Modulation of Inflammation and Immune Responses by Heme Oxygenase-1: Implications for Infection with Intracellular Pathogens


Abstract
:1. Introduction
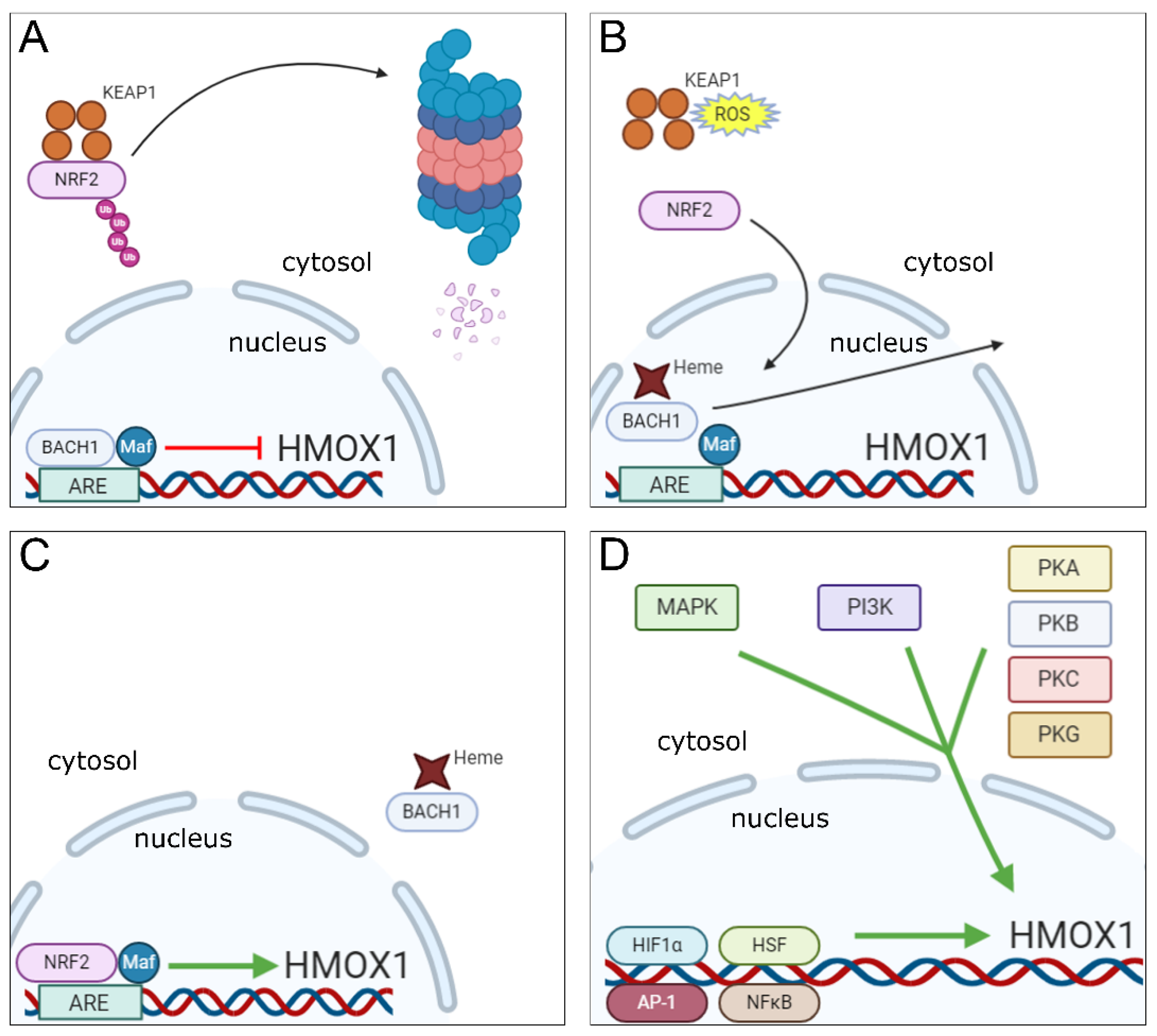
2. Mechanisms of Induction of HO-1 Expression
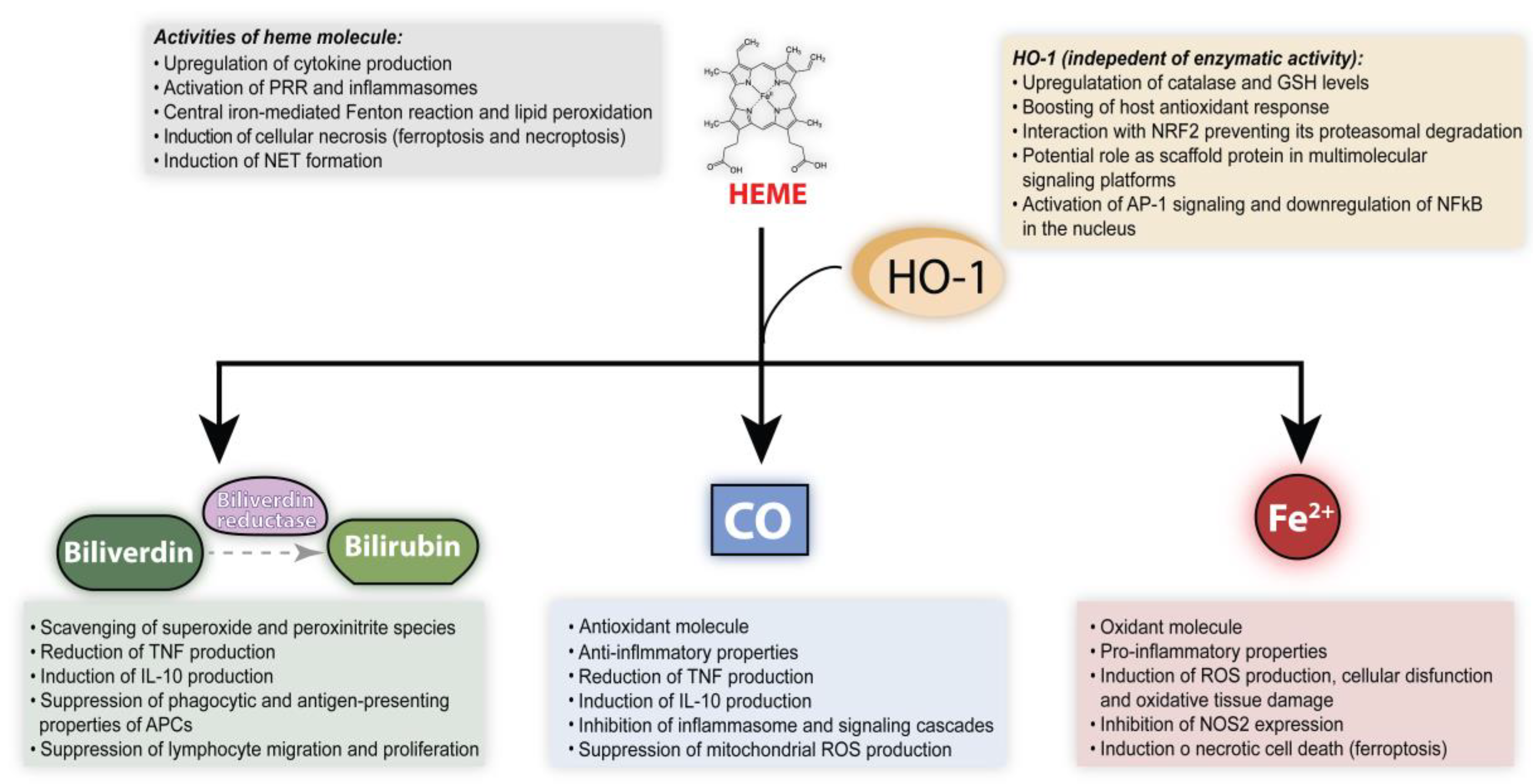
3. Pro-Oxidant and Proinflammatory Effects of Heme
4. Modulation of Oxidative Stress, Immune Responses and Inflammation by HO-1
4.1. Carbon Monoxide
4.2. Biliverdin, Biliverdin Reductase and Bilirubin
4.3. Iron
4.4. Functions of HO-1 Independent of Its Enzymatic Activity
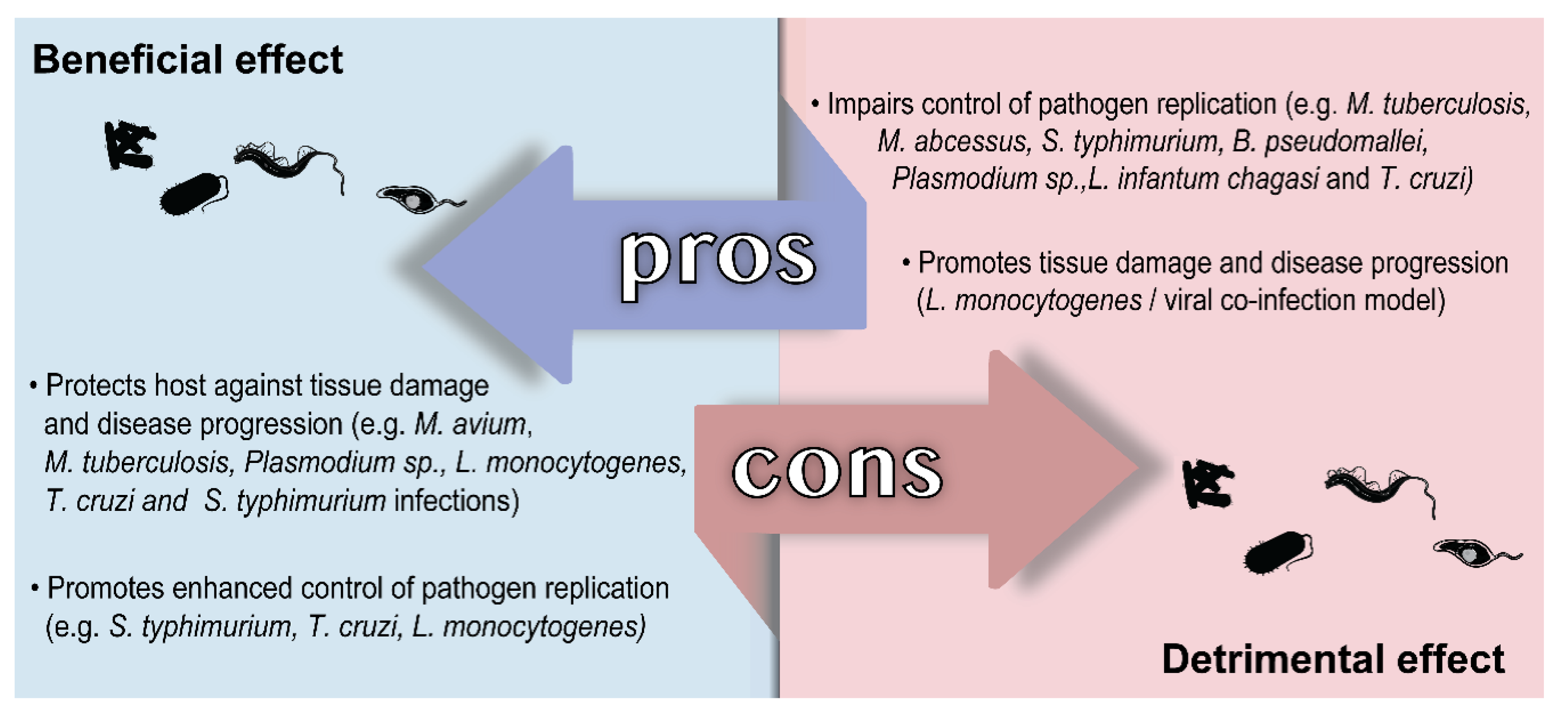
5. Role of HO-1 in Infectious Diseases Caused by Intracellular Pathogens
5.1. Diseases Caused by Bacteria
5.1.1. Tuberculosis and Other Mycobacterial Infections
5.1.2. Salmonellosis
5.1.3. Listeriosis
5.1.4. Melioidosis
5.2. Diseases Caused by Protozoan Parasites
5.2.1. Malaria
5.2.2. Leishmaniasis
5.2.3. Chagas Disease
5.2.4. Toxoplasmosis
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Morse, D.; Choi, A.M. Heme oxygenase-1: From bench to bedside. Am. J. Resp. Crit. Care 2005, 172, 660–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenhunen, R.; Marver, H.S.; Schmid, R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA 1968, 61, 748–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maines, M.D.; Ibrahim, N.G.; Kappas, A. Solubilization and partial purification of heme oxygenase from rat liver. J. Biol. Chem. 1977, 252, 5900–5903. [Google Scholar] [PubMed]
- Maines, M.D.; Trakshel, G.M.; Kutty, R.K. Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J. Biol. Chem. 1986, 261, 411–419. [Google Scholar] [PubMed]
- Hiwasa, T.; Sakiyama, S. Increase in the synthesis of a Mr 32,000 protein in BALB/c 3T3 cells after treatment with tumor promoters, chemical carcinogens, metal salts, and heat shock. Cancer Res. 1986, 46, 2474–2481. [Google Scholar] [PubMed]
- Caltabiano, M.M.; Koestler, T.P.; Poste, G.; Greig, R.G. Induction of 32- and 34-kDa stress proteins by sodium arsenite, heavy metals, and thiol-reactive agents. J. Biol. Chem. 1986, 261, 13381–13386. [Google Scholar]
- Caltabiano, M.M.; Koestler, T.P.; Poste, G.; Greig, R.G. Induction of mammalian stress proteins by a triethylphosphine gold compound used in the therapy of rheumatoid arthritis. Biochem. Biophys. Res. 1986, 138, 1074–1080. [Google Scholar] [CrossRef]
- Keyse, S.M.; Tyrrell, R.M. Both near ultraviolet radiation and the oxidizing agent hydrogen peroxide induce a 32-kDa stress protein in normal human skin fibroblasts. J. Biol. Chem. 1987, 262, 14821–14825. [Google Scholar]
- Caltabiano, M.M.; Poste, G.; Greig, R.G. Induction of the 32-kD human stress protein by auranofin and related triethylphosphine gold analogs. Biochem. Pharmacol. 1988, 37, 4089–4093. [Google Scholar] [CrossRef]
- Caltabiano, M.M.; Poste, G.; Greig, R.G. Isolation and immunological characterization of the mammalian 32-/34-kDa stress protein. Exp. Cell Res. 1988, 178, 31–40. [Google Scholar] [CrossRef]
- Taketani, S.; Kohno, H.; Yoshinaga, T.; Tokunaga, R. The human 32-kDa stress protein induced by exposure to arsenite and cadmium ions is heme oxygenase. FEBS Lett. 1989, 245, 173–176. [Google Scholar] [CrossRef] [Green Version]
- Keyse, S.M.; Tyrrell, R.M. Heme oxygenase is the major 32-kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc. Natl. Acad. Sci. USA 1989, 86, 99–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoubrey, W.K., Jr.; Huang, T.J.; Maines, M.D. Isolation and characterization of a cDNA from the rat brain that encodes hemoprotein heme oxygenase-3. Eur. J. Biochem. 1997, 247, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.; Omata, Y.; Sakamoto, H.; Higashimoto, Y.; Hara, T.; Sagara, Y.; Noguchi, M. Characterization of rat heme oxygenase-3 gene. Implication of processed pseudogenes derived from heme oxygenase-2 gene. Gene 2004, 336, 241–250. [Google Scholar] [CrossRef]
- Wilks, A. Heme oxygenase: Evolution, structure, and mechanism. Antioxid. Redox Signal. 2002, 4, 603–614. [Google Scholar] [CrossRef]
- Munoz-Sanchez, J.; Chanez-Cardenas, M.E. A review on hemeoxygenase-2: Focus on cellular protection and oxygen response. Oxid. Med. Cell. Longev. 2014, 2014, 604981. [Google Scholar] [CrossRef] [Green Version]
- Fleischhacker, A.S.; Gunawan, A.L.; Kochert, B.A.; Liu, L.; Wales, T.E.; Borowy, M.C.; Engen, J.R.; Ragsdale, S.W. The heme-regulatory motifs of heme oxygenase-2 contribute to the transfer of heme to the catalytic site for degradation. J. Biol. Chem. 2020, 295, 5177–5191. [Google Scholar] [CrossRef] [Green Version]
- Montellano, P.R. The mechanism of heme oxygenase. Curr. Opin. Chem. Biol. 2000, 4, 221–227. [Google Scholar] [CrossRef]
- Maines, M.D. The heme oxygenase system: A regulator of second messenger gases. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 517–554. [Google Scholar] [CrossRef]
- Maines, M.D. Heme oxygenase: Function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988, 2, 2557–2568. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.W.; Hall, S.R.; Perrella, M.A. Role of haem oxygenase-1 in microbial host defence. Cell Microbiol. 2009, 11, 199–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayan, V.; Wagener, F.; Immenschuh, S. The macrophage heme-heme oxygenase-1 system and its role in inflammation. Biochem. Pharmacol. 2018, 153, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Contreras, H.; Chim, N.; Credali, A.; Goulding, C.W. Heme uptake in bacterial pathogens. Curr. Opin. Chem. Biol. 2014, 19, 34–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozza, M.T.; Jeney, V. Pro-inflammatory Actions of Heme and Other Hemoglobin-Derived DAMPs. Front. Immunol. 2020, 11, 1323. [Google Scholar] [CrossRef]
- Waza, A.A.; Hamid, Z.; Ali, S.; Bhat, S.A.; Bhat, M.A. A review on heme oxygenase-1 induction: Is it a necessary evil. Inflamm. Res. 2018, 67, 579–588. [Google Scholar] [CrossRef]
- Singh, N.; Ahmad, Z.; Baid, N.; Kumar, A. Host heme oxygenase-1: Friend or foe in tackling pathogens? IUBMB Life 2018, 70, 869–880. [Google Scholar] [CrossRef] [Green Version]
- Fraser, S.T.; Midwinter, R.G.; Berger, B.S.; Stocker, R. Heme Oxygenase-1: A Critical Link between Iron Metabolism, Erythropoiesis, and Development. Adv. Hematol. 2011, 2011, 473709. [Google Scholar] [CrossRef] [Green Version]
- Reedy, C.J.; Gibney, B.R. Heme protein assemblies. Chem. Rev. 2004, 104, 617–649. [Google Scholar] [CrossRef]
- Hvidberg, V.; Maniecki, M.B.; Jacobsen, C.; Hojrup, P.; Moller, H.J.; Moestrup, S.K. Identification of the receptor scavenging hemopexin-heme complexes. Blood 2005, 106, 2572–2579. [Google Scholar] [CrossRef]
- Kristiansen, M.; Graversen, J.H.; Jacobsen, C.; Sonne, O.; Hoffman, H.J.; Law, S.K.; Moestrup, S.K. Identification of the haemoglobin scavenger receptor. Nature 2001, 409, 198–201. [Google Scholar] [CrossRef]
- Rajagopal, A.; Rao, A.U.; Amigo, J.; Tian, M.; Upadhyay, S.K.; Hall, C.; Uhm, S.; Mathew, M.K.; Fleming, M.D.; Paw, B.H.; et al. Haem homeostasis is regulated by the conserved and concerted functions of HRG-1 proteins. Nature 2008, 453, 1127–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, C.; Yuan, X.; Schmidt, P.J.; Bresciani, E.; Samuel, T.K.; Campagna, D.; Hall, C.; Bishop, K.; Calicchio, M.L.; Lapierre, A.; et al. HRG1 is essential for heme transport from the phagolysosome of macrophages during erythrophagocytosis. Cell Metab. 2013, 17, 261–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tudor, C.; Lerner-Marmarosh, N.; Engelborghs, Y.; Gibbs, P.E.; Maines, M.D. Biliverdin reductase is a transporter of haem into the nucleus and is essential for regulation of HO-1 gene expression by haematin. Biochem. J. 2008, 413, 405–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Guo, J.; Wei, X.; Niu, C.; Jia, M.; Li, Q.; Meng, D. Bach1: Function, Regulation, and Involvement in Disease. Oxid. Med. Cell Longev. 2018, 2018, 1347969. [Google Scholar] [CrossRef] [PubMed]
- Zenke-Kawasaki, Y.; Dohi, Y.; Katoh, Y.; Ikura, T.; Ikura, M.; Asahara, T.; Tokunaga, F.; Iwai, K.; Igarashi, K. Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol. Cell Biol. 2007, 27, 6962–6971. [Google Scholar] [CrossRef] [Green Version]
- Igarashi, K.; Kataoka, K.; Itoh, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Regulation of transcription by dimerization of erythroid factor NF-E2 p45 with small Maf proteins. Nature 1994, 367, 568–572. [Google Scholar] [CrossRef]
- Reichard, J.F.; Motz, G.T.; Puga, A. Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1. Nucleic Acids Res. 2007, 35, 7074–7086. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; O’Connor, T.; Yamamoto, M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 2003, 8, 379–391. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kang, M.I.; Watai, Y.; Tong, K.I.; Shibata, T.; Uchida, K.; Yamamoto, M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol. Cell Biol. 2006, 26, 221–229. [Google Scholar] [CrossRef] [Green Version]
- Baird, L.; Dinkova-Kostova, A.T. The cytoprotective role of the Keap1-Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–272. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Yamamoto, M. Molecular basis of the Keap1-Nrf2 system. Free Radic. Biol. Med. 2015, 88, 93–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, J.; Stewart, D.; Touchard, C.; Boinapally, S.; Choi, A.M.; Cook, J.L. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J. Biol. Chem. 1999, 274, 26071–26078. [Google Scholar] [CrossRef] [Green Version]
- Hajjar, D.P.; Gotto, A.M., Jr. Biological relevance of inflammation and oxidative stress in the pathogenesis of arterial diseases. Am. J. Pathol. 2013, 182, 1474–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binder, C.J.; Papac-Milicevic, N.; Witztum, J.L. Innate sensing of oxidation-specific epitopes in health and disease. Nat. Rev. Immunol. 2016, 16, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Paine, A.; Eiz-Vesper, B.; Blasczyk, R.; Immenschuh, S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem. Pharmacol. 2010, 80, 1895–1903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hull, T.D.; Agarwal, A.; George, J.F. The mononuclear phagocyte system in homeostasis and disease: A role for heme oxygenase-1. Antioxid. Redox Signal. 2014, 20, 1770–1788. [Google Scholar] [CrossRef] [Green Version]
- Alam, J.; Cook, J.L. How many transcription factors does it take to turn on the heme oxygenase-1 gene? Am. J. Respir. Cell. Mol. Biol. 2007, 36, 166–174. [Google Scholar] [CrossRef]
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef]
- Newton, K.; Dixit, V.M. Signaling in innate immunity and inflammation. Cold Spring Harb. Perspect. Biol. 2012, 4, a006049. [Google Scholar] [CrossRef]
- Bloom, D.A.; Jaiswal, A.K. Phosphorylation of Nrf2 at Ser40 by protein kinase C in response to antioxidants leads to the release of Nrf2 from INrf2, but is not required for Nrf2 stabilization/accumulation in the nucleus and transcriptional activation of antioxidant response element-mediated NAD(P)H:quinone oxidoreductase-1 gene expression. J. Biol. Chem. 2003, 278, 44675–44682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Huang, Z.; Zhang, D.D. Phosphorylation of Nrf2 at multiple sites by MAP kinases has a limited contribution in modulating the Nrf2-dependent antioxidant response. PLoS ONE 2009, 4, e6588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaiahgari, S.; Kleeberger, S.R.; Cho, H.Y.; Kalvakolanu, D.V.; Reddy, S.P. NADPH oxidase and ERK signaling regulates hyperoxia-induced Nrf2-ARE transcriptional response in pulmonary epithelial cells. J. Biol. Chem. 2004, 279, 42302–42312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pae, H.O.; Oh, G.S.; Jeong, S.O.; Jeong, G.S.; Lee, B.S.; Choi, B.M.; Lee, H.S.; Chung, H.T. 1,2,3,4,6-penta-O-galloyl-beta-D-glucose up-regulates heme oxygenase-1 expression by stimulating Nrf2 nuclear translocation in an extracellular signal-regulated kinase-dependent manner in HepG2 cells. World J. Gastroenterol. 2006, 12, 214–221. [Google Scholar] [CrossRef]
- Xu, C.; Yuan, X.; Pan, Z.; Shen, G.; Kim, J.H.; Yu, S.; Khor, T.O.; Li, W.; Ma, J.; Kong, A.N. Mechanism of action of isothiocyanates: The induction of ARE-regulated genes is associated with activation of ERK and JNK and the phosphorylation and nuclear translocation of Nrf2. Mol. Cancer Ther. 2006, 5, 1918–1926. [Google Scholar] [CrossRef] [Green Version]
- Manandhar, S.; Cho, J.M.; Kim, J.A.; Kensler, T.W.; Kwak, M.K. Induction of Nrf2-regulated genes by 3H-1, 2-dithiole-3-thione through the ERK signaling pathway in murine keratinocytes. Eur. J. Pharmacol. 2007, 577, 17–27. [Google Scholar] [CrossRef]
- Medina, M.V.; Sapochnik, D.; Garcia Sola, M.; Coso, O. Regulation of the Expression of Heme Oxygenase-1: Signal Transduction, Gene Promoter Activation, and Beyond. Antioxid. Redox Signal. 2020, 32, 1033–1044. [Google Scholar] [CrossRef]
- Okinaga, S.; Shibahara, S. Identification of a nuclear protein that constitutively recognizes the sequence containing a heat-shock element. Its binding properties and possible function modulating heat-shock induction of the rat heme oxygenase gene. Eur. J. Biochem. 1993, 212, 167–175. [Google Scholar] [CrossRef]
- Inouye, S.; Hatori, Y.; Kubo, T.; Saito, S.; Kitamura, H.; Akagi, R. NRF2 and HSF1 coordinately regulate heme oxygenase-1 expression. Biochem. Biophys. Res. 2018, 506, 7–11. [Google Scholar] [CrossRef]
- Bejjani, F.; Evanno, E.; Zibara, K.; Piechaczyk, M.; Jariel-Encontre, I. The AP-1 transcriptional complex: Local switch or remote command? Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 11–23. [Google Scholar] [CrossRef]
- Alam, J.; Den, Z. Distal AP-1 binding sites mediate basal level enhancement and TPA induction of the mouse heme oxygenase-1 gene. J. Biol. Chem. 1992, 267, 21894–21900. [Google Scholar] [PubMed]
- Immenschuh, S.; Hinke, V.; Ohlmann, A.; Gifhorn-Katz, S.; Katz, N.; Jungermann, K.; Kietzmann, T. Transcriptional activation of the haem oxygenase-1 gene by cGMP via a cAMP response element/activator protein-1 element in primary cultures of rat hepatocytes. Biochem. J. 1998, 334 Pt 1, 141–146. [Google Scholar] [CrossRef] [Green Version]
- Hock, T.D.; Liby, K.; Wright, M.M.; McConnell, S.; Schorpp-Kistner, M.; Ryan, T.M.; Agarwal, A. JunB and JunD regulate human heme oxygenase-1 gene expression in renal epithelial cells. J. Biol. Chem. 2007, 282, 6875–6886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camhi, S.L.; Alam, J.; Otterbein, L.; Sylvester, S.L.; Choi, A.M. Induction of heme oxygenase-1 gene expression by lipopolysaccharide is mediated by AP-1 activation. Am. J. Respir. Cell Mol. Biol. 1995, 13, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Stewart, D.; Hu, B.; Vinson, C.; Alam, J. Multiple basic-leucine zipper proteins regulate induction of the mouse heme oxygenase-1 gene by arsenite. Arch. Biochem. Biophys. 2002, 405, 265–274. [Google Scholar] [CrossRef]
- Wright, M.M.; Kim, J.; Hock, T.D.; Leitinger, N.; Freeman, B.A.; Agarwal, A. Human haem oxygenase-1 induction by nitro-linoleic acid is mediated by cAMP, AP-1 and E-box response element interactions. Biochem. J. 2009, 422, 353–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samoylenko, A.; Dimova, E.Y.; Horbach, T.; Teplyuk, N.; Immenschuh, S.; Kietzmann, T. Opposite expression of the antioxidant heme oxygenase-1 in primary cells and tumor cells: Regulation by interaction of USF-2 and Fra-1. Antioxid. Redox Signal. 2008, 10, 1163–1174. [Google Scholar] [CrossRef]
- Deshane, J.; Kim, J.; Bolisetty, S.; Hock, T.D.; Hill-Kapturczak, N.; Agarwal, A. Sp1 regulates chromatin looping between an intronic enhancer and distal promoter of the human heme oxygenase-1 gene in renal cells. J. Biol. Chem. 2010, 285, 16476–16486. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.Y.; Yang, Y.C.; Li, C.C.; Liu, K.L.; Lii, C.K.; Chen, H.W. Andrographolide inhibits TNFalpha-induced ICAM-1 expression via suppression of NADPH oxidase activation and induction of HO-1 and GCLM expression through the PI3K/Akt/Nrf2 and PI3K/Akt/AP-1 pathways in human endothelial cells. Biochem. Pharmacol. 2014, 91, 40–50. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFkappaB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naidu, S.; Wijayanti, N.; Santoso, S.; Kietzmann, T.; Immenschuh, S. An atypical NF-kappa B-regulated pathway mediates phorbol ester-dependent heme oxygenase-1 gene activation in monocytes. J. Immunol. 2008, 181, 4113–4123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Guo, Y.; Ou, Q.; Cui, C.; Wu, W.J.; Tan, W.; Zhu, X.; Lanceta, L.B.; Sanganalmath, S.K.; Dawn, B.; et al. Gene transfer of inducible nitric oxide synthase affords cardioprotection by upregulating heme oxygenase-1 via a nuclear factor-{kappa}B-dependent pathway. Circulation 2009, 120, 1222–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyssonnaux, C.; Zinkernagel, A.S.; Schuepbach, R.A.; Rankin, E.; Vaulont, S.; Haase, V.H.; Nizet, V.; Johnson, R.S. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J. Clin. Investig. 2007, 117, 1926–1932. [Google Scholar] [CrossRef] [Green Version]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF transcription factors, inflammation, and immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Lin, N.; Simon, M.C. Hypoxia-inducible factors: Key regulators of myeloid cells during inflammation. J. Clin. Investig. 2016, 126, 3661–3671. [Google Scholar] [CrossRef]
- Lee, P.J.; Jiang, B.H.; Chin, B.Y.; Iyer, N.V.; Alam, J.; Semenza, G.L.; Choi, A.M. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J. Biol. Chem. 1997, 272, 5375–5381. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.Z.; Zou, A.P. Transcriptional regulation of heme oxygenases by HIF-1alpha in renal medullary interstitial cells. Am. J. Physiol. Renal. Physiol. 2001, 281, F900–F908. [Google Scholar] [CrossRef] [Green Version]
- Ockaili, R.; Natarajan, R.; Salloum, F.; Fisher, B.J.; Jones, D.; Fowler, A.A., 3rd; Kukreja, R.C. HIF-1 activation attenuates postischemic myocardial injury: Role for heme oxygenase-1 in modulating microvascular chemokine generation. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H542–H548. [Google Scholar] [CrossRef] [Green Version]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in pathophysiology: A matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharmacol. 2014, 5, 61. [Google Scholar] [CrossRef] [Green Version]
- Soares, M.P.; Bozza, M.T. Red alert: Labile heme is an alarmin. Curr. Opin. Immunol. 2016, 38, 94–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadrzadeh, S.M.; Graf, E.; Panter, S.S.; Hallaway, P.E.; Eaton, J.W. Hemoglobin. A biologic fenton reagent. J. Biol. Chem. 1984, 259, 14354–14356. [Google Scholar] [PubMed]
- Klouche, K.; Morena, M.; Canaud, B.; Descomps, B.; Beraud, J.J.; Cristol, J.P. Mechanism of in vitro heme-induced LDL oxidation: Effects of antioxidants. Eur. J. Clin. Investig. 2004, 34, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Tappel, A.L. Unsaturated lipide oxidation catalyzed by hematin compounds. J. Biol. Chem. 1955, 217, 721–733. [Google Scholar]
- Van der Zee, J.; Barr, D.P.; Mason, R.P. ESR spin trapping investigation of radical formation from the reaction between hematin and tert-Butyl hydroperoxide. Free Radic. Biol. Med. 1996, 20, 199–206. [Google Scholar] [CrossRef]
- Ryter, S.W.; Tyrrell, R.M. The heme synthesis and degradation pathways: Role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic. Biol. Med. 2000, 28, 289–309. [Google Scholar] [CrossRef]
- Imoto, S.; Kono, M.; Suzuki, T.; Shibuya, Y.; Sawamura, T.; Mizokoshi, Y.; Sawada, H.; Ohbuchi, A.; Saigo, K. Haemin-induced cell death in human monocytic cells is consistent with ferroptosis. Transfus. Apher. Sci. 2018, 57, 524–531. [Google Scholar] [CrossRef]
- NaveenKumar, S.K.; Hemshekhar, M.; Kemparaju, K.; Girish, K.S. Hemin-induced platelet activation and ferroptosis is mediated through ROS-driven proteasomal activity and inflammasome activation: Protection by Melatonin. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2303–2316. [Google Scholar] [CrossRef]
- Graca-Souza, A.V.; Arruda, M.A.; de Freitas, M.S.; Barja-Fidalgo, C.; Oliveira, P.L. Neutrophil activation by heme: Implications for inflammatory processes. Blood 2002, 99, 4160–4165. [Google Scholar] [CrossRef] [Green Version]
- Arruda, M.A.; Barcellos-de-Souza, P.; Sampaio, A.L.; Rossi, A.G.; Graca-Souza, A.V.; Barja-Fidalgo, C. NADPH oxidase-derived ROS: Key modulators of heme-induced mitochondrial stability in human neutrophils. Exp. Cell Res. 2006, 312, 3939–3948. [Google Scholar] [CrossRef]
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn. Rev. 2010, 4, 118–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueiredo, R.T.; Fernandez, P.L.; Mourao-Sa, D.S.; Porto, B.N.; Dutra, F.F.; Alves, L.S.; Oliveira, M.F.; Oliveira, P.L.; Graca-Souza, A.V.; Bozza, M.T. Characterization of heme as activator of Toll-like receptor 4. J. Biol. Chem. 2007, 282, 20221–20229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belcher, J.D.; Zhang, P.; Nguyen, J.; Kiser, Z.M.; Nath, K.A.; Hu, J.; Trent, J.O.; Vercellotti, G.M. Identification of a Heme Activation Site on the MD-2/TLR4 Complex. Front. Immunol. 2020, 11, 1370. [Google Scholar] [CrossRef] [PubMed]
- Fortes, G.B.; Alves, L.S.; de Oliveira, R.; Dutra, F.F.; Rodrigues, D.; Fernandez, P.L.; Souto-Padron, T.; De Rosa, M.J.; Kelliher, M.; Golenbock, D.; et al. Heme induces programmed necrosis on macrophages through autocrine TNF and ROS production. Blood 2012, 119, 2368–2375. [Google Scholar] [CrossRef] [Green Version]
- Dutra, F.F.; Alves, L.S.; Rodrigues, D.; Fernandez, P.L.; de Oliveira, R.B.; Golenbock, D.T.; Zamboni, D.S.; Bozza, M.T. Hemolysis-induced lethality involves inflammasome activation by heme. Proc. Natl. Acad. Sci. USA 2014, 111, E4110–E4118. [Google Scholar] [CrossRef] [Green Version]
- Nyakundi, B.B.; Toth, A.; Balogh, E.; Nagy, B.; Erdei, J.; Ryffel, B.; Paragh, G.; Cordero, M.D.; Jeney, V. Oxidized hemoglobin forms contribute to NLRP3 inflammasome-driven IL-1beta production upon intravascular hemolysis. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 464–475. [Google Scholar] [CrossRef]
- Wagener, F.A.; Feldman, E.; de Witte, T.; Abraham, N.G. Heme induces the expression of adhesion molecules ICAM-1, VCAM-1, and E selectin in vascular endothelial cells. Proc. Soc. Exp. Biol. Med. 1997, 216, 456–463. [Google Scholar] [CrossRef]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Milbauer, L.; Abdulla, F.; Alayash, A.I.; Smith, A.; Nath, K.A.; Hebbel, R.P.; Vercellotti, G.M. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 2014, 123, 377–390. [Google Scholar] [CrossRef] [Green Version]
- Merle, N.S.; Paule, R.; Leon, J.; Daugan, M.; Robe-Rybkine, T.; Poillerat, V.; Torset, C.; Fremeaux-Bacchi, V.; Dimitrov, J.D.; Roumenina, L.T. P-selectin drives complement attack on endothelium during intravascular hemolysis in TLR-4/heme-dependent manner. Proc. Natl. Acad. Sci. USA 2019, 116, 6280–6285. [Google Scholar] [CrossRef] [Green Version]
- Erdei, J.; Toth, A.; Balogh, E.; Nyakundi, B.B.; Banyai, E.; Ryffel, B.; Paragh, G.; Cordero, M.D.; Jeney, V. Induction of NLRP3 Inflammasome Activation by Heme in Human Endothelial Cells. Oxid. Med. Cell. Longev. 2018, 2018, 4310816. [Google Scholar] [CrossRef] [Green Version]
- Porto, B.N.; Alves, L.S.; Fernandez, P.L.; Dutra, T.P.; Figueiredo, R.T.; Graca-Souza, A.V.; Bozza, M.T. Heme induces neutrophil migration and reactive oxygen species generation through signaling pathways characteristic of chemotactic receptors. J. Biol. Chem. 2007, 282, 24430–24436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Zhang, D.; Fuchs, T.A.; Manwani, D.; Wagner, D.D.; Frenette, P.S. Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood 2014, 123, 3818–3827. [Google Scholar] [CrossRef] [PubMed]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poss, K.D.; Tonegawa, S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc. Natl. Acad. Sci. USA 1997, 94, 10919–10924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kartikasari, A.E.; Wagener, F.A.; Yachie, A.; Wiegerinck, E.T.; Kemna, E.H.; Swinkels, D.W. Hepcidin suppression and defective iron recycling account for dysregulation of iron homeostasis in heme oxygenase-1 deficiency. J. Cell Mol. Med. 2009, 13, 3091–3102. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, N.; Yadav, S.P.; Sachdeva, A.; Pruthi, P.K.; Sawhney, S.; Piplani, T.; Wada, T.; Yachie, A. Human heme oxygenase-1 deficiency presenting with hemolysis, nephritis, and asplenia. J. Pediatr. Hematol. Oncol. 2011, 33, 74–78. [Google Scholar] [CrossRef]
- Yachie, A.; Niida, Y.; Wada, T.; Igarashi, N.; Kaneda, H.; Toma, T.; Ohta, K.; Kasahara, Y.; Koizumi, S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J. Clin. Investig. 1999, 103, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Wu, Y.; Tang, W. Heme Catabolic Pathway in Inflammation and Immune Disorders. Front. Pharmacol. 2019, 10, 825. [Google Scholar] [CrossRef] [Green Version]
- Vanella, L.; Barbagallo, I.; Tibullo, D.; Forte, S.; Zappala, A.; Li Volti, G. The non-canonical functions of the heme oxygenases. Oncotarget 2016, 7, 69075–69086. [Google Scholar] [CrossRef] [Green Version]
- Motterlini, R.; Foresti, R. Biological signaling by carbon monoxide and carbon monoxide-releasing molecules. Am. J. Physiol. Cell Physiol. 2017, 312, C302–C313. [Google Scholar] [CrossRef] [Green Version]
- Ryter, S.W. Heme oxygenase-1/carbon monoxide as modulators of autophagy and inflammation. Arch. Biochem. Biophys. 2019, 678, 108186. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Kim, H.P.; Geng, X.H.; Nakao, A.; Wang, X.; Murase, N.; Drain, P.F.; Wang, X.; Sasidhar, M.; Nabel, E.G.; et al. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J. Exp. Med. 2006, 203, 2377–2389. [Google Scholar] [CrossRef] [PubMed]
- Otterbein, L.E.; Bach, F.H.; Alam, J.; Soares, M.; Tao Lu, H.; Wysk, M.; Davis, R.J.; Flavell, R.A.; Choi, A.M. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat. Med. 2000, 6, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Morse, D.; Pischke, S.E.; Zhou, Z.; Davis, R.J.; Flavell, R.A.; Loop, T.; Otterbein, S.L.; Otterbein, L.E.; Choi, A.M. Suppression of inflammatory cytokine production by carbon monoxide involves the JNK pathway and AP-1. J. Biol. Chem. 2003, 278, 36993–36998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pae, H.O.; Oh, G.S.; Choi, B.M.; Chae, S.C.; Kim, Y.M.; Chung, K.R.; Chung, H.T. Carbon monoxide produced by heme oxygenase-1 suppresses T cell proliferation via inhibition of IL-2 production. J. Immunol. 2004, 172, 4744–4751. [Google Scholar] [CrossRef] [Green Version]
- Megias, J.; Busserolles, J.; Alcaraz, M.J. The carbon monoxide-releasing molecule CORM-2 inhibits the inflammatory response induced by cytokines in Caco-2 cells. Br. J. Pharmacol. 2007, 150, 977–986. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.S.; Moon, J.S.; Xu, J.F.; Ifedigbo, E.; Ryter, S.W.; Choi, A.M.; Nakahira, K. Carbon monoxide negatively regulates NLRP3 inflammasome activation in macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L1058–L1067. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.K.; Joe, Y.; Chen, Y.; Ryu, J.; Lee, J.H.; Cho, G.J.; Ryter, S.W.; Chung, H.T. Carbon monoxide decreases interleukin-1beta levels in the lung through the induction of pyrin. Cell. Mol. Immunol. 2017, 14, 349–359. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Joe, Y.; Jeong, S.O.; Zheng, M.; Back, S.H.; Park, S.W.; Ryter, S.W.; Chung, H.T. Endoplasmic reticulum stress is sufficient for the induction of IL-1beta production via activation of the NF-kappaB and inflammasome pathways. Innate Immun. 2014, 20, 799–815. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Fei, D.; Gong, R.; Yang, W.; Yu, W.; Pan, S.; Zhao, M.; Zhao, M. CORM-2 inhibits TXNIP/NLRP3 inflammasome pathway in LPS-induced acute lung injury. Inflamm. Res. 2016, 65, 905–915. [Google Scholar] [CrossRef]
- Lee, D.W.; Shin, H.Y.; Jeong, J.H.; Han, J.; Ryu, S.; Nakahira, K.; Moon, J.S. Carbon monoxide regulates glycolysis-dependent NLRP3 inflammasome activation in macrophages. Biochem. Biophys. Res. 2017, 493, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Tao, A.; Lan, T.; Cepinskas, G.; Kao, R.; Martin, C.M.; Rui, T. Carbon monoxide releasing molecule-3 improves myocardial function in mice with sepsis by inhibiting NLRP3 inflammasome activation in cardiac fibroblasts. Basic Res. Cardiol. 2017, 112, 16. [Google Scholar] [CrossRef] [PubMed]
- Mackern-Oberti, J.P.; Llanos, C.; Carreno, L.J.; Riquelme, S.A.; Jacobelli, S.H.; Anegon, I.; Kalergis, A.M. Carbon monoxide exposure improves immune function in lupus-prone mice. Immunology 2013, 140, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Mackern-Oberti, J.P.; Obreque, J.; Mendez, G.P.; Llanos, C.; Kalergis, A.M. Carbon monoxide inhibits T cell activation in target organs during systemic lupus erythematosus. Clin. Exp. Immunol. 2015, 182, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, T.; Naito, Y.; Inoue, M.; Akagiri, S.; Mizushima, K.; Handa, O.; Kokura, S.; Ichikawa, H.; Yoshikawa, T. Inhalation of carbon monoxide ameliorates collagen-induced arthritis in mice and regulates the articular expression of IL-1beta and MCP-1. Inflammation 2009, 32, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, M.; Savitskaya, A.; Steiner, C.W.; Rath, E.; Bilban, M.; Wagner, O.; Bach, F.H.; Smolen, J.S.; Scheinecker, C. Heme oxygenase-1 end-products carbon monoxide and biliverdin ameliorate murine collagen induced arthritis. Clin. Exp. Rheumatol. 2012, 30, 73–78. [Google Scholar]
- Fagone, P.; Mangano, K.; Quattrocchi, C.; Motterlini, R.; Di Marco, R.; Magro, G.; Penacho, N.; Romao, C.C.; Nicoletti, F. Prevention of clinical and histological signs of proteolipid protein (PLP)-induced experimental allergic encephalomyelitis (EAE) in mice by the water-soluble carbon monoxide-releasing molecule (CORM)-A1. Clin. Exp. Immunol. 2011, 163, 368–374. [Google Scholar] [CrossRef]
- Jansen, T.; Daiber, A. Direct Antioxidant Properties of Bilirubin and Biliverdin. Is there a Role for Biliverdin Reductase? Front. Pharmacol. 2012, 3, 30. [Google Scholar] [CrossRef] [Green Version]
- Wegiel, B.; Otterbein, L.E. Go green: The anti-inflammatory effects of biliverdin reductase. Front. Pharmacol. 2012, 3, 47. [Google Scholar] [CrossRef] [Green Version]
- Sarady-Andrews, J.K.; Liu, F.; Gallo, D.; Nakao, A.; Overhaus, M.; Ollinger, R.; Choi, A.M.; Otterbein, L.E. Biliverdin administration protects against endotoxin-induced acute lung injury in rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L1131–L1137. [Google Scholar] [CrossRef]
- Gibbs, P.E.; Maines, M.D. Biliverdin inhibits activation of NF-kappaB: Reversal of inhibition by human biliverdin reductase. Int. J. Cancer 2007, 121, 2567–2574. [Google Scholar] [CrossRef] [PubMed]
- Bisht, K.; Wegiel, B.; Tampe, J.; Neubauer, O.; Wagner, K.H.; Otterbein, L.E.; Bulmer, A.C. Biliverdin modulates the expression of C5aR in response to endotoxin in part via mTOR signaling. Biochem. Biophys. Res. 2014, 449, 94–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canesin, G.; Hejazi, S.M.; Swanson, K.D.; Wegiel, B. Heme-Derived Metabolic Signals Dictate Immune Responses. Front. Immunol. 2020, 11, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegiel, B.; Baty, C.J.; Gallo, D.; Csizmadia, E.; Scott, J.R.; Akhavan, A.; Chin, B.Y.; Kaczmarek, E.; Alam, J.; Bach, F.H.; et al. Cell surface biliverdin reductase mediates biliverdin-induced anti-inflammatory effects via phosphatidylinositol 3-kinase and Akt. J. Biol. Chem. 2009, 284, 21369–21378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegiel, B.; Gallo, D.; Csizmadia, E.; Roger, T.; Kaczmarek, E.; Harris, C.; Zuckerbraun, B.S.; Otterbein, L.E. Biliverdin inhibits Toll-like receptor-4 (TLR4) expression through nitric oxide-dependent nuclear translocation of biliverdin reductase. Proc. Natl. Acad. Sci. USA 2011, 108, 18849–18854. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, Z.; Salim, M.; Maines, M.D. Human biliverdin reductase is a leucine zipper-like DNA-binding protein and functions in transcriptional activation of heme oxygenase-1 by oxidative stress. J. Biol. Chem. 2002, 277, 9226–9232. [Google Scholar] [CrossRef] [Green Version]
- Kravets, A.; Hu, Z.; Miralem, T.; Torno, M.D.; Maines, M.D. Biliverdin reductase, a novel regulator for induction of activating transcription factor-2 and heme oxygenase-1. J. Biol. Chem. 2004, 279, 19916–19923. [Google Scholar] [CrossRef] [Green Version]
- Basiglio, C.L.; Arriaga, S.M.; Pelusa, H.F.; Almara, A.M.; Roma, M.G.; Mottino, A.D. Protective role of unconjugated bilirubin on complement-mediated hepatocytolysis. Biochim. Biophys. Acta 2007, 1770, 1003–1010. [Google Scholar] [CrossRef]
- Vetvicka, V.; Miler, I.; Sima, P.; Taborsky, L.; Fornusek, L. The effect of bilirubin on the Fc receptor expression and phagocytic activity of mouse peritoneal macrophages. Folia Microbiol. 1985, 30, 373–380. [Google Scholar] [CrossRef]
- Arai, T.; Yoshikai, Y.; Kamiya, J.; Nagino, M.; Uesaka, K.; Yuasa, N.; Oda, K.; Sano, T.; Nimura, Y. Bilirubin impairs bactericidal activity of neutrophils through an antioxidant mechanism in vitro. J. Surg. Res. 2001, 96, 107–113. [Google Scholar] [CrossRef]
- Thong, Y.H.; Ferrante, A.; Ness, D. Neutrophil phagocytic and bactericidal dysfunction induced by bilirubin. Aust. Paediatr. J. 1977, 13, 287–289. [Google Scholar] [CrossRef]
- Miler, I.; Vondracek, J.; Hromadkova, L. Bilirubin inhibits the chemotactic activity of human polymorphonuclear leukocytes in vitro. Folia Microbiol. 1981, 26, 413–416. [Google Scholar] [CrossRef]
- Svejcar, J.; Miler, I.; Pekarek, J. Effect of bilirubin on an in vitro correlate of cell-mediated immunity—The migration inhibition test. J. Clin. Lab. Immunol. 1984, 13, 145–149. [Google Scholar]
- Hayashi, S.; Takamiya, R.; Yamaguchi, T.; Matsumoto, K.; Tojo, S.J.; Tamatani, T.; Kitajima, M.; Makino, N.; Ishimura, Y.; Suematsu, M. Induction of heme oxygenase-1 suppresses venular leukocyte adhesion elicited by oxidative stress: Role of bilirubin generated by the enzyme. Circ. Res. 1999, 85, 663–671. [Google Scholar] [CrossRef] [Green Version]
- Mazzone, G.L.; Rigato, I.; Ostrow, J.D.; Bossi, F.; Bortoluzzi, A.; Sukowati, C.H.; Tedesco, F.; Tiribelli, C. Bilirubin inhibits the TNFalpha-related induction of three endothelial adhesion molecules. Biochem. Biophys. Res. 2009, 386, 338–344. [Google Scholar] [CrossRef]
- Haga, Y.; Tempero, M.A.; Kay, D.; Zetterman, R.K. Intracellular accumulation of unconjugated bilirubin inhibits phytohemagglutin-induced proliferation and interleukin-2 production of human lymphocytes. Dig. Dis. Sci. 1996, 41, 1468–1474. [Google Scholar] [CrossRef]
- Liu, Y.; Li, P.; Lu, J.; Xiong, W.; Oger, J.; Tetzlaff, W.; Cynader, M. Bilirubin possesses powerful immunomodulatory activity and suppresses experimental autoimmune encephalomyelitis. J. Immunol. 2008, 181, 1887–1897. [Google Scholar] [CrossRef] [Green Version]
- Longhi, M.S.; Vuerich, M.; Kalbasi, A.; Kenison, J.E.; Yeste, A.; Csizmadia, E.; Vaughn, B.; Feldbrugge, L.; Mitsuhashi, S.; Wegiel, B.; et al. Bilirubin suppresses Th17 immunity in colitis by upregulating CD39. JCI Insight 2017, 2, e92791. [Google Scholar] [CrossRef] [Green Version]
- Adin, C.A.; VanGundy, Z.C.; Papenfuss, T.L.; Xu, F.; Ghanem, M.; Lakey, J.; Hadley, G.A. Physiologic Doses of Bilirubin Contribute to Tolerance of Islet Transplants by Suppressing the Innate Immune Response. Cell Transplant. 2017, 26, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Wessling-Resnick, M. Iron homeostasis and the inflammatory response. Annu. Rev. Nutr. 2010, 30, 105–122. [Google Scholar] [CrossRef] [Green Version]
- Gozzelino, R.; Soares, M.P. Coupling heme and iron metabolism via ferritin H chain. Antioxid. Redox Signal. 2014, 20, 1754–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, N.J.; Quinlan, G.J.; Mumby, S.; Evans, T.W.; Gutteridge, J.M. Haem oxygenase shows pro-oxidant activity in microsomal and cellular systems: Implications for the release of low-molecular-mass iron. Biochem. J. 1999, 344 Pt 1, 153–158. [Google Scholar] [CrossRef]
- Suttner, D.M.; Dennery, P.A. Reversal of HO-1 related cytoprotection with increased expression is due to reactive iron. FASEB J. 1999, 13, 1800–1809. [Google Scholar] [CrossRef] [PubMed]
- Duvigneau, J.C.; Piskernik, C.; Haindl, S.; Kloesch, B.; Hartl, R.T.; Huttemann, M.; Lee, I.; Ebel, T.; Moldzio, R.; Gemeiner, M.; et al. A novel endotoxin-induced pathway: Upregulation of heme oxygenase 1, accumulation of free iron, and free iron-mediated mitochondrial dysfunction. Lab. Investig. 2008, 88, 70–77. [Google Scholar] [CrossRef] [Green Version]
- Kwon, M.Y.; Park, E.; Lee, S.J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef] [Green Version]
- Fibach, E.; Rachmilewitz, E.A. Iron overload in hematological disorders. Presse Med. 2017, 46, e296–e305. [Google Scholar] [CrossRef]
- White, B.C.; Krause, G.S.; Aust, S.D.; Eyster, G.E. Postischemic tissue injury by iron-mediated free radical lipid peroxidation. Ann. Emerg. Med. 1985, 14, 804–809. [Google Scholar] [CrossRef]
- Bou-Abdallah, F.; Paliakkara, J.J.; Melman, G.; Melman, A. Reductive Mobilization of Iron from Intact Ferritin: Mechanisms and Physiological Implication. Pharmaceuticals 2018, 11, 120. [Google Scholar] [CrossRef] [Green Version]
- Weiss, G.; Werner-Felmayer, G.; Werner, E.R.; Grunewald, K.; Wachter, H.; Hentze, M.W. Iron regulates nitric oxide synthase activity by controlling nuclear transcription. J. Exp. Med. 1994, 180, 969–976. [Google Scholar] [CrossRef] [Green Version]
- Komarov, A.M.; Mattson, D.L.; Mak, I.T.; Weglicki, W.B. Iron attenuates nitric oxide level and iNOS expression in endotoxin-treated mice. FEBS Lett. 1998, 424, 253–256. [Google Scholar] [CrossRef] [Green Version]
- Agoro, R.; Taleb, M.; Quesniaux, V.F.J.; Mura, C. Cell iron status influences macrophage polarization. PLoS ONE 2018, 13, e0196921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dlaska, M.; Weiss, G. Central role of transcription factor NF-IL6 for cytokine and iron-mediated regulation of murine inducible nitric oxide synthase expression. J. Immunol. 1999, 162, 6171–6177. [Google Scholar] [PubMed]
- Fritsche, G.; Nairz, M.; Werner, E.R.; Barton, H.C.; Weiss, G. Nramp1-functionality increases iNOS expression via repression of IL-10 formation. Eur. J. Immunol. 2008, 38, 3060–3067. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.T.; Yen, C.J.; Chang, C.C.; Huang, K.T.; Chen, K.H.; Zhang, R.Y.; Lee, P.Y.; Miaw, S.C.; Huang, J.W.; Chiang, C.K.; et al. Ferritin heavy chain mediates the protective effect of heme oxygenase-1 against oxidative stress. Biochim. Biophys. Acta 2015, 1850, 2506–2517. [Google Scholar] [CrossRef]
- Cai, C.X.; Birk, D.E.; Linsenmayer, T.F. Nuclear ferritin protects DNA from UV damage in corneal epithelial cells. Mol. Biol. Cell 1998, 9, 1037–1051. [Google Scholar] [CrossRef] [Green Version]
- Linsenmayer, T.F.; Cai, C.X.; Millholland, J.M.; Beazley, K.E.; Fitch, J.M. Nuclear ferritin in corneal epithelial cells: Tissue-specific nuclear transport and protection from UV-damage. Prog. Retin. Eye Res. 2005, 24, 139–159. [Google Scholar] [CrossRef]
- Fan, Y.; Zhang, J.; Cai, L.; Wang, S.; Liu, C.; Zhang, Y.; You, L.; Fu, Y.; Shi, Z.; Yin, Z.; et al. The effect of anti-inflammatory properties of ferritin light chain on lipopolysaccharide-induced inflammatory response in murine macrophages. Biochim. Biophys. Acta 2014, 1843, 2775–2783. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, K.; Oseko, F.; Morikawa, S. A role for ferritin in hematopoiesis and the immune system. Leuk. Lymphoma 1995, 18, 429–433. [Google Scholar] [CrossRef]
- Zandman-Goddard, G.; Shoenfeld, Y. Ferritin in autoimmune diseases. Autoimmun. Rev. 2007, 6, 457–463. [Google Scholar] [CrossRef]
- Hori, R.; Kashiba, M.; Toma, T.; Yachie, A.; Goda, N.; Makino, N.; Soejima, A.; Nagasawa, T.; Nakabayashi, K.; Suematsu, M. Gene transfection of H25A mutant heme oxygenase-1 protects cells against hydroperoxide-induced cytotoxicity. J. Biol. Chem. 2002, 277, 10712–10718. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Weis, S.; Yang, G.; Weng, Y.H.; Helston, R.; Rish, K.; Smith, A.; Bordner, J.; Polte, T.; Gaunitz, F.; et al. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J. Biol. Chem. 2007, 282, 20621–20633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Q.S.; Weis, S.; Yang, G.; Zhuang, T.; Abate, A.; Dennery, P.A. Catalytic inactive heme oxygenase-1 protein regulates its own expression in oxidative stress. Free Radic. Biol. Med. 2008, 44, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Biswas, C.; Shah, N.; Muthu, M.; La, P.; Fernando, A.P.; Sengupta, S.; Yang, G.; Dennery, P.A. Nuclear heme oxygenase-1 (HO-1) modulates subcellular distribution and activation of Nrf2, impacting metabolic and anti-oxidant defenses. J. Biol. Chem. 2014, 289, 26882–26894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paez, A.V.; Pallavicini, C.; Schuster, F.; Valacco, M.P.; Giudice, J.; Ortiz, E.G.; Anselmino, N.; Labanca, E.; Binaghi, M.; Salierno, M.; et al. Heme oxygenase-1 in the forefront of a multi-molecular network that governs cell-cell contacts and filopodia-induced zippering in prostate cancer. Cell Death Dis. 2016, 7, e2570. [Google Scholar] [CrossRef] [PubMed]
- Weng, Y.H.; Yang, G.; Weiss, S.; Dennery, P.A. Interaction between heme oxygenase-1 and -2 proteins. J. Biol. Chem. 2003, 278, 50999–51005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. Top 10 Causes of Death. Available online: https://www.who.int/gho/mortality_burden_disease/causes_death/top_10/en/ (accessed on 9 September 2020).
- Hutchings, M.I.; Truman, A.W.; Wilkinson, B. Antibiotics: Past, present and future. Curr. Opin. Microbiol. 2019, 51, 72–80. [Google Scholar] [CrossRef]
- Collignon, P.; Beggs, J.J.; Walsh, T.R.; Gandra, S.; Laxminarayan, R. Anthropological and socioeconomic factors contributing to global antimicrobial resistance: A univariate and multivariable analysis. Lancet Planet. Health 2018, 2, e398–e405. [Google Scholar] [CrossRef]
- WHO. Global Tuberculosis Report 2018; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- WHO. Guidelines for Treatment of Tuberculosis, 4th ed.; WHO Press: Geneva, Switzerland, 2010. [Google Scholar]
- Young, C.; Walzl, G.; Du Plessis, N. Therapeutic host-directed strategies to improve outcome in tuberculosis. Mucosal Immunol. 2020, 13, 190–204. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Deshane, J.S.; Crossman, D.K.; Bolisetty, S.; Yan, B.S.; Kramnik, I.; Agarwal, A.; Steyn, A.J. Heme oxygenase-1-derived carbon monoxide induces the Mycobacterium tuberculosis dormancy regulon. J. Biol. Chem. 2008, 283, 18032–18039. [Google Scholar] [CrossRef] [Green Version]
- Shiloh, M.U.; Manzanillo, P.; Cox, J.S. Mycobacterium tuberculosis senses host-derived carbon monoxide during macrophage infection. Cell Host Microbe 2008, 3, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Rockwood, N.; Costa, D.L.; Amaral, E.P.; Du Bruyn, E.; Kubler, A.; Gil-Santana, L.; Fukutani, K.F.; Scanga, C.A.; Flynn, J.L.; Jackson, S.H.; et al. Mycobacterium tuberculosis Induction of Heme Oxygenase-1 Expression Is Dependent on Oxidative Stress and Reflects Treatment Outcomes. Front. Immunol. 2017, 8, 542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wayne, L.G.; Sohaskey, C.D. Nonreplicating persistence of mycobacterium tuberculosis. Annu. Rev. Microbiol. 2001, 55, 139–163. [Google Scholar] [CrossRef] [PubMed]
- Voskuil, M.I.; Schnappinger, D.; Visconti, K.C.; Harrell, M.I.; Dolganov, G.M.; Sherman, D.R.; Schoolnik, G.K. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J. Exp. Med. 2003, 198, 705–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacMicking, J.D.; North, R.J.; LaCourse, R.; Mudgett, J.S.; Shah, S.K.; Nathan, C.F. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc. Natl. Acad. Sci. USA 1997, 94, 5243–5248. [Google Scholar] [CrossRef] [Green Version]
- Regev, D.; Surolia, R.; Karki, S.; Zolak, J.; Montes-Worboys, A.; Oliva, O.; Guroji, P.; Saini, V.; Steyn, A.J.; Agarwal, A.; et al. Heme oxygenase-1 promotes granuloma development and protects against dissemination of mycobacteria. Lab. Investig. 2012, 92, 1541–1552. [Google Scholar] [CrossRef] [Green Version]
- Silva-Gomes, S.; Appelberg, R.; Larsen, R.; Soares, M.P.; Gomes, M.S. Heme catabolism by heme oxygenase-1 confers host resistance to Mycobacterium infection. Infect. Immun. 2013, 81, 2536–2545. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Kansal, P.; Ahmad, Z.; Baid, N.; Kushwaha, H.; Khatri, N.; Kumar, A. Antimycobacterial effect of IFNG (interferon gamma)-induced autophagy depends on HMOX1 (heme oxygenase 1)-mediated increase in intracellular calcium levels and modulation of PPP3/calcineurin-TFEB (transcription factor EB) axis. Autophagy 2018, 14, 972–991. [Google Scholar] [CrossRef] [Green Version]
- Chinta, K.C.; Rahman, M.A.; Saini, V.; Glasgow, J.N.; Reddy, V.P.; Lever, J.M.; Nhamoyebonde, S.; Leslie, A.; Wells, R.M.; Traylor, A.; et al. Microanatomic Distribution of Myeloid Heme Oxygenase-1 Protects against Free Radical-Mediated Immunopathology in Human Tuberculosis. Cell Rep. 2018, 25, 1938–1952. [Google Scholar] [CrossRef] [Green Version]
- Abdalla, M.Y.; Ahmad, I.M.; Switzer, B.; Britigan, B.E. Induction of heme oxygenase-1 contributes to survival of Mycobacterium abscessus in human macrophages-like THP-1 cells. Redox Biol. 2015, 4, 328–339. [Google Scholar] [CrossRef] [Green Version]
- Scharn, C.R.; Collins, A.C.; Nair, V.R.; Stamm, C.E.; Marciano, D.K.; Graviss, E.A.; Shiloh, M.U. Heme Oxygenase-1 Regulates Inflammation and Mycobacterial Survival in Human Macrophages during Mycobacterium tuberculosis Infection. J. Immunol. 2016, 196, 4641–4649. [Google Scholar] [CrossRef] [Green Version]
- Costa, D.L.; Namasivayam, S.; Amaral, E.P.; Arora, K.; Chao, A.; Mittereder, L.R.; Maiga, M.; Boshoff, H.I.; Barry, C.E., 3rd; Goulding, C.W.; et al. Pharmacological Inhibition of Host Heme Oxygenase-1 Suppresses Mycobacterium tuberculosis Infection In Vivo by a Mechanism Dependent on T Lymphocytes. MBio 2016, 7, e01675-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, D.L.; Amaral, E.P.; Namasivayam, S.; Mittereder, L.R.; Fisher, L.; Bonfim, C.C.; Sardinha-Silva, A.; Thompson, R.W.; Hieny, S.E.; Andrade, B.B.; et al. Heme oxygenase-1 inhibition promotes IFNgamma- and NOS2-mediated control of Mycobacterium tuberculosis infection. Mucosal Immunol. 2020, 1–14. [Google Scholar] [CrossRef]
- Won, G.; Lee, J.H. Salmonella Typhimurium, the major causative agent of foodborne illness inactivated by a phage lysis system provides effective protection against lethal challenge by induction of robust cell-mediated immune responses and activation of dendritic cells. Vet. Res. 2017, 48, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaki, M.H.; Fujii, S.; Okamoto, T.; Islam, S.; Khan, S.; Ahmed, K.A.; Sawa, T.; Akaike, T. Cytoprotective function of heme oxygenase 1 induced by a nitrated cyclic nucleotide formed during murine salmonellosis. J. Immunol. 2009, 182, 3746–3756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onyiah, J.C.; Sheikh, S.Z.; Maharshak, N.; Steinbach, E.C.; Russo, S.M.; Kobayashi, T.; Mackey, L.C.; Hansen, J.J.; Moeser, A.J.; Rawls, J.F.; et al. Carbon monoxide and heme oxygenase-1 prevent intestinal inflammation in mice by promoting bacterial clearance. Gastroenterology 2013, 144, 789–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitterstiller, A.M.; Haschka, D.; Dichtl, S.; Nairz, M.; Demetz, E.; Talasz, H.; Soares, M.; Einwallner, E.; Esterbauer, H.; Fang, F.C.; et al. Heme oxygenase 1 controls early innate immune response of macrophages to Salmonella typhimurium infection. Cell Microbiol. 2016, 18, 1374–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nairz, M.; Theurl, I.; Ludwiczek, S.; Theurl, M.; Mair, S.M.; Fritsche, G.; Weiss, G. The co-ordinated regulation of iron homeostasis in murine macrophages limits the availability of iron for intracellular Salmonella typhimurium. Cell Microbiol. 2007, 9, 2126–2140. [Google Scholar] [CrossRef]
- de Noordhout, C.M.; Devleesschauwer, B.; Angulo, F.J.; Verbeke, G.; Haagsma, J.; Kirk, M.; Havelaar, A.; Speybroeck, N. The global burden of listeriosis: A systematic review and meta-analysis. Lancet Infect. Dis. 2014, 14, 1073–1082. [Google Scholar] [CrossRef] [Green Version]
- Tzima, S.; Victoratos, P.; Kranidioti, K.; Alexiou, M.; Kollias, G. Myeloid heme oxygenase-1 regulates innate immunity and autoimmunity by modulating IFN-beta production. J. Exp. Med. 2009, 206, 1167–1179. [Google Scholar] [CrossRef]
- Tachibana, M.; Hashino, M.; Nishida, T.; Shimizu, T.; Watarai, M. Protective role of heme oxygenase-1 in Listeria monocytogenes-induced abortion. PLoS ONE 2011, 6, e25046. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Sun, D.; Chen, G.; Li, G.; Dou, S.; Wang, R.; Xiao, H.; Hou, C.; Li, Y.; Feng, J.; et al. Tim-3 inhibits macrophage control of Listeria monocytogenes by inhibiting Nrf2. Sci Rep. 2017, 7, 42095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiersinga, W.J.; Currie, B.J.; Peacock, S.J. Melioidosis. N. Engl. J. Med. 2012, 367, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Stolt, C.; Schmidt, I.H.; Sayfart, Y.; Steinmetz, I.; Bast, A. Heme Oxygenase-1 and Carbon Monoxide Promote Burkholderia pseudomallei Infection. J. Immunol. 2016, 197, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Nithichanon, A.; Tussakhon, I.; Samer, W.; Kewcharoenwong, C.; Ato, M.; Bancroft, G.J.; Lertmemongkolchai, G. Immune responses in beta-thalassaemia: Heme oxygenase 1 reduces cytokine production and bactericidal activity of human leucocytes. Sci. Rep. 2020, 10, 10297. [Google Scholar] [CrossRef] [PubMed]
- Ashley, E.A.; Pyae Phyo, A.; Woodrow, C.J. Malaria. Lancet 2018, 391, 1608–1621. [Google Scholar] [CrossRef]
- Varikuti, S.; Jha, B.K.; Volpedo, G.; Ryan, N.M.; Halsey, G.; Hamza, O.M.; McGwire, B.S.; Satoskar, A.R. Host-Directed Drug Therapies for Neglected Tropical Diseases Caused by Protozoan Parasites. Front. Microbiol. 2018, 9, 2655. [Google Scholar] [CrossRef]
- Montoya, J.G.; Liesenfeld, O. Toxoplasmosis. Lancet 2004, 363, 1965–1976. [Google Scholar] [CrossRef]
- Andrews, K.T.; Fisher, G.; Skinner-Adams, T.S. Drug repurposing and human parasitic protozoan diseases. Int. J. Parasitol. Drugs Drug Resist. 2014, 4, 95–111. [Google Scholar] [CrossRef] [Green Version]
- WHO. World Malaria Report 2019; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Pamplona, A.; Ferreira, A.; Balla, J.; Jeney, V.; Balla, G.; Epiphanio, S.; Chora, A.; Rodrigues, C.D.; Gregoire, I.P.; Cunha-Rodrigues, M.; et al. Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat. Med. 2007, 13, 703–710. [Google Scholar] [CrossRef]
- Pena, A.C.; Penacho, N.; Mancio-Silva, L.; Neres, R.; Seixas, J.D.; Fernandes, A.C.; Romao, C.C.; Mota, M.M.; Bernardes, G.J.; Pamplona, A. A novel carbon monoxide-releasing molecule fully protects mice from severe malaria. Antimicrob. Agents Chemother. 2012, 56, 1281–1290. [Google Scholar] [CrossRef] [Green Version]
- Pereira, M.L.; Ortolan, L.S.; Sercundes, M.K.; Debone, D.; Murillo, O.; Lima, F.A.; Marinho, C.R.; Epiphanio, S. Association of Heme Oxygenase 1 with Lung Protection in Malaria-Associated ALI/ARDS. Mediat. Inflamm. 2016, 2016, 4158698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seixas, E.; Gozzelino, R.; Chora, A.; Ferreira, A.; Silva, G.; Larsen, R.; Rebelo, S.; Penido, C.; Smith, N.R.; Coutinho, A.; et al. Heme oxygenase-1 affords protection against noncerebral forms of severe malaria. Proc. Natl. Acad. Sci. USA 2009, 106, 15837–15842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epiphanio, S.; Mikolajczak, S.A.; Goncalves, L.A.; Pamplona, A.; Portugal, S.; Albuquerque, S.; Goldberg, M.; Rebelo, S.; Anderson, D.G.; Akinc, A.; et al. Heme oxygenase-1 is an anti-inflammatory host factor that promotes murine plasmodium liver infection. Cell Host Microbe 2008, 3, 331–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walther, M.; De Caul, A.; Aka, P.; Njie, M.; Amambua-Ngwa, A.; Walther, B.; Predazzi, I.M.; Cunnington, A.; Deininger, S.; Takem, E.N.; et al. HMOX1 gene promoter alleles and high HO-1 levels are associated with severe malaria in Gambian children. PLoS Pathog 2012, 8, e1002579. [Google Scholar] [CrossRef] [Green Version]
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef]
- Gebremichael Tedla, D.; Bariagabr, F.H.; Abreha, H.H. Incidence and Trends of Leishmaniasis and Its Risk Factors in Humera, Western Tigray. J. Parasitol. Res. 2018, 2018, 8463097. [Google Scholar] [CrossRef] [Green Version]
- Pham, N.K.; Mouriz, J.; Kima, P.E. Leishmania pifanoi amastigotes avoid macrophage production of superoxide by inducing heme degradation. Infect. Immun. 2005, 73, 8322–8333. [Google Scholar] [CrossRef] [Green Version]
- Luz, N.F.; Andrade, B.B.; Feijo, D.F.; Araujo-Santos, T.; Carvalho, G.Q.; Andrade, D.; Abanades, D.R.; Melo, E.V.; Silva, A.M.; Brodskyn, C.I.; et al. Heme oxygenase-1 promotes the persistence of Leishmania chagasi infection. J. Immunol. 2012, 188, 4460–4467. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Basu, M.; Guin, S.; Gupta, P.; Mitterstiller, A.M.; Weiss, G.; Jana, K.; Ukil, A. Leishmania donovani Exploits Macrophage Heme Oxygenase-1 To Neutralize Oxidative Burst and TLR Signaling-Dependent Host Defense. J. Immunol. 2019, 202, 827–840. [Google Scholar] [CrossRef] [Green Version]
- Moreno, J.; Alvar, J. Canine leishmaniasis: Epidemiological risk and the experimental model. Trends Parasitol. 2002, 18, 399–405. [Google Scholar] [CrossRef]
- Almeida, B.F.M.; Silva, K.L.O.; Venturin, G.L.; Chiku, V.M.; Leal, A.A.C.; Bosco, A.M.; Ciarlini, P.C.; Lima, V.M.F. Induction of haem oxygenase-1 increases infection of dog macrophages by L. infantum. Parasite Immunol. 2017, 39, e12494. [Google Scholar] [CrossRef] [PubMed]
- Luz, N.F.; DeSouza-Vieira, T.; De Castro, W.; Vivarini, A.C.; Pereira, L.; Franca, R.R.; Silveira-Mattos, P.S.; Costa, D.L.; Teixeira, C.; Meneses, C.; et al. Lutzomyia longipalpis Saliva Induces Heme Oxygenase-1 Expression at Bite Sites. Front. Immunol. 2018, 9, 2779. [Google Scholar] [CrossRef] [PubMed]
- Justi, S.A.; Galvao, C. The Evolutionary Origin of Diversity in Chagas Disease Vectors. Trends Parasitol. 2017, 33, 42–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lidani, K.C.F.; Andrade, F.A.; Bavia, L.; Damasceno, F.S.; Beltrame, M.H.; Messias-Reason, I.J.; Sandri, T.L. Chagas Disease: From Discovery to a Worldwide Health Problem. Front. Public Health 2019, 7, 166. [Google Scholar] [CrossRef]
- Kirchhoff, L.V. Epidemiology of American trypanosomiasis (Chagas disease). Adv. Parasitol. 2011, 75, 1–18. [Google Scholar] [CrossRef]
- Frade, A.F.; Pissetti, C.W.; Ianni, B.M.; Saba, B.; Lin-Wang, H.T.; Nogueira, L.G.; de Melo Borges, A.; Buck, P.; Dias, F.; Baron, M.; et al. Genetic susceptibility to Chagas disease cardiomyopathy: Involvement of several genes of the innate immunity and chemokine-dependent migration pathways. BMC Infect. Dis. 2013, 13, 587. [Google Scholar] [CrossRef]
- Paiva, C.N.; Feijo, D.F.; Dutra, F.F.; Carneiro, V.C.; Freitas, G.B.; Alves, L.S.; Mesquita, J.; Fortes, G.B.; Figueiredo, R.T.; Souza, H.S.; et al. Oxidative stress fuels Trypanosoma cruzi infection in mice. J. Clin. Investig. 2012, 122, 2531–2542. [Google Scholar] [CrossRef]
- Gutierrez, F.R.; Pavanelli, W.R.; Medina, T.S.; Silva, G.K.; Mariano, F.S.; Guedes, P.M.; Mineo, T.W.; Rossi, M.A.; Cunha, F.Q.; Silva, J.S. Haeme oxygenase activity protects the host against excessive cardiac inflammation during experimental Trypanosoma cruzi infection. Microbes Infect. 2014, 16, 28–39. [Google Scholar] [CrossRef]
- Araujo, E.C.; Barbosa, B.F.; Coutinho, L.B.; Barenco, P.V.; Sousa, L.A.; Milanezi, C.M.; Bonfa, G.; Pavanelli, W.R.; Silva, J.S.; Ferro, E.A.; et al. Heme oxygenase-1 activity is involved in the control of Toxoplasma gondii infection in the lung of BALB/c and C57BL/6 and in the small intestine of C57BL/6 mice. Vet. Res. 2013, 44, 89. [Google Scholar] [CrossRef] [Green Version]
- Thakur, A.; Mikkelsen, H.; Jungersen, G. Intracellular Pathogens: Host Immunity and Microbial Persistence Strategies. J. Immunol. Res. 2019, 2019, 1356540. [Google Scholar] [CrossRef]
- Soares, M.P.; Teixeira, L.; Moita, L.F. Disease tolerance and immunity in host protection against infection. Nat. Rev. Immunol. 2017, 17, 83–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vreman, H.J.; Ekstrand, B.C.; Stevenson, D.K. Selection of metalloporphyrin heme oxygenase inhibitors based on potency and photoreactivity. Pediatr. Res. 1993, 33, 195–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appleton, S.D.; Chretien, M.L.; McLaughlin, B.E.; Vreman, H.J.; Stevenson, D.K.; Brien, J.F.; Nakatsu, K.; Maurice, D.H.; Marks, G.S. Selective inhibition of heme oxygenase, without inhibition of nitric oxide synthase or soluble guanylyl cyclase, by metalloporphyrins at low concentrations. Drug Metab. Dispos. 1999, 27, 1214–1219. [Google Scholar] [PubMed]
- Salerno, L.; Floresta, G.; Ciaffaglione, V.; Gentile, D.; Margani, F.; Turnaturi, R.; Rescifina, A.; Pittala, V. Progress in the development of selective heme oxygenase-1 inhibitors and their potential therapeutic application. Eur. J. Med. Chem. 2019, 167, 439–453. [Google Scholar] [CrossRef]
- Motterlini, R.; Otterbein, L.E. The therapeutic potential of carbon monoxide. Nat. Rev. Drug Discov. 2010, 9, 728–743. [Google Scholar] [CrossRef]
- Ollinger, R.; Wang, H.; Yamashita, K.; Wegiel, B.; Thomas, M.; Margreiter, R.; Bach, F.H. Therapeutic applications of bilirubin and biliverdin in transplantation. Antioxid. Redox Signal. 2007, 9, 2175–2185. [Google Scholar] [CrossRef]
- Ferrandiz, M.L.; Devesa, I. Inducers of heme oxygenase-1. Curr. Pharm. Des. 2008, 14, 473–486. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Compound Structure | Compound Name and Function | Infections with Intracellular Pathogens in which Compound was Tested |
|---|---|---|
 | Tin Protoporphyrin IX (dichloride) SnPPIX HO-1 activity inhibitor | M. abcessus—improves control of pathogen replication in vitro in THP-1 cells [192]. M. tuberculosis—improves control of pathogen replication in vivo in mice [194,195] and in vitro in mouse [195] and human [193] macrophages. L. donovani—improves control of pathogen replication in vivo in mice [223]. L. infantum chagasi—improves control of pathogen replication in vitro in dog macrophages [225]. T. cruzi—impairs control of pathogen replication in mice in vivo and in vitro in macrophages [231]. |
 | Tin Mesoporphyrin IX (dichloride) SnMP HO-1 activity inhibitor | L. Mexicana pifanoi—increases ROS production in vitro in infected mouse macrophages [221]. |
 | Zinc Protoporphyrin IX ZnPPIX HO-1 activity inhibitor | S. typhymurium—impairs control of pathogen replication in mice in vivo and in vitro in peritoneal macrophages and RAW 267 cells [197]. Improves control of pathogen replication in vitro in RAW 267 cells [199]. P. berghei ANKA—impairs survival in murine model of cerebral malaria [213]. T. cruzi—improves control of pathogen replication in mice in vivo [232]. T. gondii—impairs control of parasite replication in mice in vivo [233]. |
 | Cobalt Protoporphyrin XI (chloride) CoPPIX HO-1 inducer | S. typhymurium—improves control of pathogen replication in mice in vivo [198]. L. monocytogenes—decreases the rate of infection-induced abortion in mice [203]. B. pseudomallei—impairs control of pathogen replication in mice in vivo and in vitro in macrophages [206]. P. berghei ANKA—improves survival in murine model of cerebral malaria [213]. L. mexicana pifanoi—decreases ROS production in vitro in infected mouse macrophages [221]. L. infantum chagasi—impairs control of pathogen replication in vitro in mouse and human macrophages [222]. T. cruzi—improves control of pathogen replication in mice in vivo and in vitro in macrophages [231]. |
 | Hemin Ferriprotoporphyrin XI (chloride) HO-1 inducer | P. berghei ANKA—decreases inflammatory tissue damage in lungs of infected mice [215]. T. gondii—improves control of pathogen replication in mice in vivo [233]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, D.L.; Amaral, E.P.; Andrade, B.B.; Sher, A. Modulation of Inflammation and Immune Responses by Heme Oxygenase-1: Implications for Infection with Intracellular Pathogens. Antioxidants 2020, 9, 1205. https://doi.org/10.3390/antiox9121205
Costa DL, Amaral EP, Andrade BB, Sher A. Modulation of Inflammation and Immune Responses by Heme Oxygenase-1: Implications for Infection with Intracellular Pathogens. Antioxidants. 2020; 9(12):1205. https://doi.org/10.3390/antiox9121205
Chicago/Turabian StyleCosta, Diego L., Eduardo P. Amaral, Bruno B. Andrade, and Alan Sher. 2020. "Modulation of Inflammation and Immune Responses by Heme Oxygenase-1: Implications for Infection with Intracellular Pathogens" Antioxidants 9, no. 12: 1205. https://doi.org/10.3390/antiox9121205