 | 20 | ↑↑ | [30] | ||||||
| MM | Saliva | 30 | 7 | ns | [31] | ||||||
| MM | Plasma | 24 | 20 | ↑↑ | [32] | ||||||
| MM | Serum | 19 | 16 | ↓↓ | [33] | ||||||
| ALL | Plasma | 80 | 50 | ↑↑↑ | [3] | ||||||
| ALL | Serum | 80 | 50 | ↑↑↑ | [3] | ||||||
| ALL | Plasma | 16 | 15 | ns | [34] | ||||||
| ALL | Plasma | 7 | 20 | ns | [35] | ||||||
| B-CLL | Serum | 48 | 30 | ↑↑↑ | [36] | ||||||
| B-CLL | Serum | 60 | 23 | ↑↑↑ | ↑↑↑ | ↑↑ | [19] | ||||
| B-CLL | Plasma | 50 | 31 | ↑↑ | [37] | ||||||
| B-CLL | Serum | 20 | 15 | ↑ | [38] | ||||||
| AML | Plasma | 11 | 15 | ns | [34] | ||||||
| AML | Plasma | 34 | 20 | ns | [35] | ||||||
| CML | Plasma | 3 | 15 | ns | [34] | ||||||
| CML | Plasma | 8 | 20 | ns | [35] | ||||||
| CML | Plasma | 20 | 10 | ↑ | ↑ | [4] | |||||
| CML | Plasma | 47 | 20 | ↑ | ↑ | [39] | |||||
| CML | Plasma | 40 | 20 | ↑ | ↑ | [1] | |||||
| CML | Plasma | 128 | 50 | ↑↑ | [5] | ||||||
| MDS | Plasma | 32 | 20 | ↑ | [40] | ||||||
| MDS | Plasma | 76 | 45 | ↑ | [41] | ||||||
| MDS | Plasma | 78 | 87 | ↑↑ | [22] | ||||||
| MDS | Serum | 33 | 10 | ↑↑ | [42] | ||||||
| MDS | Plasma | 61 | 23 | ns | [43] | ||||||
| MDS | BM | 21 | 13 | ↑↑ | ns | [44] | |||||
| MPN | Plasma | 73 | 10 | ↑↑ | ↑ | [45] | |||||
| MPN | Serum | 34 | 23 | ↑↑ | ↑↑↑ | ns | [36] | ||||
| Malignancy | Chemotherapy Regimen | Measurement | N | Biomarkers | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|
| MDA | TBARS | AOPPs | Ascorbic Acid | Carbonyl Groups | |||||
| HL | ABVD | Plasma | 34 | ↑↑↑ | [46] | ||||
| HL | CHOP | Plasma | 5 | ↓↓↓ | [47] | ||||
| NHL | CHOP | Serum | 146 | ↓↓↓ | [25] | ||||
| NHL | CHOP | Plasma | 6 | ↓↓↓ | [47] | ||||
| NHL * | CHOP | Serum | 25 | ↑↑↑ | ↑↑ | [48] | |||
| MM | VAD | Plasma | 14 | ↓ | [49] | ||||
| MM | IT | Plasma | 30 | ↑↑ | ↓ | [50] | |||
| ALL | GBTLI LLA-99 protocol | Plasma | 80 | ns | [3] | ||||
| CML ALL | DOX-TRF | Cell lines (K562 and CCRF-CEM) | 6 | ↑ ↑ | [51] | ||||
| AML | Cytarabine and daunorubicin | Plasma | 38 | ↑ | [52] | ||||
| APL | Cisplatin | Cell lines (HL-60, NB4 and KG-1a) | 3 | ↑↑ | [53] | ||||
| APL | Arsenic trioxide | Cell line (HL-60) | 3 | ↑ | [54] | ||||
| APL ** | Arsenic trioxide | Cardiac tissue | 6 | ↑ | [55] | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-García, A.; García-Vicente, R.; Morales, M.L.; Ortiz-Ruiz, A.; Martínez-López, J.; Linares, M. Protein Carbonylation and Lipid Peroxidation in Hematological Malignancies. Antioxidants 2020, 9, 1212. https://doi.org/10.3390/antiox9121212
Rodríguez-García A, García-Vicente R, Morales ML, Ortiz-Ruiz A, Martínez-López J, Linares M. Protein Carbonylation and Lipid Peroxidation in Hematological Malignancies. Antioxidants. 2020; 9(12):1212. https://doi.org/10.3390/antiox9121212
Chicago/Turabian StyleRodríguez-García, Alba, Roberto García-Vicente, María Luz Morales, Alejandra Ortiz-Ruiz, Joaquín Martínez-López, and María Linares. 2020. "Protein Carbonylation and Lipid Peroxidation in Hematological Malignancies" Antioxidants 9, no. 12: 1212. https://doi.org/10.3390/antiox9121212