Vasoconstrictor Mechanisms in Chronic Hypoxia-Induced Pulmonary Hypertension: Role of Oxidant Signaling


Abstract
:1. Introduction
2. ROS in the Pathogenesis of PH
2.1. Increased ROS Production in PH
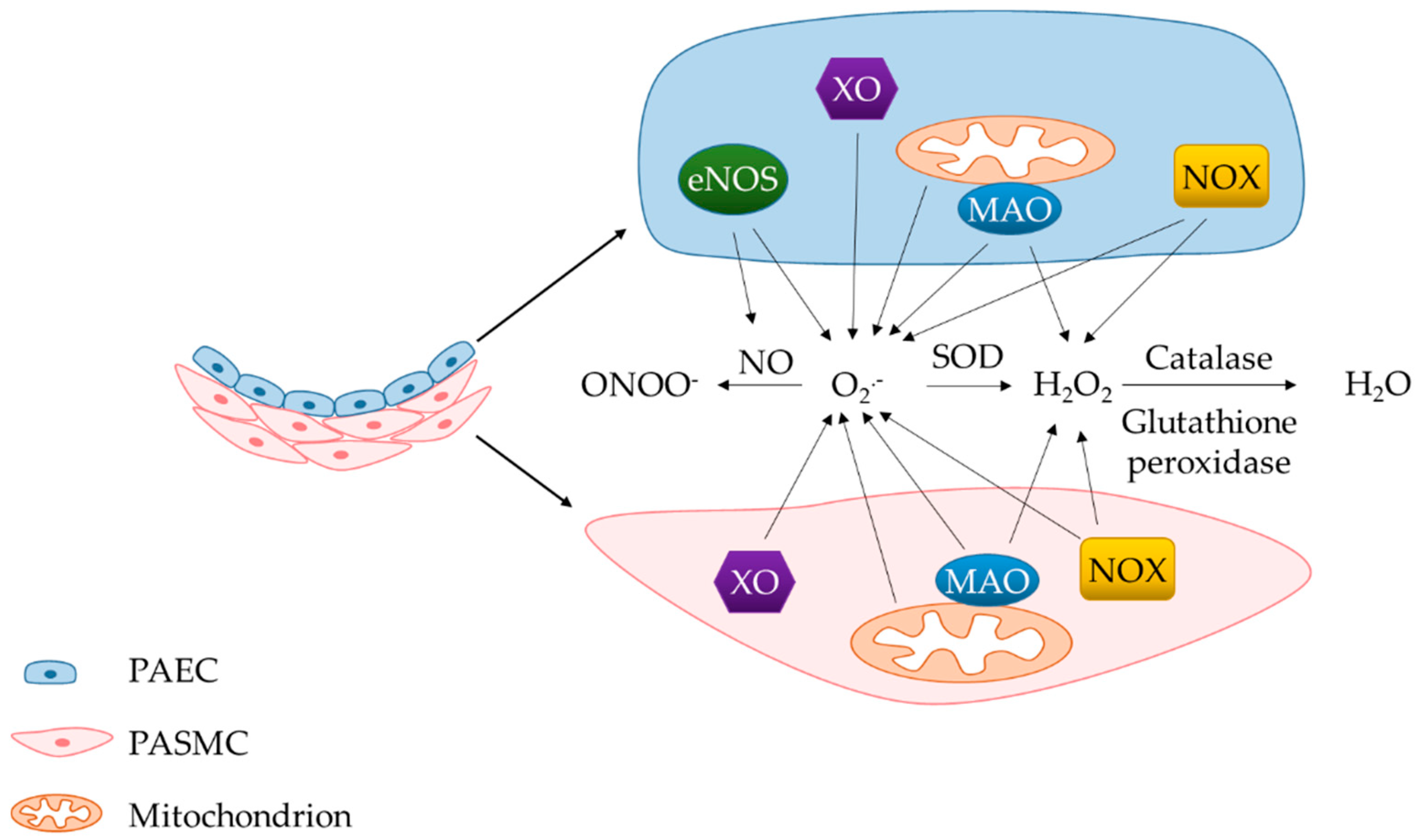
2.1.1. NADPH Oxidase Family
2.1.2. Mitochondria
2.1.3. Endothelial Nitric Oxide Synthase
2.1.4. Xanthine Oxidase
2.1.5. Monoamine Oxidases
2.2. Decreased Antioxidant Capacity in PH
2.2.1. SOD1
2.2.2. SOD2
2.2.3. SOD3
3. ROS Modulation of Augmented PA Constriction
3.1. ROS Modulation of Ca2+-Dependent Vasoconstriction
3.1.1. Ca2+ Influx
Voltage-Gated Calcium Channels
Transient Receptor Potential Canonical 1 and 6 (TRPC 1 and 6) Channels
Transient Receptor Potential Vanilloid 4 (TRPV4) Channels
Acid-Sensing Ion Channel 1 (ASIC1)
Orai/STIM
Mechanosensitive Channels (Aka Stretch-Activated Channels)
3.1.2. K+ Efflux

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Model | CH Protocol | Down-Regulation | Functional Outcomes | Ref. |
|---|---|---|---|---|
| Primary PASMCs from normal rats, unspecified strain | Hypoxia (3% O2, 5% CO2, and 92% N2), Normoxia (5% CO2 in air), 48–72 h | KV1.2, KV1.5 | N/A | [247] |
| Male Wistar rats | Hypoxia (10±0.5 % O2), Normoxia (room air), 21 days | KV1.1, KV1.5, KV1.6, KV2.1, and KV4.3 | N/A | [248] |
| Primary PASMCs from male SD rats | Hypoxia (1% O2, 5% CO2, balance N2), Normoxia (5% CO2 in air), 48–72 h | KV1.5 and KV2.1 | Decreased KV currents | [249] |
| Primary PASMCs from normal SD rats | Hypoxia (3% O2, 5% CO2, and 92% N2), Normoxia (5% CO2 in air), 60–72 h | KV1.1, KV1.5, KV2.1, KV4.3, KV9.3 | Loss of channels causes decreased KV currents, membrane depolarization, increase cytosolic Ca2+ | [250] |
| Male Wistar rats | Hypoxia (10% O2, balance N2), Normoxia (air), 21 days | KV1.5 and KV2.1 in PAs | Expression restoration prevents elevation in mPAP, RV hypertrophy | [251] |
| Male SD rats | Hypoxia (10 % O2), Normoxia (room air), 14–21 days | KV1.5 and KV2.1 in PAs | Expression restoration reverses and prevents PH indices following CH, including mPAP, PVR, RV hypertrophy, PA remodeling | [252] |
| Male SD rats | Hypoxia (380 mmHg), Normoxia (718 mmHg), 28 days | KV1.5 and KV2.1 in PAs | Expression restoration (1) rescues CH-induced suppression of KV currents in PASMCs and (2) prevents RVSP elevation, RV hypertrophy and PA remodeling following CH | [253] |
| Female wild-type control mice (C57BL/6XCBA strain) | Hypoxia (10% O2), Normoxia (air), 14 days | N/A | KV7 activator dilates pre-constricted PAs and prevents PH indices following CH, including mRVP, RV hypertrophy and PA remodeling | [255] |
3.2 ROS Participate in Ca2+ Sensitization
3.2.1 Rho Kinase Mediates Enhanced Ca2+ Sensitization
3.2.2 ROS Regulation of RhoA/ROK Signaling
4. Conclusion/Perspective
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| [Ca2+]i | Intracellular Ca2+ level |
| 5-HT | Serotonin |
| ASIC | Acid-sensing ion channel |
| BH2 | Dihydrobiopterin |
| BH4 | Tetrahydrobiopterin |
| BKCa | Large conductance Ca2+-activated K+ channels |
| BNP | Brain natriuretic peptide |
| Ca2+ | Calcium |
| CH | Chronic hypoxia |
| COPD | Chronic obstructive pulmonary disease |
| CoQ | Coenzyme Q |
| DAG | Diacylglycerol |
| DTNB | 5,5′-Dithiobis(2-nitrobenzoic acid) |
| DTT | Dithiothreitol |
| EC | Endothelial cell |
| EGFR | Epidermal growth factor receptor |
| eNOS | Endothelial nitric oxide synthase |
| ET-1 | Endothelin-1 |
| ETC | Electron transport chain |
| FAD | Flavin adenine dinucleotide |
| GPCR | G protein coupled receptor |
| GSH | Reduced glutathione |
| GSSG | Oxidized glutathione |
| H2O2 | Hydrogen peroxide |
| HPV | Hypoxic pulmonary vasoconstriction |
| IKCa | Intermediate conductance Ca2+-activated K+ channels |
| IP3 | Inositol triphosphate |
| K+ | Potassium |
| K2P | Four transmembrane segments-2 pores K+ channels |
| KATP | ATP-sensitive K+ channels |
| KCa | Ca2+-activated K+ channels |
| KO | Knockout |
| KV | Voltage-gated K+ channels |
| MAO | Monoamine oxidase |
| MitoROS | Mitochondria-derived ROS |
| MLCK | Myosin light chain kinase |
| MLCP | Myosin light chain phosphatase |
| mPAP | Mean pulmonary arterial pressure |
| MSC | Mechanosensitive channel |
| MYPT1 | Myosin phosphatase target subunit 1 |
| Na+ | Sodium |
| NADPH | Reduced nicotinamide adenine dinucleotide phosphate |
| NO | Nitric oxide |
| NOX | NADPH oxidase |
| O2.− | Superoxide anion |
| ONOO− | Peroxynitrite ion |
| PA | Pulmonary artery |
| PAEC | Pulmonary arterial endothelial cell |
| PAH | Pulmonary arterial hypertension |
| PASMC | Pulmonary arterial smooth muscle cell |
| PEG-SOD | Polyethylene glycol-conjugated SOD |
| PH | Pulmonary hypertension |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| PLC | Phospholipase C |
| PVR | Pulmonary vascular resistance |
| rhACE2 | Recombinant human angiotensin converting enzyme type 2 |
| ROC | Receptor-operated channel |
| ROCE | Receptor-operated Ca2+ entry |
| ROK | Rho kinase |
| ROS | Reactive oxygen species |
| RV | Right ventricular |
| RVSP | Right ventricular systolic pressure |
| SKCa | Small conductance Ca2+-activated K+ channels |
| SMC | Smooth muscle cell |
| SOC | Store-operated channel |
| SOCE | Store-operated Ca2+ entry |
| SOD | Superoxide dismutase |
| SR | Sarcoplasmic reticulum |
| STIM | Stromal interaction molecule |
| TRPC | Transient receptor potential canonical |
| TRPV4 | Transient receptor potential vanilloid 4 |
| VGCC | Voltage-gated calcium channel |
| WHO | World health organization |
| WT | Wild-type |
| XDH | Xanthine dehydrogenase |
| XO | Xanthine oxidase |
| XOR | Xanthine oxidoreductase |
References
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Brown, L.M.; Chen, H.; Halpern, S.; Taichman, D.; McGoon, M.D.; Farber, H.W.; Frost, A.E.; Liou, T.G.; Turner, M.; Feldkircher, K.; et al. Delay in recognition of pulmonary arterial hypertension: Factors identified from the REVEAL Registry. Chest 2011, 140, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Jernigan, N.L.; Naik, J.S.; Weise-Cross, L.; Detweiler, N.D.; Herbert, L.M.; Yellowhair, T.R.; Resta, T.C. Contribution of reactive oxygen species to the pathogenesis of pulmonary arterial hypertension. PLoS ONE 2017, 12, e0180455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenmark, K.R.; Fagan, K.A.; Frid, M.G. Hypoxia-Induced Pulmonary Vascular Remodeling. Circ. Res. 2006, 99, 675–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, J.M.; Williams, D.R.; Thompson, A.A.R. Thin Air, Thick Vessels: Historical and Current Perspectives on Hypoxic Pulmonary Hypertension. Front. Med. (Lausanne) 2019, 6, 93. [Google Scholar] [CrossRef] [Green Version]
- Cahill, E.; Rowan, S.C.; Sands, M.; Banahan, M.; Ryan, D.; Howell, K.; McLoughlin, P. The pathophysiological basis of chronic hypoxic pulmonary hypertension in the mouse: Vasoconstrictor and structural mechanisms contribute equally. Exp. Physiol. 2012, 97, 796–806. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, L.A.; Sham, J.S.; Sylvester, J.T. Altered pulmonary vasoreactivity in the chronically hypoxic lung. Physiol. Res. 2000, 49, 549–560. [Google Scholar] [PubMed]
- Jernigan, N.L.; Resta, T.C. Calcium Homeostasis and Sensitization in Pulmonary Arterial Smooth Muscle. Microcirculation 2014, 21, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Resta, T.C.; Broughton, B.R.S.; Jernigan, N.L. Reactive oxygen species and RhoA signaling in vascular smooth muscle: Role in chronic hypoxia-induced pulmonary hypertension. Adv. Exp. Med. Biol. 2010, 661, 355–373. [Google Scholar] [CrossRef]
- Stenmark, K.R.; McMurtry, I.F. Vascular remodeling versus vasoconstriction in chronic hypoxic pulmonary hypertension: A time for reappraisal? Circ. Res. 2005, 97, 95–98. [Google Scholar] [CrossRef] [Green Version]
- Undem, C.; Luke, T.; Shimoda, L.A. Contribution of elevated intracellular calcium to pulmonary arterial myocyte alkalinization during chronic hypoxia. Pulm. Circ. 2016, 6, 93–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jernigan, N.L.; Walker, B.R.; Resta, T.C. Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+ sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L515–L529. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, G.; Ducret, T.; Marthan, R.; Savineau, J.-P.; Quignard, J.-F. Stretch-induced Ca2+ signalling in vascular smooth muscle cells depends on Ca2+ store segregation. Cardiovasc. Res. 2014, 103, 313–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Xu, C.; Zheng, Q.; Yang, K.; Lai, N.; Wang, T.; Wang, J.; Lu, W. Orai1, 2, 3 and STIM1 promote store-operated calcium entry in pulmonary arterial smooth muscle cells. Cell Death Discov. 2017, 3, 17074. [Google Scholar] [CrossRef] [Green Version]
- Jernigan, N.L.; Paffett, M.L.; Walker, B.R.; Resta, T.C. ASIC1 contributes to pulmonary vascular smooth muscle store-operated Ca2+ entry. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L271–L285. [Google Scholar] [CrossRef] [Green Version]
- Herbert, L.M.; Resta, T.C.; Jernigan, N.L. RhoA increases ASIC1a plasma membrane localization and calcium influx in pulmonary arterial smooth muscle cells following chronic hypoxia. Am. J. Physiol. Cell. Physiol. 2018, 314, C166–C176. [Google Scholar] [CrossRef]
- Jernigan, N.L.; Herbert, L.M.; Walker, B.R.; Resta, T.C. Chronic hypoxia upregulates pulmonary arterial ASIC1: A novel mechanism of enhanced store-operated Ca2+ entry and receptor-dependent vasoconstriction. Am. J. Physiol. Cell. Physiol. 2012, 302, C931–C940. [Google Scholar] [CrossRef]
- Norton, C.E.; Broughton, B.R.; Jernigan, N.L.; Walker, B.R.; Resta, T.C. Enhanced depolarization-induced pulmonary vasoconstriction following chronic hypoxia requires EGFR-dependent activation of NAD(P)H oxidase 2. Antioxid. Redox Signal. 2013, 18, 1777–1788. [Google Scholar] [CrossRef] [Green Version]
- Weise-Cross, L.; Sands, M.A.; Sheak, J.R.; Broughton, B.R.S.; Snow, J.B.; Bosc, L.V.G.; Jernigan, N.L.; Walker, B.R.; Resta, T.C. Actin polymerization contributes to enhanced pulmonary vasoconstrictor reactivity after chronic hypoxia. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H1011–H1021. [Google Scholar] [CrossRef]
- Broughton, B.R.S.; Jernigan, N.L.; Norton, C.E.; Walker, B.R.; Resta, T.C. Chronic hypoxia augments depolarization-induced Ca2+ sensitization in pulmonary vascular smooth muscle through superoxide-dependent stimulation of RhoA. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, L232–L242. [Google Scholar] [CrossRef] [Green Version]
- Broughton, B.R.S.; Walker, B.R.; Resta, T.C. Chronic hypoxia induces Rho kinase-dependent myogenic tone in small pulmonary arteries. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L797–L806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norton, C.E.; Sheak, J.R.; Yan, S.; Weise-Cross, L.; Jernigan, N.L.; Walker, B.R.; Resta, T.C. Augmented Pulmonary Vasoconstrictor Reactivity after Chronic Hypoxia Requires Src Kinase and Epidermal Growth Factor Receptor Signaling. Am. J. Respir. Cell Mol. Biol. 2020, 62, 61–73. [Google Scholar] [CrossRef]
- Budhiraja, R.; Tuder, R.M.; Hassoun, P.M. Endothelial Dysfunction in Pulmonary Hypertension. Circulation 2004, 109, 159–165. [Google Scholar] [CrossRef]
- Liu, J.Q.; Erbynn, E.M.; Folz, R.J. Chronic hypoxia-enhanced murine pulmonary vasoconstriction: Role of superoxide and gp91phox. Chest 2005, 128 (Suppl. S6), 594S–596S. [Google Scholar] [CrossRef]
- Mackay, C.E.; Shaifta, Y.; Snetkov, V.V.; Francois, A.A.; Ward, J.P.; A Knock, G. ROS-dependent activation of RhoA/Rho-kinase in pulmonary artery: Role of Src-family kinases and ARHGEF1. Free Radic. Biol. Med. 2017, 110, 316–331. [Google Scholar] [CrossRef] [Green Version]
- Jernigan, N.L.; Walker, B.R.; Resta, T.C. Endothelium-derived reactive oxygen species and endothelin-1 attenuate NO-dependent pulmonary vasodilation following chronic hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L801–L808. [Google Scholar] [CrossRef] [Green Version]
- Knock, G.A.; Snetkov, V.A.; Shaifta, Y.; Connolly, M.; Drndarski, S.; Noah, A.; Pourmahram, G.E.; Becker, S.; Aaronson, P.I.; Ward, J.P. Superoxide constricts rat pulmonary arteries via Rho-kinase-mediated Ca(2+) sensitization. Free Radic. Biol. Med. 2009, 46, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Plomaritas, D.R.; Herbert, L.M.; Yellowhair, T.R.; Resta, T.C.; Bosc, L.V.G.; Walker, B.R.; Jernigan, N.L. Chronic hypoxia limits H2O2-induced inhibition of ASIC1-dependent store-operated calcium entry in pulmonary arterial smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Adesina, S.E.; Kang, B.-Y.; Bijli, K.M.; Ma, J.; Cheng, J.; Murphy, T.C.; Hart, C.M.; Sutliff, R.L. Targeting mitochondrial reactive oxygen species to modulate hypoxia-induced pulmonary hypertension. Free Radic. Biol. Med. 2015, 87, 36–47. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.N.; Zhang, Y.; Codipilly, C.; Zaghloul, N.; Patel, D.; Wolin, M.; Miller, E.J. Extracellular Superoxide Dismutase Overexpression Can Reverse the Course of Hypoxia-Induced Pulmonary Hypertension. Mol. Med. 2012, 18, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.; Aschner, J.L.; Kaplowitz, M.R.; Summar, M.; Fike, C.D. Tetrahydrobiopterin oral therapy recouples eNOS and ameliorates chronic hypoxia-induced pulmonary hypertension in newborn pigs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L743–L753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmedal, B.; De Dam, M.Y.; Mulvany, M.J.; Simonsen, U. The superoxide dismutase mimetic, tempol, blunts right ventricular hypertrophy in chronic hypoxic rats. Br. J. Pharmacol. 2004, 141, 105–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fike, C.D.; Dikalova, A.; Slaughter, J.C.; Kaplowitz, M.; Zhang, Y.; Aschner, J.L. Reactive Oxygen Species-Reducing Strategies Improve Pulmonary Arterial Responses to Nitric Oxide in Piglets with Chronic Hypoxia-Induced Pulmonary Hypertension. Antioxid. Redox Signal. 2013, 18, 1727–1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, B.N.; Hale, A.; Channon, K.M.; Wilkins, M.R.; Zhao, L. Effects of tetrahydrobiopterin oral treatment in hypoxia-induced pulmonary hypertension in rat. Pulm. Circ. 2014, 4, 462–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshikawa, Y.; Ono, S.; Suzuki, S.; Tanita, T.; Chida, M.; Song, C.; Noda, M.; Tabata, T.; Voelkel, N.F.; Fujimura, S. Generation of oxidative stress contributes to the development of pulmonary hypertension induced by hypoxia. J. Appl. Physiol. 2001, 90, 1299–1306. [Google Scholar] [CrossRef] [Green Version]
- Jankov, R.P.; Kantores, C.; Pan, J.; Belik, J. Contribution of xanthine oxidase-derived superoxide to chronic hypoxic pulmonary hypertension in neonatal rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L233–L245. [Google Scholar] [CrossRef]
- Villegas, L.R.; Kluck, D.; Field, C.; Oberley-Deegan, R.E.; Woods, C.; Yeager, M.E.; El Kasmi, K.C.; Savani, R.C.; Bowler, R.P.; Nozik-Grayck, E. Superoxide Dismutase Mimetic, MnTE-2-PyP, Attenuates Chronic Hypoxia-Induced Pulmonary Hypertension, Pulmonary Vascular Remodeling, and Activation of the NALP3 Inflammasome. Antioxid. Redox Signal. 2013, 18, 1753–1764. [Google Scholar] [CrossRef] [Green Version]
- Fukai, T.; Ushio-Fukai, M. Superoxide Dismutases: Role in Redox Signaling, Vascular Function, and Diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petry, A.; Djordjevic, T.; Weitnauer, M.; Kietzmann, T.; Hess, J.; Görlach, A. NOX2 and NOX4 Mediate Proliferative Response in Endothelial Cells. Antioxid. Redox Signal. 2006, 8, 1473–1484. [Google Scholar] [CrossRef]
- Abid, M.R.; Kachra, Z.; Spokes, K.C.; Aird, W.C. NADPH oxidase activity is required for endothelial cell proliferation and migration. FEBS Lett. 2000, 486, 252–256. [Google Scholar] [CrossRef] [Green Version]
- Clempus, R.E.; Sorescu, D.; Dikalova, A.E.; Pounkova, L.; Jo, P.; Sorescu, G.P.; Schmidt, H.H.H.; Lassègue, B.; Griendling, K.K. Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arter. Thromb. Vasc. Biol. 2007, 27, 42–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yada, T. Hydrogen Peroxide, an Endogenous Endothelium-Derived Hyperpolarizing Factor, Plays an Important Role in Coronary Autoregulation In Vivo. Circulation 2003, 107, 1040–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sindler, A.L.; Reyes, R.; Chen, B.; Ghosh, P.; Gurovich, A.N.; Kang, L.S.; Cardounel, A.J.; Delp, M.D.; Muller-Delp, J. Age and exercise training alter signaling through reactive oxygen species in the endothelium of skeletal muscle arterioles. J. Appl. Physiol. 2013, 114, 681–693. [Google Scholar] [CrossRef]
- Muñoz, M.; Martínez, M.P.; López-Oliva, M.E.; Rodríguez, C.; Corbacho, C.; Carballido, J.; García-Sacristán, A.; Hernández, M.; Rivera, L.; Medina, J.S.; et al. Hydrogen peroxide derived from NADPH oxidase 4- and 2 contributes to the endothelium-dependent vasodilatation of intrarenal arteries. Redox Biol. 2018, 19, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Drouin, A.; Thorin, E. Flow-induced dilation is mediated by Akt-dependent activation of endothelial nitric oxide synthase-derived hydrogen peroxide in mouse cerebral arteries. Stroke 2009, 40, 1827–1833. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, M.; Kelly, M.R.; Zhao, X.; Kandhi, S.; Wolin, M.S. Roles for Nox4 in the contractile response of bovine pulmonary arteries to hypoxia. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1879–H1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murtaza, G.; Paddenberg, R.; Pfeil, U.; Goldenberg, A.; Mermer, P.; Kummer, W. Hypoxia-induced pulmonary vasoconstriction of intra-acinar arteries is impaired in NADPH oxidase 4 gene-deficient mice. Pulm. Circ. 2018, 8, 2045894018808240. [Google Scholar] [CrossRef] [Green Version]
- Mak, S.; Egri, Z.; Tanna, G.; Colman, R.; Newton, G.E. Vitamin C prevents hyperoxia-mediated vasoconstriction and impairment of endothelium-dependent vasodilation. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H2414–H2421. [Google Scholar] [CrossRef] [Green Version]
- Rousseau, A.; Tesselaar, E.; Henricson, J.; Sjöberg, F. Prostaglandins and Radical Oxygen Species Are Involved in Microvascular Effects of Hyperoxia. J. Vasc. Res. 2010, 47, 441–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhilyaev, S.Y.; Moskvin, A.N.; Platonova, T.F.; Gutsaeva, D.R.; Churilina, I.V.; Demchenko, I.T. Hyperoxic vasoconstriction in the brain is mediated by inactivation of nitric oxide by superoxide anions. Neurosci. Behav. Physiol. 2003, 33, 783–787. [Google Scholar] [CrossRef]
- Burke, T.M.; Wolin, M.S. Hydrogen peroxide elicits pulmonary arterial relaxation and guanylate cyclase activation. Am. J. Physiol. Heart Circ. Physiol. 1987, 252 Pt 2, H721–H732. [Google Scholar] [CrossRef]
- Michelakis, E.D.; Hampl, V.; Nsair, A.; Wu, X.; Harry, G.; Haromy, A.; Gurtu, R.; Archer, S.L. Diversity in Mitochondrial Function Explains Differences in Vascular Oxygen Sensing. Circ. Res. 2002, 90, 1307–1315. [Google Scholar] [CrossRef]
- Neo, B.H.; Patel, D.; Kandhi, S.; Wolin, M.S. Roles for cytosolic NADPH redox in regulating pulmonary artery relaxation by thiol oxidation-elicited subunit dimerization of protein kinase G1α. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H330–H343. [Google Scholar] [CrossRef] [Green Version]
- Neo, B.H.; Kandhi, S.; Wolin, M.S. Roles for soluble guanylate cyclase and a thiol oxidation-elicited subunit dimerization of protein kinase G in pulmonary artery relaxation to hydrogen peroxide. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1235–H1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, D.; Alhawaj, R.; Wolin, M.S. Exposure of mice to chronic hypoxia attenuates pulmonary arterial contractile responses to acute hypoxia by increases in extracellular hydrogen peroxide. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R426–R433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudyk, O.; Rowan, A.; Prysyazhna, O.; Krasemann, S.; Hartmann, K.; Zhang, M.; Shah, A.M.; Ruppert, C.; Weiss, A.; Schermuly, R.T.; et al. Oxidation of PKGIα mediates an endogenous adaptation to pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2019, 116, 13016–13025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wedgwood, S.; Lakshminrusimha, S.; Fukai, T.; Russell, J.A.; Schumacker, P.T.; Steinhorn, R. Hydrogen Peroxide Regulates Extracellular Superoxide Dismutase Activity and Expression in Neonatal Pulmonary Hypertension. Antioxid. Redox Signal. 2011, 15, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Wedgwood, S.; Steinhorn, R.; Bunderson, M.; Wilham, J.; Lakshminrusimha, S.; Brennan, L.A.; Black, S.M. Increased hydrogen peroxide downregulates soluble guanylate cyclase in the lungs of lambs with persistent pulmonary hypertension of the newborn. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L660–L666. [Google Scholar] [CrossRef] [Green Version]
- Jernigan, N.L.; Resta, T.C.; Walker, B.R. Contribution of oxygen radicals to altered NO-dependent pulmonary vasodilation in acute and chronic hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L947–L955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nozik-Grayck, E.; Stenmark, K.R. Role of reactive oxygen species in chronic hypoxia-induced pulmonary hypertension and vascular remodeling. Adv. Exp. Med. Biol. 2007, 618, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Ravi, Y.; Selvendiran, K.; Naidu, S.K.; Meduru, S.; Citro, L.A.; Bognár, B.; Khan, M.; Kálai, T.; Hideg, K.; Kuppusamy, M.L.; et al. Pulmonary hypertension secondary to left-heart failure involves peroxynitrite-induced downregulation of PTEN in the lung. Hypertension 2013, 61, 593–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.-Y.; Zhao, Y.D.; Mirza, M.K.; Huang, J.H.; Potula, H.-H.S.; Vogel, S.M.; Brovkovych, V.; Yuan, J.X.-J.; Wharton, J.; Malik, A.B. Persistent eNOS activation secondary to caveolin-1 deficiency induces pulmonary hypertension in mice and humans through PKG nitration. J. Clin. Investig. 2009, 119, 2009–2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oishi, P.; Grobe, A.; Benavidez, E.; Ovadia, B.; Harmon, C.; Ross, G.A.; Hendricks-Munoz, K.; Xu, J.; Black, S.M.; Fineman, J.R. Inhaled nitric oxide induced NOS inhibition and rebound pulmonary hypertension: A role for superoxide and peroxynitrite in the intact lamb. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L359–L366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vara, D.; Pula, G. Reactive oxygen species: Physiological roles in the regulation of vascular cells. Curr. Mol. Med. 2014, 14, 1103–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Vizcaino, F.; Cogolludo, A.; Moreno, L. Reactive oxygen species signaling in pulmonary vascular smooth muscle. Respir. Physiol. Neurobiol. 2010, 174, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Raaz, U.; Toh, R.; Maegdefessel, L.; Adam, M.; Nakagami, F.; Emrich, F.C.; Spin, J.M.; Tsao, P.S. Hemodynamic Regulation of Reactive Oxygen Species: Implications for Vascular Diseases. Antioxid. Redox Signal. 2014, 20, 914–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krylatov, A.V.; Maslov, L.N.; Voronkov, N.; Boshchenko, A.; Popov, S.V.; Gomez, L.; Wang, H.; Jaggi, A.S.; Downey, J.M. Reactive Oxygen Species as Intracellular Signaling Molecules in the Cardiovascular System. Curr. Cardiol. Rev. 2018, 14, 290–300. [Google Scholar] [CrossRef]
- Burgoyne, J.R.; Mongue-Din, H.; Eaton, P.; Shah, A.M. Redox Signaling in Cardiac Physiology and Pathology. Circ. Res. 2012, 111, 1091–1106. [Google Scholar] [CrossRef]
- Burgoyne, J.R.; Oka, S.-I.; Ale-Agha, N.; Eaton, P. Hydrogen Peroxide Sensing and Signaling by Protein Kinases in the Cardiovascular System. Antioxid. Redox Signal. 2013, 18, 1042–1052. [Google Scholar] [CrossRef] [Green Version]
- Spickett, C.M.; Pitt, A.R.; Morrice, N.; Kolch, W. Proteomic analysis of phosphorylation, oxidation and nitrosylation in signal transduction. Biochim. Biophys. Acta 2006, 1764, 1823–1841. [Google Scholar] [CrossRef]
- Tang, H.; Viola, H.M.; Filipovska, A.; Hool, L.C. Cav1.2 calcium channel is glutathionylated during oxidative stress in guinea pig and ischemic human heart. Free Radic. Biol. Med. 2011, 51, 1501–1511. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, V.P.A.; Hool, L.C. Glutathionylation of the L-type Ca2+ Channel in Oxidative Stress-Induced Pathology of the Heart. Int. J. Mol. Sci. 2014, 15, 19203–19225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, B.J.; Irrinki, K.M.; Mallilankaraman, K.; Lien, Y.-C.; Wang, Y.; Bhanumathy, C.D.; Subbiah, R.; Ritchie, M.F.; Soboloff, J.; Baba, Y.; et al. S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J. Cell Biol. 2010, 190, 391–405. [Google Scholar] [CrossRef] [Green Version]
- Zha, X.-M.; Wang, R.; Collier, D.M.; Snyder, P.M.; Wemmie, J.A.; Welsh, M.J. Oxidant regulated inter-subunit disulfide bond formation between ASIC1a subunits. Proc. Natl. Acad. Sci. USA 2009, 106, 3573–3578. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Hernandez-Enriquez, B.; Banas, M.; Xu, R.; Sesti, F. Molecular Mechanisms Underlying the Apoptotic Effect of KCNB1 K+ Channel Oxidation. J. Biol. Chem. 2013, 288, 4128–4134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotella, D.; Hernandez-Enriquez, B.; Wu, X.; Li, R.; Pan, Z.; Leveille, J.; Link, C.D.; Oddo, S.; Sesti, F. Toxic Role of K+ Channel Oxidation in Mammalian Brain. J. Neurosci. 2012, 32, 4133–4144. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.; Raines, K.W.; Mocanu, V.; Campbell, S.L. Redox Regulation of RhoA. Biochemistry 2006, 45, 14481–14489. [Google Scholar] [CrossRef]
- Svoboda, L.K.; Reddie, K.G.; Zhang, L.; Vesely, E.D.; Williams, E.S.; Schumacher, S.M.; O’Connell, R.P.; Shaw, R.M.; Day, S.M.; Anumonwo, J.M.; et al. Redox-sensitive sulfenic acid modification regulates surface expression of the cardiovascular voltage-gated potassium channel Kv1.5. Circ. Res. 2012, 111, 842–853. [Google Scholar] [CrossRef] [Green Version]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front. Cell Infect. Microbiol. 2017, 7, 373. [Google Scholar] [CrossRef] [PubMed]
- Menden, H.; Tate, E.; Hogg, N.; Sampath, V. LPS-mediated endothelial activation in pulmonary endothelial cells: Role of Nox2-dependent IKK-β phosphorylation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L445–L455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pendyala, S.; A Gorshkova, I.; Usatyuk, P.V.; He, D.; Pennathur, A.; Lambeth, J.D.; Thannickal, V.J.; Natarajan, V. Role of Nox4 and Nox2 in Hyperoxia-Induced Reactive Oxygen Species Generation and Migration of Human Lung Endothelial Cells. Antioxid. Redox Signal. 2009, 11, 747–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghouleh, I.A.; Sahoo, S.; Meijles, D.N.; Amaral, J.H.; De Jesus, D.S.; Sembrat, J.; Rojas, M.; Goncharov, D.A.; Goncharova, E.A.; Pagano, P.J. Endothelial Nox1 oxidase assembly in human pulmonary arterial hypertension; driver of Gremlin1-mediated proliferation. Clin. Sci. (Lond.) 2017, 131, 2019–2035. [Google Scholar] [CrossRef] [PubMed]
- Chignalia, A.Z.; Weinberg, G.; Dull, R. Norepinephrine Induces Lung Microvascular Endothelial Cell Death by NADPH Oxidase-Dependent Activation of Caspase-3. Oxidative Med. Cell. Longev. 2020, 2020, 2563764. [Google Scholar] [CrossRef]
- Hood, K.Y.; Mair, K.M.; Harvey, A.P.; Montezano, A.C.; Touyz, R.M.; MacLean, M.R. Serotonin Signaling Through the 5-HT(1B) Receptor and NADPH Oxidase 1 in Pulmonary Arterial Hypertension. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1361–1370. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Ji, W.; Mi, L.; Lin, C. Mechanisms of N-acetylcysteine in reducing monocrotaline-induced pulmonary hypertension in rats: Inhibiting the expression of Nox1 in pulmonary vascular smooth muscle cells. Mol. Med. Rep. 2017, 16, 6148–6155. [Google Scholar] [CrossRef] [Green Version]
- Veit, F.; Pak, O.; Egemnazarov, B.; Roth, M.; Kosanovic, D.; Seimetz, M.; Sommer, N.; Ghofrani, H.-A.; Seeger, W.; Grimminger, F.; et al. Function of NADPH Oxidase 1 in Pulmonary Arterial Smooth Muscle Cells After Monocrotaline-Induced Pulmonary Vascular Remodeling. Antioxid. Redox Signal. 2013, 19, 2213–2231. [Google Scholar] [CrossRef]
- Iwata, K.; Ikami, K.; Matsuno, K.; Yamashita, T.; Shiba, D.; Ibi, M.; Misaki, M.; Masato, K.; Wenhao, C.; Jia, Z.; et al. Deficiency of NOX1/Nicotinamide Adenine Dinucleotide Phosphate, Reduced Form Oxidase Leads to Pulmonary Vascular Remodeling. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Hendricks, K.; To, E.; Vlahos, R.; Broughton, B.; Peshavariya, H.; Selemidis, S. Influenza A virus causes vascular endothelial cell oxidative stress via NOX2 oxidase. Eur. Respir. J. 2016, 48 (Suppl. S60), PA3967. [Google Scholar] [CrossRef]
- Mittal, M.; Roth, M.; König, P.; Hofmann, S.; Dony, E.; Goyal, P.; Selbitz, A.-C.; Schermuly, R.T.; Ghofrani, H.A.; Kwapiszewska, G.; et al. Hypoxia-Dependent Regulation of Nonphagocytic NADPH Oxidase Subunit NOX4 in the Pulmonary Vasculature. Circ. Res. 2007, 101, 258–267. [Google Scholar] [CrossRef]
- Gandhirajan, R.K.; Meng, S.; Chandramoorthy, H.C.; Mallilankaraman, K.; Mancarella, S.; Gao, H.; Razmpour, R.; Yang, X.-F.; Houser, S.R.; Chen, J.; et al. Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation. J. Clin. Investig. 2013, 123, 887–902. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Luo, X.-J.; Wang, E.-L.; Li, N.-S.; Zhang, X.-J.; Song, F.-L.; Yang, J.-F.; Liu, B.; Peng, J. Magnesium lithospermate B prevents phenotypic transformation of pulmonary arteries in rats with hypoxic pulmonary hypertension through suppression of NADPH oxidase. Eur. J. Pharmacol. 2019, 847, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Bánfi, B.; Malgrange, B.; Knisz, J.; Steger, K.; Dubois-Dauphin, M.; Krause, K.-H. NOX3, a Superoxide-generating NADPH Oxidase of the Inner Ear. J. Biol. Chem. 2004, 279, 46065–46072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goitre, L.; Distefano, P.V.; Moglia, A.; Nobiletti, N.; Baldini, E.; Trabalzini, L.; Keubel, J.; Trapani, E.; Shuvaev, V.V.; Muzykantov, V.R.; et al. Up-regulation of NADPH oxidase-mediated redox signaling contributes to the loss of barrier function in KRIT1 deficient endothelium. Sci. Rep. 2017, 7, 8296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schröder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 Is a Protective Reactive Oxygen Species Generating Vascular NADPH Oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ismail, S.; Sturrock, A.; Wu, P.; Cahill, B.; Norman, K.; Huecksteadt, T.; Sanders, K.; Kennedy, T.; Hoidal, J. NOX4 mediates hypoxia-induced proliferation of human pulmonary artery smooth muscle cells: The role of autocrine production of transforming growth factor-β1 and insulin-like growth factor binding protein-3. Am. J. Physchol. Lung Cell. Mol. Physchol. 2009, 296, L489–L499. [Google Scholar] [CrossRef] [PubMed]
- Diebold, I.; Petry, A.; Hess, J.; Görlach, A. The NADPH Oxidase Subunit NOX4 Is a New Target Gene of the Hypoxia-inducible Factor-1. Mol. Biol. Cell 2010, 21, 2087–2096. [Google Scholar] [CrossRef] [Green Version]
- Hood, K.Y.; Montezano, A.C.; Harvey, A.P.; Nilsen, M.; MacLean, M.R.; Touyz, R.M. Nicotinamide Adenine Dinucleotide Phosphate Oxidase-Mediated Redox Signaling and Vascular Remodeling by 16α-Hydroxyestrone in Human Pulmonary Artery Cells: Implications in Pulmonary Arterial Hypertension. Hypertension 2016, 68, 796–808. [Google Scholar] [CrossRef]
- Martyn, K.D.; Frederick, L.M.; Von Loehneysen, K.; Dinauer, M.C.; Knaus, U.G. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell. Signal. 2006, 18, 69–82. [Google Scholar] [CrossRef]
- Nisimoto, Y.; Jackson, H.M.; Ogawa, H.; Kawahara, T.; Lambeth, J.D. Constitutive NADPH-Dependent Electron Transferase Activity of the Nox4 Dehydrogenase Domain. Biochemistry 2010, 49, 2433–2442. [Google Scholar] [CrossRef]
- Nisimoto, Y.; Diebold, B.A.; Cosentino-Gomes, D.; Constentino-Gomes, D.; Lambeth, J.D. Nox4: A Hydrogen Peroxide-Generating Oxygen Sensor. Biochemistry 2014, 53, 5111–5120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Fan, Y.; Cui, J.; Hao, B.; Zhu, L.; Sun, X.; He, J.; Yang, J.; Dong, J.; Wang, Y.; et al. NOX4 expression and distal arteriolar remodeling correlate with pulmonary hypertension in COPD. BMC Pulm. Med. 2018, 18, 111. [Google Scholar] [CrossRef]
- Veith, C.; Kraut, S.; Wilhelm, J.; Sommer, N.; Quanz, K.; Seeger, W.; Brandes, R.P.; Weissmann, N.; Schröder, K. NADPH oxidase 4 is not involved in hypoxia-induced pulmonary hypertension. Pulm. Circ. 2016, 6, 397–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, R.; Murdoch, C.E.; Wang, M.; Santos, C.X.; Zhang, M.; Alom-Ruiz, S.; Anilkumar, N.; Ouattara, A.; Cave, A.C.; Walker, S.J.; et al. Endothelial Nox4 NADPH Oxidase Enhances Vasodilatation and Reduces Blood Pressure In Vivo. Arter. Thromb. Vasc. Biol. 2011, 31, 1368–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Mongue-Din, H.; Martin, D.; Catibog, N.; Smyrnias, I.; Zhang, X.; Yu, B.; Wang, M.; Brandes, R.P.; Schröder, K.; et al. Both cardiomyocyte and endothelial cell Nox4 mediate protection against hemodynamic overload-induced remodelling. Cardiovasc. Res. 2018, 114, 401–408. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J.A. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Miriyala, S.; Holley, A.K.; Clair, D.K.S. Mitochondrial superoxide dismutase--signals of distinction. Anti Cancer Agents Med. Chem. 2011, 11, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Al Shahrani, M.; Heales, S.J.; Hargreaves, I.; Orford, M. Oxidative Stress: Mechanistic Insights into Inherited Mitochondrial Disorders and Parkinson’s Disease. J. Clin. Med. 2017, 6, 100. [Google Scholar] [CrossRef]
- Handy, D.E.; Lubos, E.; Yang, Y.; Galbraith, J.D.; Kelly, N.; Zhang, Y.-Y.; Leopold, J.A.; Loscalzo, J. Glutathione Peroxidase-1 Regulates Mitochondrial Function to Modulate Redox-dependent Cellular Responses. J. Biol. Chem. 2009, 284, 11913–11921. [Google Scholar] [CrossRef] [Green Version]
- Yu, E.; Mercer, J.; Bennett, M.R. Mitochondria in vascular disease. Cardiovasc. Res. 2012, 95, 173–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, J.; Cederbaum, A.I. Mitochondrial Catalase and Oxidative Injury. Biol. Signals Recept. 2001, 10, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Suresh, K.; Servinsky, L.; Jiang, H.; Bigham, Z.; Yun, X.; Kliment, C.; Huetsch, J.; Damarla, M.; Shimoda, L.A. Reactive oxygen species induced Ca(2+) influx via TRPV4 and microvascular endothelial dysfunction in the SU5416/hypoxia model of pulmonary arterial hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L893–L907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheak, J.R.; Yan, S.; Weise-Cross, L.; Ahmadian, R.; Walker, B.R.; Jernigan, N.L.; Resta, T.C. PKCβ and reactive oxygen species mediate enhanced pulmonary vasoconstrictor reactivity following chronic hypoxia in neonatal rats. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H470–H483. [Google Scholar] [CrossRef] [PubMed]
- Coggins, M.P.; Bloch, K.D.; Zadelaar, S.; Kleemann, R.; Verschuren, L.; Weij, J.D.V.-V.D.; Van Der Hoorn, J.; Princen, H.M.; Kooistra, T. Nitric Oxide in the Pulmonary Vasculature. Arter. Thromb. Vasc. Biol. 2007, 27, 1877–1885. [Google Scholar] [CrossRef] [Green Version]
- Vásquez-Vivar, J.; Kalyanaraman, B.; Martásek, P.; Hogg, N.; Masters, B.S.S.; Karoui, H.; Tordo, P.; Pritchard, K.A. Superoxide generation by endothelial nitric oxide synthase: The influence of cofactors. Proc. Natl. Acad. Sci. USA 1998, 95, 9220–9225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, T.S.; Alp, N.J. Mechanisms for the role of tetrahydrobiopterin in endothelial function and vascular disease. Clin. Sci. (Lond.) 2007, 113, 47–63. [Google Scholar] [CrossRef] [Green Version]
- Griffith, O.W.; Stuehr, D.J. Nitric oxide synthases: Properties and catalytic mechanism. Annu. Rev. Physiol. 1995, 57, 707–736. [Google Scholar] [CrossRef] [PubMed]
- Habib, S.; Ali, A. Biochemistry of Nitric Oxide. Indian J. Clin. Biochem. IJCB 2011, 26, 3–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vásquez-Vivar, J.; Kalyanaraman, B.; Martasek, P. The role of tetrahydrobiopterin in superoxide generation from eNOS: Enzymology and physiological implications. Free Radic. Res. 2003, 37, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, M.J.; Tatham, A.L.; Al-Wakeel, Y.; Warrick, N.; Hale, A.B.; Cai, S.; Channon, K.M.; Alp, N.J. Quantitative Regulation of Intracellular Endothelial Nitric-oxide Synthase (eNOS) Coupling by Both Tetrahydrobiopterin-eNOS Stoichiometry and Biopterin Redox Status: Insights From Cells With Tet-Regulated Gtp Cyclohydrolase I Expression. J. Biol. Chem. 2009, 284, 1136–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crabtree, M.J.; Channon, K.M. Synthesis and recycling of tetrahydrobiopterin in endothelial function and vascular disease. Nitric Oxide Biol. Chem. 2011, 25, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Vásquez-Vivar, J.; Martásek, P.; Whitsett, J.; Joseph, J.; Kalyanaraman, B. The ratio between tetrahydrobiopterin and oxidized tetrahydrobiopterin analogues controls superoxide release from endothelial nitric oxide synthase: An EPR spin trapping study. Biochem. J. 2002, 362 Pt 3, 733–739. [Google Scholar] [CrossRef]
- Khoo, J.P.; Zhao, L.; Alp, N.J.; Bendall, J.K.; Nicoli, T.; Rockett, K.; Wilkins, M.R.; Channon, K.M.; Woo, A.; Williams, W.G.; et al. Pivotal Role for Endothelial Tetrahydrobiopterin in Pulmonary Hypertension. Circulation 2005, 111, 2126–2133. [Google Scholar] [CrossRef] [Green Version]
- Bevers, L.M.; Braam, B.; Post, J.A.; Van Zonneveld, A.J.; Rabelink, T.J.; Koomans, H.A.; Verhaar, M.; Joles, J.A. Tetrahydrobiopterin, but Not l -Arginine, Decreases NO Synthase Uncoupling in Cells Expressing High Levels of Endothelial NO Synthase. Hypertension 2006, 47, 87–94. [Google Scholar] [CrossRef] [Green Version]
- Edgar, K.S.; Galvin, O.M.; Collins, A.; Katusic, Z.S.; McDonald, D.M. BH4-Mediated Enhancement of Endothelial Nitric Oxide Synthase Activity Reduces Hyperoxia-Induced Endothelial Damage and Preserves Vascular Integrity in the Neonate. Investig. Opthalmology Vis. Sci. 2017, 58, 230–241. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, C.; Eilenberg, M.S.; Panzenboeck, A.; Winter, M.P.; Bergmeister, H.; Herzog, R.; Mascherbauer, J.; Lang, I.M.; Bonderman, D. Combined oral administration of L-arginine and tetrahydrobiopterin in a rat model of pulmonary arterial hypertension. Pulm. Circ. 2017, 7, 89–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheehy, A.M.; Burson, M.A.; Black, S.M. Nitric oxide exposure inhibits endothelial NOS activity but not gene expression: A role for superoxide. Am. J. Physiol. Lung Cell. Mol. Physiol. 1998, 274, L833–L841. [Google Scholar] [CrossRef] [PubMed]
- Wedgwood, S.; McMullan, D.M.; Bekker, J.M.; Fineman, J.R.; Black, S.M. Role for endothelin-1-induced superoxide and peroxynitrite production in rebound pulmonary hypertension associated with inhaled nitric oxide therapy. Circ. Res. 2001, 89, 357–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, O.; Tang, S.; Keech, A.; Celermajer, D. Rebound pulmonary hypertension on withdrawal from inhaled nitric oxide. Lancet 1995, 346, 51–52. [Google Scholar] [CrossRef]
- Atz, A.M.; Adatia, I.; Wessel, D.L. Rebound Pulmonary Hypertension After Inhalation of Nitric Oxide. Ann. Thorac. Surg. 1996, 62, 1759–1764. [Google Scholar] [CrossRef]
- Kostić, D.A.; Dimitrijević, D.S.; Stojanović, G.S.; Palić, I.R.; Đorđević, A.; Ickovski, J. Xanthine Oxidase: Isolation, Assays of Activity, and Inhibition. J. Chem. 2015, 2015, 294858. [Google Scholar] [CrossRef] [Green Version]
- Cantu-Medellin, N.; Kelley, E.E. Xanthine oxidoreductase-catalyzed reactive species generation: A process in critical need of reevaluation. Redox Biol. 2013, 1, 353–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwabara, Y.; Nishino, T.; Okamoto, K.; Matsumura, T.; Eger, B.T.; Pai, E.F.; Nishino, T. Unique amino acids cluster for switching from the dehydrogenase to oxidase form of xanthine oxidoreductase. Proc. Natl. Acad. Sci. USA 2003, 100, 8170–8175. [Google Scholar] [CrossRef] [Green Version]
- Battelli, M.G.; Polito, L.; Bortolotti, M.; Bolognesi, A. Xanthine Oxidoreductase in Drug Metabolism: Beyond a Role as a Detoxifying Enzyme. Curr. Med. Chem. 2016, 23, 4027–4036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Lü, J.-M.; Yao, Q. Hyperuricemia-Related Diseases and Xanthine Oxidoreductase (XOR) Inhibitors: An Overview. Med. Sci. Monit. 2016, 22, 2501–2512. [Google Scholar] [CrossRef] [Green Version]
- Kelley, E.E.; Khoo, N.K.; Hundley, N.J.; Malik, U.Z.; Freeman, B.A.; Tarpey, M.M. Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free Radic. Biol. Med. 2010, 48, 493–498. [Google Scholar] [CrossRef] [Green Version]
- Terada, L.S.; Guidot, D.M.; Leff, J.A.; Willingham, I.R.; Hanley, M.E.; Piermattei, D.; Repine, J.E. Hypoxia injures endothelial cells by increasing endogenous xanthine oxidase activity. Proc. Natl. Acad. Sci. USA 1992, 89, 3362–3366. [Google Scholar] [CrossRef] [Green Version]
- Cheong, P.L.S. Effects of Xanthine Oxidase Inhibitors in Pulmonary Hypertension Associated with Chronic Lung Disease. Ph.D. Thesis, University of Dundee, Dundee, UK, 2019. [Google Scholar]
- Thorpe, L.W.; Westlund, K.N.; Kochersperger, L.M.; Abell, C.W.; Denney, R.M. Immunocytochemical localization of monoamine oxidases A and B in human peripheral tissues and brain. J. Histochem. Cytochem. 1987, 35, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Shih, J.C.; Chen, K.; Ridd, M.J. Monoamine Oxidase: From Genes to Behavior. Annu. Rev. Neurosci. 1999, 22, 197–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowler, C.J.; Mantle, T.J.; Tipton, K.F. The nature of the inhibition of rat liver monoamine oxidase types A and B by the acetylenic inhibitors clorgyline, l-deprenyl and pargyline. Biochem. Pharmacol. 1982, 31, 3555–3561. [Google Scholar] [CrossRef]
- Green, A.R.; Youdim, M.B. Effects of Monoamine Oxidase Inhibition by Clorgyline, Deprenil or Tranylcypromine on 5-Hydroxytryptamine Concentrations in Rat Brain and Hyperactivity Following Subsequent Tryptophan Administration. Br. J. Pharmacol. 1975, 55, 415–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowler, C.J.; Benedetti, M.S. The Metabolism of Dopamine by Both Forms of Monoamine Oxidase in the Rat Brain and Its Inhibition by Cimoxatone. J. Neurochem. 1983, 40, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- O’Carroll, A.-M.; Fowler, C.J.; Phillips, J.P.; Tobbia, I.; Tipton, K.F. The deamination of dopamine by human brain monoamine oxidase. Specificity for the two enzyme forms in seven brain regions. Naunyn Schmiedeberg’s Arch. Pharmacol. 1983, 322, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Tsugeno, Y.; Ito, A. A Key Amino Acid Responsible for Substrate Selectivity of Monoamine Oxidase A and B. J. Biol. Chem. 1997, 272, 14033–14036. [Google Scholar] [CrossRef] [Green Version]
- Gaweska, H.; Fitzpatrick, P.F. Structures and mechanism of the monoamine oxidase family. Biomol. Concepts 2011, 2, 365–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peña-Silva, R.A.; Miller, J.D.; Chu, Y.; Heistad, D.D. Serotonin produces monoamine oxidase-dependent oxidative stress in human heart valves. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1354–H1360. [Google Scholar] [CrossRef] [Green Version]
- Zang, L.Y.; Misra, H.P. Generation of reactive oxygen species during the monoamine oxidase-catalyzed oxidation of the neurotoxicant, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J. Biol. Chem. 1993, 268, 16504–16512. [Google Scholar]
- Sandri, G.; Panfili, E.; Ernster, L. Hydrogen peroxide production by monoamine oxidase in isolated rat-brain mitochondria: Its effect on glutathione levels and Ca2+ efflux. Biochim. Biophys. Acta 1990, 1035, 300–305. [Google Scholar] [CrossRef]
- Maurel-Ribes, A.; Hernandez, C.; Kunduzova, O.; Bompart, G.; Cambon, C.; Parini, A.; Francès, B. Age-dependent increase in hydrogen peroxide production by cardiac monoamine oxidase A in rats. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1460–H1467. [Google Scholar] [CrossRef]
- Kunduzova, O.R.; Bianchi, P.; Parini, A.; Cambon, C. Hydrogen peroxide production by monoamine oxidase during ischemia/reperfusion. Eur. J. Pharmacol. 2002, 448, 225–230. [Google Scholar] [CrossRef]
- Simonson, S.G.; Zhang, J.; Canada, A.T.; Su, Y.-F.; Benveniste, H.; Piantadosi, C.A. Hydrogen Peroxide Production by Monoamine Oxidase during Ischemia-Reperfusion in the Rat Brain. J. Cereb. Blood Flow Metab. 1993, 13, 125–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Edmondson, D.E. Topological probes of monoamine oxidases A and B in rat liver mitochondria: Inhibition by TEMPO-substituted pargyline analogues and inactivation by proteolysis. Biochemistry 2011, 50, 2499–2505. [Google Scholar] [CrossRef] [Green Version]
- Mitoma, J.-Y.; Ito, A. Mitochondrial Targeting Signal of Rat Liver Monoamine Oxidase B Is Located at Its Carboxy Terminus. J. Biochem. 1992, 111, 20–24. [Google Scholar] [CrossRef] [Green Version]
- Son, S.-Y.; Ma, J.; Kondou, Y.; Yoshimura, M.; Yamashita, E.; Tsukihara, T. Structure of human monoamine oxidase A at 2.2-A resolution: The control of opening the entry for substrates/inhibitors. Proc. Natl. Acad. Sci. USA 2008, 105, 5739–5744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorato, E.; Menazza, S.; Zulian, A.; Sabatelli, P.; Gualandi, F.; Merlini, L.; Bonaldo, P.; Canton, M.; Bernardi, P.; Di Lisa, F. Monoamine oxidase inhibition prevents mitochondrial dysfunction and apoptosis in myoblasts from patients with collagen VI myopathies. Free Radic. Biol. Med. 2014, 75, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Kaludercic, N.; Carpi, A.; Nagayama, T.; Sivakumaran, V.; Zhu, G.; Lai, E.W.; Bedja, D.; De Mario, A.; Chen, K.; Gabrielson, K.L.; et al. Monoamine Oxidase B Prompts Mitochondrial and Cardiac Dysfunction in Pressure Overloaded Hearts. Antioxid. Redox Signal 2014, 20, 267–280. [Google Scholar] [CrossRef] [Green Version]
- Antonucci, S.; Di Sante, M.; Tonolo, F.; Pontarollo, L.; Scalcon, V.; Alanova, P.; Menabo’, R.; Carpi, A.; Bindoli, A.; Rigobello, M.P.; et al. The Determining Role of Mitochondrial Reactive Oxygen Species Generation and Monoamine Oxidase Activity in Doxorubicin-Induced Cardiotoxicity. Antioxid. Redox Signal. 2020. [Google Scholar] [CrossRef]
- Jana, S.; Sinha, M.; Chanda, D.; Roy, T.; Banerjee, K.; Munshi, S.; Patro, B.S.; Chakrabarti, S. Mitochondrial dysfunction mediated by quinone oxidation products of dopamine: Implications in dopamine cytotoxicity and pathogenesis of Parkinson’s disease. Biochim. Biophys. Acta 2011, 1812, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Peters, E.; Schalij, I.; Andersen, S.; Bos, D.D.S.G.; Noordegraaf, A.V.; De Man, F.; Van Der Laarse, W.; Bogaard, H. Inhibition of Monoamine Oxidase-A Reduces Pulmonary Vascular Remodeling in Experimentally Induced Pulmonary Arterial Hypertension, in B108. In Under Pressure: The Role of Cellular Stress in Pulmonary Vascular Remodeling; American Thoracic Society: New York, NY, USA, 2019; p. A4205. [Google Scholar] [CrossRef]
- Sun, X.; Peters, E.; Schalij, I.; Andersen, S.; Bos, D.D.S.G.; Vonk-Noordegraaf, A.; De Man, F.; Van Der Laarse, W.; Bogaard, H. The effect of Monoamine oxidase A inhibition on experimentally induced pulmonary arterial hypertension. Eur. Respir. J. 2018, 52 (Suppl. S62), PA3072. [Google Scholar] [CrossRef]
- Archer, S.L.; Marsboom, G.; Kim, G.H.; Zhang, H.J.; Toth, P.T.; Svensson, E.C.; Dyck, J.R.; Gomberg-Maitland, M.; Thébaud, B.; Husain, A.N.; et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: A basis for excessive cell proliferation and a new therapeutic target. Circulation 2010, 121, 2661–2671. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, S.; Michelakis, E.D.; Porter, C.J.; Andrade-Navarro, M.A.; Thébaud, B.; Bonnet, S.; Haromy, A.; Harry, G.; Moudgil, R.; McMurtry, M.S.; et al. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: Similarities to human pulmonary arterial hypertension. Circulation 2006, 113, 2630–2641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennis, K.E.; Aschner, J.L.; Milatović, D.; Schmidt, J.W.; Aschner, M.; Kaplowitz, M.R.; Zhang, Y.; Fike, C.D. NADPH oxidases and reactive oxygen species at different stages of chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L596–L607. [Google Scholar] [CrossRef] [PubMed]
- Ramiro-Diaz, J.M.; Nitta, C.H.; Maston, L.D.; Codianni, S.; Giermakowska, W.; Resta, T.C.; Bosc, L.V.G. NFAT is required for spontaneous pulmonary hypertension in superoxide dismutase 1 knockout mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L613–L625. [Google Scholar] [CrossRef] [Green Version]
- Brennan, L.A.; Steinhorn, R.; Wedgwood, S.; Mata-Greenwood, E.; Roark, E.A.; Russell, J.A.; Black, S.M. Increased Superoxide Generation Is Associated With Pulmonary Hypertension in Fetal Lambs. Circ. Res. 2003, 92, 683–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowers, R.; Cool, C.; Murphy, R.C.; Tuder, R.M.; Hopken, M.W.; Flores, S.C.; Voelkel, N.F. Oxidative Stress in Severe Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2004, 169, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Alhawaj, R.; Patel, D.; Kelly, M.R.; Sun, N.; Wolin, M.S. Heme biosynthesis modulation via δ-aminolevulinic acid administration attenuates chronic hypoxia-induced pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L719–L728. [Google Scholar] [CrossRef] [Green Version]
- Afolayan, A.J.; Eis, A.; Teng, R.-J.; Bakhutashvili, I.; Kaul, S.; Davis, J.M.; Konduri, G.G. Decreases in manganese superoxide dismutase expression and activity contribute to oxidative stress in persistent pulmonary hypertension of the newborn. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L870–L879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Xu, J. miR-140-5p regulates hypoxia-mediated human pulmonary artery smooth muscle cell proliferation, apoptosis and differentiation by targeting Dnmt1 and promoting SOD2 expression. Biochem. Biophys. Res. Commun. 2016, 473, 342–348. [Google Scholar] [CrossRef]
- Tseng, V.; Ni, K.; Allawzi, A.; Prohaska, C.; Hernandez-Lagunas, L.; Elajaili, H.; Cali, V.; Midura, R.; Hascall, V.; Triggs-Raine, B.; et al. Extracellular Superoxide Dismutase Regulates Early Vascular Hyaluronan Remodeling in Hypoxic Pulmonary Hypertension. Sci. Rep. 2020, 10, 280. [Google Scholar] [CrossRef] [Green Version]
- Hartney, T.; Birari, R.; Venkataraman, S.; Villegas, L.; Martinez, M.; Black, S.M.; Stenmark, K.R.; Nozik-Gryck, E. Xanthine oxidase-derived ROS upregulate Egr-1 via ERK1/2 in PA smooth muscle cells; model to test impact of extracellular ROS in chronic hypoxia. PLoS ONE 2011, 6, e27531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nozik-Grayck, E.; Woods, C.; Taylor, J.M.; Benninger, R.K.P.; Johnson, R.D.; Villegas, L.R.; Stenmark, K.R.; Harrison, D.G.; Majka, S.M.; Irwin, D.; et al. Selective depletion of vascular EC-SOD augments chronic hypoxic pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L868–L876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Guo, H.; Xu, X.; Chen, Y.; Fassett, J.; Hu, X.; Xu, Y.; Tang, Q.; Hu, D.; Somani, A.; et al. Exacerbated pulmonary arterial hypertension and right ventricular hypertrophy in animals with loss of function of extracellular superoxide dismutase. Hypertension 2011, 58, 303–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducret, T.; El Arrouchi, J.; Courtois, A.; Quignard, J.-F.; Marthan, R.; Savineau, J.-P. Stretch-activated channels in pulmonary arterial smooth muscle cells from normoxic and chronically hypoxic rats. Cell Calcium 2010, 48, 251–259. [Google Scholar] [CrossRef]
- Wang, J.; Weigand, L.; Lu, W.; Sylvester, J.; Semenza, G.L.; Shimoda, L.A. Hypoxia Inducible Factor 1 Mediates Hypoxia-Induced TRPC Expression and Elevated Intracellular Ca 2+ in Pulmonary Arterial Smooth Muscle Cells. Circ. Res. 2006, 98, 1528–1537. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Leung, G.P.; Zhang, W.-M.; Yang, X.-R.; Yip, K.-P.; Tse, C.-M.; Sham, J.S. Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: A novel mechanism of hypoxic pulmonary hypertension. Circ. Res. 2004, 95, 496–505. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, M.; Gilbert, G.; Roux, E.; Lory, P.; Marthan, R.; Savineau, J.-P.; Quignard, J.-F. T-type calcium channels are involved in hypoxic pulmonary hypertension. Cardiovasc. Res. 2014, 103, 597–606. [Google Scholar] [CrossRef] [Green Version]
- Shimoda, L.A.; Sham, J.S.K.; Shimoda, T.H.; Sylvester, J.T. L-type Ca(2+) channels, resting [Ca(2+)](i), and ET-1-induced responses in chronically hypoxic pulmonary myocytes. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L884–L894. [Google Scholar] [CrossRef]
- Nagaoka, T.; Morio, Y.; Casanova, N.; Bauer, N.; Gebb, S.; McMurtry, I.; Oka, M. Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L665–L672. [Google Scholar] [CrossRef]
- Weigand, L.; Sylvester, J.T.; Shimoda, L.A. Mechanisms of endothelin-1-induced contraction in pulmonary arteries from chronically hypoxic rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L284–L290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catterall, W.A. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef] [PubMed]
- Firth, A.L.; Remillard, C.V.; Platoshyn, O.; Fantozzi, I.; Ko, E.A.; Yuan, J.X.-J. Functional Ion Channels in Human Pulmonary Artery Smooth Muscle Cells: Voltage-Dependent Cation Channels. Pulm. Circ. 2011, 1, 48–71. [Google Scholar] [CrossRef] [Green Version]
- Makino, A.; Firth, A.L.; Yuan, J.X.-J. Endothelial and smooth muscle cell ion channels in pulmonary vasoconstriction and vascular remodeling. Compr. Physiol. 2011, 1, 1555–1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimoda, L.A.; Sylvester, J.T.; Sham, J.S.K. Chronic hypoxia alters effects of endothelin and angiotensin on K+ currents in pulmonary arterial myocytes. Am. J. Physiol. 1999, 277, L431–L439. [Google Scholar] [CrossRef]
- Reeve, H.L.; Michelakis, E.; Nelson, D.P.; Weir, E.K.; Archer, S.L. Alterations in a redox oxygen sensing mechanism in chronic hypoxia. J. Appl. Physiol. 2001, 90, 2249–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.X.-J.; Aldinger, A.M.; Juhaszova, M.; Wang, J.; Conte, J.V.; Gaine, S.P.; Orens, J.B.; Rubin, L.J. Dysfunctional Voltage-Gated K + Channels in Pulmonary Artery Smooth Muscle Cells of Patients With Primary Pulmonary Hypertension. Circulation 1998, 98, 1400–1406. [Google Scholar] [CrossRef] [Green Version]
- Lai, N.; Lu, W.; Wang, J. Ca2+ and ion channels in hypoxia-mediated pulmonary hypertension. Int. J. Clin. Exp. Pathol. 2015, 8, 1081–1092. [Google Scholar]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E.; Meyer, T. STIM Is a Ca2+ Sensor Essential for Ca2+-Store-Depletion-Triggered Ca2+ Influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [Green Version]
- Hewavitharana, T.; Deng, X.; Soboloff, J.; Gill, D.L. Role of STIM and Orai proteins in the store-operated calcium signaling pathway. Cell Calcium 2007, 42, 173–182. [Google Scholar] [CrossRef]
- Garcia, S.M.; Herbert, L.M.; Walker, B.R.; Resta, T.C.; Jernigan, N.L. Coupling of store-operated calcium entry to vasoconstriction is acid-sensing ion channel 1a dependent in pulmonary but not mesenteric arteries. PLoS ONE 2020, 15, e0236288. [Google Scholar] [CrossRef]
- Salido, G.M.; Jardin, I.; Rosado, J.A. The TRPC ion channels: Association with Orai1 and STIM1 proteins and participation in capacitative and non-capacitative calcium entry. Adv. Exp. Med. Biol. 2011, 704, 413–433. [Google Scholar] [CrossRef] [PubMed]
- Snow, J.B.; Kanagy, N.L.; Walker, B.R.; Resta, T.C. Rat Strain Differences in Pulmonary Artery Smooth Muscle Ca2+Entry Following Chronic Hypoxia. Microcirculation 2009, 16, 603–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirenallur-S, D.K.; Haworth, S.T.; Leming, J.T.; Chang, J.; Hernández, G.; Gordon, J.B.; Rusch, N.J. Upregulation of vascular calcium channels in neonatal piglets with hypoxia-induced pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L915–L924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, J.; Yamamura, A.; Zimnicka, A.M.; Voiriot, G.; Smith, K.A.; Tang, H.; Ayon, R.J.; Choudhury, M.S.R.; Ko, E.A.; Wang, J.; et al. Chronic hypoxia selectively enhances L- and T-type voltage-dependent Ca2+ channel activity in pulmonary artery by upregulating Cav1.2 and Cav3.2. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L154–L164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oka, M.; Morris, K.G.; McMurtry, I.F. NIP-121 is more effective than nifedipine in acutely reversing chronic pulmonary hypertension. J. Appl. Physiol. 1993, 75, 1075–1080. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.C.; Joshi, R.C.; Mehta, R.; Cunnington, A.R. Acute and long term effect of nifedipine on pulmonary hypertension secondary to chronic obstructive airways disease. Eur. J. Respir. Dis. Suppl. 1986, 146, 495–502. [Google Scholar]
- Brown, S.E.; Linden, G.S.; King, R.R.; Blair, G.P.; Stansbury, D.W.; Light, R.W. Effects of verapamil on pulmonary haemodynamics during hypoxaemia, at rest, and during exercise in patients with chronic obstructive pulmonary disease. Thorax 1983, 38, 840–844. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.-X.; Xu, X.-Y.; Zhao, Z.; Zhao, F.-Y.; Gao, Y.-M.; Yan, X.-H.; Wan, Y. Hydrogen peroxide is a critical regulator of the hypoxia-induced alterations of store-operated Ca2+ entry into rat pulmonary arterial smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L477–L487. [Google Scholar] [CrossRef]
- Malczyk, M.; Veith, C.; Fuchs, B.; Hofmann, K.; Storch, U.; Schermuly, R.T.; Witzenrath, M.; Ahlbrecht, K.; Fecher-Trost, C.; Flockerzi, V.; et al. Classical Transient Receptor Potential Channel 1 in Hypoxia-induced Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2013, 188, 1451–1459. [Google Scholar] [CrossRef]
- Xia, Y.; Yang, X.R.; Fu, Z.; Paudel, O.; Abramowitz, J.; Birnbaumer, L.; Sham, J.S. Classical transient receptor potential 1 and 6 contribute to hypoxic pulmonary hypertension through differential regulation of pulmonary vascular functions. Hypertension 2014, 63, 173–180. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.A.; Voiriot, G.; Tang, H.; Fraidenburg, D.R.; Song, S.; Yamamura, H.; Yamamura, A.; Guo, Q.; Wan, J.; Pohl, N.M.; et al. Notch Activation of Ca2+Signaling in the Development of Hypoxic Pulmonary Vasoconstriction and Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2015, 53, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.-R.; Lin, A.H.Y.; Hughes, J.M.; Flavahan, N.A.; Cao, Y.-N.; Liedtke, W.; Sham, J.S.K. Upregulation of osmo-mechanosensitive TRPV4 channel facilitates chronic hypoxia-induced myogenic tone and pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L555–L568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahan, D.; Ducret, T.; Quignard, J.-F.; Marthan, R.; Savineau, J.-P.; Estève, E. Implication of the ryanodine receptor in TRPV4-induced calcium response in pulmonary arterial smooth muscle cells from normoxic and chronically hypoxic rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L824–L833. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Fu, Z.; Hu, J.; Huang, C.; Paudel, O.; Cai, S.; Liedtke, W.; Sham, J.S.K. TRPV4 channel contributes to serotonin-induced pulmonary vasoconstriction and the enhanced vascular reactivity in chronic hypoxic pulmonary hypertension. Am. J. Physiol. Cell Physiol. 2013, 305, C704–C715. [Google Scholar] [CrossRef] [Green Version]
- Nitta, C.H.; Osmond, D.A.; Herbert, L.M.; Beasley, B.F.; Resta, T.C.; Walker, B.R.; Jernigan, N.L. Role of ASIC1 in the development of chronic hypoxia-induced pulmonary hypertension. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H41–H52. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Song, S.; Ayon, R.J.; Balisterieri, A.; Black, S.M.; Makino, A.; Gil Wier, W.; Zang, W.-J.; Yuan, J.X.-J. Hypoxia selectively upregulates cation channels and increases cytosolic [Ca2+] in pulmonary, but not coronary, arterial smooth muscle cells. Am. J. Physiol. Cell Physiol. 2018, 314, C504–C517. [Google Scholar] [CrossRef] [Green Version]
- Song, M.Y.; Makino, A.; Yuan, J.X.-J. STIM2 Contributes to Enhanced Store-Operated Ca2+ Entry in Pulmonary Artery Smooth Muscle Cells from Patients with Idiopathic Pulmonary Arterial Hypertension. Pulm. Circ. 2011, 1, 84–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, D.L.; Stamler, J.S.; Strauss, H.C. Redox modulation of L-type calcium channels in ferret ventricular myocytes. Dual mechanism regulation by nitric oxide and S-nitrosothiols. J. Gen. Physiol. 1996, 108, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Chiamvimonvat, N.; Yamagishi, T.; Marban, E. Direct inhibition of expressed cardiac L-type Ca2+ channels by S-nitrosothiol nitric oxide donors. Circ. Res. 1997, 81, 742–752. [Google Scholar] [CrossRef]
- Viola, H.M.; Arthur, P.G.; Hool, L.C. Transient Exposure to Hydrogen Peroxide Causes an Increase in Mitochondria-Derived Superoxide As a Result of Sustained Alteration in L-Type Ca2+ Channel Function in the Absence of Apoptosis in Ventricular Myocytes. Circ. Res. 2007, 100, 1036–1044. [Google Scholar] [CrossRef] [Green Version]
- Akaishi, T.; Nakazawa, K.; Sato, K.; Saito, H.; Ohno, Y.; Ito, Y. Hydrogen peroxide modulates whole cell Ca2+ currents through L-type channels in cultured rat dentate granule cells. Neurosci. Lett. 2004, 356, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xu, J.; Minobe, E.; Yu, L.; Feng, R.; Kameyama, A.; Yazawa, K.; Kameyama, M. Mechanisms underlying the modulation of L-type Ca2+ channel by hydrogen peroxide in guinea pig ventricular myocytes. J. Physiol. Sci. 2013, 63, 419–426. [Google Scholar] [CrossRef]
- Luke, T.; Maylor, J.; Undem, C.; Sylvester, J.T.; Shimoda, L.A. Kinase-dependent activation of voltage-gated Ca2+ channels by ET-1 in pulmonary arterial myocytes during chronic hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L1128–L1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosentino-Gomes, D.; Rocco-Machado, N.; Meyer-Fernandes, J.R. Cell Signaling through Protein Kinase C Oxidation and Activation. Int. J. Mol. Sci. 2012, 13, 10697–10721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Platoshyn, O.; Firth, A.L.; Yuan, J.X.-J. Hypoxia divergently regulates production of reactive oxygen species in human pulmonary and coronary artery smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L952–L959. [Google Scholar] [CrossRef] [Green Version]
- Wedgwood, S.; Black, S.M. Endothelin-1 decreases endothelial NOS expression and activity through ETA receptor-mediated generation of hydrogen peroxide. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 288, L480–L487. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Q.; Zhou, Q.; Yao, F.; O’Rourke, S.T.; Sun, C. Endothelin-1 Regulates Cardiac L-Type Calcium Channels via NAD(P)H Oxidase-Derived Superoxide. J. Pharmacol. Exp. Ther. 2008, 326, 732–738. [Google Scholar] [CrossRef] [Green Version]
- Weissmann, N.; Dietrich, A.; Fuchs, B.; Kalwa, H.; Ay, M.; Dumitrascu, R.; Olschewski, A.; Storch, U.; Schnitzler, M.M.Y.; Ghofrani, H.-A.; et al. Classical transient receptor potential channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proc. Natl. Acad. Sci. USA 2006, 103, 19093–19098. [Google Scholar] [CrossRef] [Green Version]
- Aires, V.; Hichami, A.; Boulay, G.; Khan, N.A. Activation of TRPC6 calcium channels by diacylglycerol (DAG)-containing arachidonic acid: A comparative study with DAG-containing docosahexaenoic acid. Biochimie 2007, 89, 926–937. [Google Scholar] [CrossRef]
- Weissmann, N.; Sydykov, A.; Kalwa, H.; Storch, U.; Fuchs, B.; Schnitzler, M.M.Y.; Brandes, R.P.; Grimminger, F.; Meissner, M.; Freichel, M.; et al. Activation of TRPC6 channels is essential for lung ischaemia–reperfusion induced oedema in mice. Nat. Commun. 2012, 3, 649. [Google Scholar] [CrossRef]
- Inoue, R.; Jensen, L.J.; Jian, Z.; Shi, J.; Hai, L.; Lurie, A.I.; Henriksen, F.H.; Salomonsson, M.; Morita, H.; Kawarabayashi, Y.; et al. Synergistic activation of vascular TRPC6 channel by receptor and mechanical stimulation via phospholipase C/diacylglycerol and phospholipase A2/omega-hydroxylase/20-HETE pathways. Circ. Res. 2009, 104, 1399–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Winters, A.; Ding, M.; Graham, S.; Akopova, I.; Muallem, S.; Wang, Y.; Hong, J.H.; Gryczynski, Z.; Yang, S.-H.; et al. Reactive Oxygen Species-mediated TRPC6 Protein Activation in Vascular Myocytes, a Mechanism for Vasoconstrictor-regulated Vascular Tone. J. Biol. Chem. 2011, 286, 31799–31809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marziano, C.; Hong, K.; Cope, E.L.; Kotlikoff, M.I.; Isakson, B.E.; Sonkusare, S.K. Nitric Oxide–Dependent Feedback Loop Regulates Transient Receptor Potential Vanilloid 4 (TRPV4) Channel Cooperativity and Endothelial Function in Small Pulmonary Arteries. J. Am. Heart Assoc. 2017, 6, e007157. [Google Scholar] [CrossRef] [Green Version]
- Sukumaran, S.V.; Singh, T.U.; Parida, S.; Reddy, C.N.; Thangamalai, R.; Kandasamy, K.; Singh, V.; Mishra, S.K. TRPV4 channel activation leads to endothelium-dependent relaxation mediated by nitric oxide and endothelium-derived hyperpolarizing factor in rat pulmonary artery. Pharmacol. Res. 2013, 78, 18–27. [Google Scholar] [CrossRef]
- Ottolini, M.; Daneva, Z.; Chen, Y.L.; Cope, E.L.; Kasetti, R.B.; Zode, G.S.; Sonkusare, S.K. Mechanisms underlying selective coupling of endothelial Ca(2+) signals with eNOS vs. IK/SK channels in systemic and pulmonary arteries. J. Physiol. 2020, 598, 3577–3596. [Google Scholar] [CrossRef] [PubMed]
- Cussac, L.-A.; Cardouat, G.; Pillai, N.T.; Campagnac, M.; Robillard, P.; Montillaud, A.; Guibert, C.; Gailly, P.; Marthan, R.; Quignard, J.-F.; et al. TRPV4 channel mediates adventitial fibroblast activation and adventitial remodeling in pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L135–L146. [Google Scholar] [CrossRef] [PubMed]
- Pankey, E.A.; Zsombok, A.; Lasker, G.F.; Kadowitz, P.J. Analysis of responses to the TRPV4 agonist GSK1016790A in the pulmonary vascular bed of the intact-chest rat. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, 33–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suresh, K.; Servinsky, L.; Reyes, J.; Baksh, S.; Undem, C.; Caterina, M.; Pearse, D.B.; Shimoda, L.A. Hydrogen peroxide-induced calcium influx in lung microvascular endothelial cells involves TRPV4. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L1467–L1477. [Google Scholar] [CrossRef] [Green Version]
- Suresh, K.; Servinsky, L.; Reyes, J.; Undem, C.; Zaldumbide, J.; Rentsendorj, O.; Modekurty, S.; Dodd-O, J.M.; Scott, A.L.; Pearse, D.B.; et al. CD36 mediates H2O2-induced calcium influx in lung microvascular endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L143–L153. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.; Anishkin, A.; Zinkevich, N.S.; Nishijima, Y.; Korishettar, A.; Wang, Z.; Fang, J.; Wilcox, D.A.; Zhang, D.X. Transient receptor potential vanilloid 4 (TRPV4) activation by arachidonic acid requires protein kinase A–mediated phosphorylation. J. Biol. Chem. 2018, 293, 5307–5322. [Google Scholar] [CrossRef] [Green Version]
- Suresh, K.; Servinsky, L.; Jiang, H.; Bigham, Z.; Zaldumbide, J.; Huetsch, J.C.; Kliment, C.; Acoba, M.G.; Kirsch, B.J.; Claypool, S.M.; et al. Regulation of mitochondrial fragmentation in microvascular endothelial cells isolated from the SU5416/hypoxia model of pulmonary arterial hypertension. Am. J. Physiol. Lung. Cell Mol. Physiol. 2019, 317, L639–L652. [Google Scholar] [CrossRef] [PubMed]
- Jernigan, N.L. Smooth muscle acid-sensing ion channel 1: Pathophysiological implication in hypoxic pulmonary hypertension. Exp. Physiol. 2015, 100, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, T.W.; Frey, E.N.; Askwith, C.C. Structure and activity of the acid-sensing ion channels. Am. J. Physiol. Cell Physiol. 2012, 303, C699–C710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, M.; Bera, A.K. Modulation of acid-sensing ion channels by hydrogen sulfide. Biochem. Biophys. Res. Commun. 2020, 527, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Andrey, F.; Tsintsadze, T.; Volkova, T.; Lozovaya, N.; Krishtal, O. Acid sensing ionic channels: Modulation by redox reagents. Biochim. Biophys. Acta 2005, 1745, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, J.-H.; Askwith, C.C. Potentiation of acid-sensing ion channels by sulfhydryl compounds. Am. J. Physiol. Cell Physiol. 2007, 292, C2161–C2174. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.-P.; Close, N.; Saugstad, J.A.; Xiong, Z.-G. ASIC1a-specific modulation of acid-sensing ion channels in mouse cortical neurons by redox reagents. J. Neurosci. 2006, 26, 5329–5339. [Google Scholar] [CrossRef] [Green Version]
- Mungai, P.T.; Waypa, G.B.; Jairaman, A.; Prakriya, M.; Dokic, D.; Ball, M.K.; Schumacker, P.T. Hypoxia Triggers AMPK Activation through Reactive Oxygen Species-Mediated Activation of Calcium Release-Activated Calcium Channels. Mol. Cell. Biol. 2011, 31, 3531–3545. [Google Scholar] [CrossRef] [Green Version]
- Dyachenko, V.; Rueckschloss, U.; Isenberg, G. Modulation of cardiac mechanosensitive ion channels involves superoxide, nitric oxide and peroxynitrite. Cell Calcium 2009, 45, 55–64. [Google Scholar] [CrossRef]
- Burg, E.D.; Remillard, C.V.; Yuan, J.X.-J. Potassium channels in the regulation of pulmonary artery smooth muscle cell proliferation and apoptosis: Pharmacotherapeutic implications. Br. J. Pharmacol. 2008, 153 (Suppl. S1), S99–S111. [Google Scholar] [CrossRef] [Green Version]
- Weise-Cross, L.; Resta, T.C.; Jernigan, N.L. Redox Regulation of Ion Channels and Receptors in Pulmonary Hypertension. Antioxid. Redox Signal. 2019, 31, 898–915. [Google Scholar] [CrossRef] [PubMed]
- Ayon, R.J.; Tang, H.; Fernandez, R.A.; Makino, A.; Yuan, J.X.-J. Smooth Muscle Cell Ion Channels in Pulmonary Arterial Hypertension: Pathogenic Role in Pulmonary Vasoconstriction and Vascular Remodeling. In Vascular Ion Channels in Physiology and Disease; Levitan, P.I., Dopico, M.D.P.A.M., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 295–324. [Google Scholar] [CrossRef]
- Doi, S.; Damron, D.S.; Ogawa, K.; Tanaka, S.; Horibe, M.; A Murray, P. K(+) channel inhibition, calcium signaling, and vasomotor tone in canine pulmonary artery smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L242–L251. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Balan, P.; Gurney, A.M. Pulmonary vasoconstrictor action of KCNQ potassium channel blockers. Respir. Res. 2006, 7, 31. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Juhaszova, M.; Rubin, L.J.; Yuan, X.J. Hypoxia inhibits gene expression of voltage-gated K+ channel alpha subunits in pulmonary artery smooth muscle cells. J. Clin. Investig. 1997, 100, 2347–2353. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Weigand, L.; Wang, W.; Sylvester, J.T.; Shimoda, L.A. Chronic hypoxia inhibits Kv channel gene expression in rat distal pulmonary artery. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 288, L1049–L1058. [Google Scholar] [CrossRef]
- Mittal, M.; Gu, X.Q.; Pak, O.; Pamenter, M.E.; Haag, D.; Fuchs, D.B.; Schermuly, R.T.; Ghofrani, H.-A.; Brandes, R.P.; Seeger, W.; et al. Hypoxia induces Kv channel current inhibition by increased NADPH oxidase-derived reactive oxygen species. Free Radic. Biol. Med. 2012, 52, 1033–1042. [Google Scholar] [CrossRef]
- Platoshyn, O.; Yu, Y.; Golovina, V.A.; McDaniel, S.S.; Krick, S.; Li, L.; Wang, J.-Y.; Rubin, L.J.; Yuan, J.X.-J. Chronic hypoxia decreases KV channel expression and function in pulmonary artery myocytes. Am. J. Physiol. Cell. Mol. Physiol. 2001, 280, L801–L812. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Liu, B.; Zhang, S.; Liu, H.; Li, Y.; Wang, N.; Liu, Y.; Li, J.; Wang, N.; Liu, Y.; et al. YM155, a selective survivin inhibitor, reverses chronic hypoxic pulmonary hypertension in rats via upregulating voltage-gated potassium channels. Clin. Exp. Hypertens. 2015, 37, 381–387. [Google Scholar] [CrossRef]
- Michelakis, E.D.; McMurtry, M.S.; Wu, X.-C.; Dyck, J.R.B.; Moudgil, R.; Hopkins, T.A.; Lopaschuk, G.D.; Puttagunta, L.; Waite, R.; Archer, S.L. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: Role of increased expression and activity of voltage-gated potassium channels. Circulation 2002, 105, 244–250. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Liu, M.; Wei, M.; Liu, Y.; Dong, M.; Luo, Y.; Zhao, P.; Dong, H.; Niu, W.; Yan, Z.; et al. Tanshinone IIA attenuates hypoxic pulmonary hypertension via modulating KV currents. Respir. Physiol. Neurobiol. 2015, 205, 120–128. [Google Scholar] [CrossRef]
- Frazzini, V.; Guarnieri, S.; Bomba, M.; Navarra, R.; Morabito, C.; A Mariggiò, M.; Sensi, S.L. Altered Kv2.1 functioning promotes increased excitability in hippocampal neurons of an Alzheimer’s disease mouse model. Cell Death Dis. 2016, 7, e2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morecroft, I.; Murray, A.; Nilsen, M.; Gurney, A.M.; MacLean, M.R. Treatment with the Kv7 potassium channel activator flupirtine is beneficial in two independent mouse models of pulmonary hypertension. Br. J. Pharmacol. 2009, 157, 1241–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, X.; Zong, F.; Wang, H.; Wang, Q.; Xie, W.; Wang, H. Iptakalim, a novel ATP-sensitive potassium channel opener, inhibits pulmonary arterial smooth muscle cell proliferation by downregulation of PKC-α. J. Biomed. Res. 2011, 25, 392–401. [Google Scholar] [CrossRef] [Green Version]
- Revermann, M.; Neofitidou, S.; Kirschning, T.; Schloss, M.; Brandes, R.P.; Hofstetter, C. Inhalation of the BK(Ca)-opener NS1619 attenuates right ventricular pressure and improves oxygenation in the rat monocrotaline model of pulmonary hypertension. PLoS ONE 2014, 9, e86636. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.; Rupp, M.; Hofmann, S.; Mittal, M.; Fuchs, B.; Sommer, N.; Parajuli, N.; Quanz, K.; Schubert, D.; Dony, E.; et al. Heme oxygenase-2 and large-conductance Ca2+-activated K+ channels: Lung vascular effects of hypoxia. Am. J. Respir. Crit. Care Med. 2009, 180, 353–364. [Google Scholar] [CrossRef]
- Nielsen, G.; Wandall-Frostholm, C.; Sadda, V.; Oliván-Viguera, A.; Lloyd, E.E.; Bryan, R.M.; Simonsen, U.; Köhler, R. Alterations of N-3 polyunsaturated fatty acid-activated K2P channels in hypoxia-induced pulmonary hypertension. Basic Clin. Pharmacol. Toxicol. 2013, 113, 250–258. [Google Scholar] [CrossRef]
- Pandit, L.M.; Lloyd, E.E.; Reynolds, J.O.; Lawrence, W.S.; Reynolds, C.; Wehrens, X.H.; Bryan, R.M. TWIK-2 Channel Deficiency Leads to Pulmonary Hypertension Through a Rho-Kinase–Mediated Process. Hypertension 2014, 64, 1260–1265. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, M.G.; Reynolds, J.O.; Durgan, D.; Rodney, G.; Karmouty-Quintana, H.; Bryan, R.; Pandit, L.M. Twik-2(-/-) mouse demonstrates pulmonary vascular heterogeneity in intracellular pathways for vasocontractility. Physiol. Rep. 2019, 7, e13950. [Google Scholar] [CrossRef] [Green Version]
- Antigny, F.; Hautefort, A.; Meloche, J.; Belacel-Ouari, M.; Manoury, B.; Rucker-Martin, C.; Péchoux, C.; Potus, F.; Nadeau, V.; Tremblay, E.; et al. Potassium Channel Subfamily K Member 3 (KCNK3) Contributes to the Development of Pulmonary Arterial Hypertension. Circulation 2016, 133, 1371–1385. [Google Scholar] [CrossRef]
- Kitagawa, M.G.; Reynolds, J.O.; Wehrens, X.H.; Bryan, R.M., Jr.; Pandit, L.M. Hemodynamic and Pathologic Characterization of the TASK-1(-/-) Mouse Does Not Demonstrate Pulmonary Hypertension. Front. Med. (Lausanne) 2017, 4, 177. [Google Scholar] [CrossRef] [Green Version]
- Tang, G.; Wu, L.; Wang, R. The Effect of Hydroxylamine on KATP Channels in Vascular Smooth Muscle and Underlying Mechanisms. Mol. Pharmacol. 2005, 67, 1723–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, T.; Kajikuri, J.; Katsuya, H.; Itoh, T. Effects of H2O2 on membrane potential of smooth muscle cells in rabbit mesenteric resistance artery. Eur. J. Pharmacol. 2003, 464, 101–109. [Google Scholar] [CrossRef]
- Wei, E.P.; Kontos, H.A.; Beckman, J.S. Mechanisms of cerebral vasodilation by superoxide, hydrogen peroxide, and peroxynitrite. Am. J. Physiol. 1996, 271 Pt 2, H1262–H1266. [Google Scholar] [CrossRef]
- Fukumoto, M.; Nakaizumi, A.; Zhang, T.; Lentz, S.I.; Shibata, M.; Puro, D.G. Vulnerability of the retinal microvasculature to oxidative stress: Ion channel-dependent mechanisms. Am. J. Physiol. Cell Physiol. 2012, 302, C1413–C1420. [Google Scholar] [CrossRef] [PubMed]
- Ichinari, K. Direct Activation of the ATP-sensitive Potassium Channel by Oxygen Free Radicals in Guinea-pig Ventricular Cells:its Potentiation by MgADP. J. Mol. Cell. Cardiol. 1996, 28, 1867–1877. [Google Scholar] [CrossRef]
- Ohashi, M.; Faraci, F.; Heistad, D. Peroxynitrite hyperpolarizes smooth muscle and relaxes internal carotid artery in rabbit via ATP-sensitive K+ channels. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2244–H2250. [Google Scholar] [CrossRef]
- Armstead, W.M. Endothelin-Induced Cyclooxygenase-Dependent Superoxide Generation Contributes to K+ Channel Functional Impairment after Brain Injury. J. Neurotrauma 2001, 18, 1039–1048. [Google Scholar] [CrossRef]
- Yasui, S.; Mawatari, K.; Morizumi, R.; Furukawa, H.; Shimohata, T.; Harada, N.; Takahashi, A.; Nakaya, Y. Hydrogen peroxide inhibits insulin-induced ATP-sensitive potassium channel activation independent of insulin signaling pathway in cultured vascular smooth muscle cells. J. Med Investig. 2012, 59, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Thengchaisri, N.; Kuo, L. Hydrogen peroxide induces endothelium-dependent and -independent coronary arteriolar dilation: Role of cyclooxygenase and potassium channels. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2255–H2263. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.X.; Borbouse, L.; Gebremedhin, D.; Mendoza, S.A.; Zinkevich, N.S.; Li, R.; Gutterman, D.D. H2O2-induced dilation in human coronary arterioles: Role of protein kinase G dimerization and large-conductance Ca2+-activated K+ channel activation. Circ. Res. 2012, 110, 471–480. [Google Scholar] [CrossRef] [Green Version]
- Hayabuchi, Y.; Nakaya, Y.; Matsuoka, S.; Kuroda, Y. Hydrogen peroxide-induced vascular relaxation in porcine coronary arteries is mediated by Ca2+-activated K+ channels. Heart Vessel. 1998, 13, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Iida, Y.; Katusic, Z.S.; Wei, E.P. Mechanisms of Cerebral Arterial Relaxations to Hydrogen Peroxide. Stroke 2000, 31, 2224–2230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, K.S.; Liu, J.; Yang, G.Y.; Jin, C.; Huang, Q.; Huang, X. Peroxynitrite leads to arteriolar smooth muscle cell membrane hyperpolarization and low vasoreactivity in severe shock. Clin. Hemorheol. Microcirc. 2000, 23, 259–267. [Google Scholar] [PubMed]
- Au, A.L.; Seto, S.W.; Chan, S.; Chan, M.; Kwan, Y. Modulation by homocysteine of the iberiotoxin-sensitive, Ca2+-activated K+ channels of porcine coronary artery smooth muscle cells. Eur. J. Pharmacol. 2006, 546, 109–119. [Google Scholar] [CrossRef]
- Brakemeier, S.; Eichler, I.; Knorr, A.; Fassheber, T.; Köhler, R.; Hoyer, J.; Köhler, R. Modulation of Ca2+-activated K+ channel in renal artery endothelium in situ by nitric oxide and reactive oxygen species. Kidney Int. 2003, 64, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Terata, K.; Chai, Q.; Li, H.; Kleinman, L.H.; Gutterman, D.D. Peroxynitrite inhibits Ca2+-activated K+ channel activity in smooth muscle of human coronary arterioles. Circ. Res. 2002, 91, 1070–1076. [Google Scholar] [CrossRef] [Green Version]
- Frisbee, J.C.; Maier, K.G.; Stepp, D.W. Oxidant stress-induced increase in myogenic activation of skeletal muscle resistance arteries in obese Zucker rats. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H2160–H2168. [Google Scholar] [CrossRef] [Green Version]
- Somlyo, A.P.; Somlyo, A.V. Ca2+ Sensitivity of Smooth Muscle and Nonmuscle Myosin II: Modulated by G Proteins, Kinases, and Myosin Phosphatase. Physiol. Rev. 2003, 83, 1325–1358. [Google Scholar] [CrossRef] [Green Version]
- Fagan, K.A.; Oka, M.; Bauer, N.R.; Gebb, S.A.; Ivy, D.D.; Morris, K.G.; McMurtry, I.F. Attenuation of acute hypoxic pulmonary vasoconstriction and hypoxic pulmonary hypertension in mice by inhibition of Rho-kinase. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L656–L664. [Google Scholar] [CrossRef]
- Kitazawa, T.; Eto, M.; Woodsome, T.P.; Brautigan, D.L. Agonists Trigger G Protein-mediated Activation of the CPI-17 Inhibitor Phosphoprotein of Myosin Light Chain Phosphatase to Enhance Vascular Smooth Muscle Contractility. J. Biol. Chem. 2000, 275, 9897–9900. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, S.; Ridley, A.J.; Lutz, S. The Function of Rho-Associated Kinases ROCK1 and ROCK2 in the Pathogenesis of Cardiovascular Disease. Front. Pharmacol. 2015, 6, 276. [Google Scholar] [CrossRef]
- Pankey, E.A.; Byun, R.J.; Smith, W.B.; Bhartiya, M.; Bueno, F.R.; Badejo, A.M.; Stasch, J.-P.; Murthy, S.N.; Nossaman, B.; Kadowitz, P.J. The Rho kinase inhibitor azaindole-1 has long-acting vasodilator activity in the pulmonary vascular bed of the intact chest rat. Can. J. Physiol. Pharmacol. 2012, 90, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Tawara, S.; Oi, K.; Hizume, T.; Uwatoku, T.; Fukumoto, Y.; Kaibuchi, K.; Shimokawa, H. Long-Term Inhibition of Rho-kinase Ameliorates Hypoxia-Induced Pulmonary Hypertension in Mice. J. Cardiovasc. Pharmacol. 2006, 48, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Oka, M.; A Fagan, K.; Jones, P.L.; McMurtry, I.F. Therapeutic potential of RhoA/Rho kinase inhibitors in pulmonary hypertension. Br. J. Pharmacol. 2008, 155, 444–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilluy, C.; Sauzeau, V.; Rolli-Derkinderen, M.; Guerin, P.; Sagan, C.; Pacaud, P.; Loirand, G. Inhibition of RhoA/Rho kinase pathway is involved in the beneficial effect of sildenafil on pulmonary hypertension. Br. J. Pharmacol. 2005, 146, 1010–1018. [Google Scholar] [CrossRef] [Green Version]
- Ziino, A.J.; Ivanovska, J.; Belcastro, R.; Kantores, C.; Xu, E.Z.; Lau, M.; McNamara, P.J.; Tanswell, A.K.; Jankov, R.P. Effects of rho-kinase inhibition on pulmonary hypertension, lung growth, and structure in neonatal rats chronically exposed to hypoxia. Pediatr. Res. 2010, 67, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Lannér, M.C.; Jin, N.; Swartz, D.; Li, L.; Rhoades, R.A. Hypoxia inhibits myosin phosphatase in pulmonary arterial smooth muscle cells: Role of Rho-kinase. Am. J. Respir. Cell Mol. Biol. 2003, 29, 465–471. [Google Scholar] [CrossRef] [Green Version]
- Woodsome, T.P.; Polzin, A.; Kitazawa, K.; Eto, M. Agonist- and depolarization-induced signals for myosin light chain phosphorylation and force generation of cultured vascular smooth muscle cells. J. Cell Sci. 2006, 119 Pt 9, 1769–1780. [Google Scholar] [CrossRef] [Green Version]
- Mita, M.; Tanaka, H.; Yanagihara, H.; Nakagawà, J.-I.; Hishinuma, S.; Sutherland, C.; Walsh, M.P.; Shoji, M. Membrane depolarization-induced RhoA/Rho-associated kinase activation and sustained contraction of rat caudal arterial smooth muscle involves genistein-sensitive tyrosine phosphorylation. J. Smooth Muscle Res. 2013, 49, 26–45. [Google Scholar] [CrossRef] [Green Version]
- Porras-González, C.; Ordoñez, A.; Castellano, A.; Ureña, J. Regulation of RhoA/ROCK and sustained arterial contraction by low cytosolic Ca2+ levels during prolonged depolarization of arterial smooth muscle. Vasc. Pharmacol. 2017, 93, 33–41. [Google Scholar] [CrossRef]
- Sakurada, S.; Takuwa, N.; Sugimoto, N.; Wang, Y.; Seto, M.; Sasaki, Y.; Takuwa, N. Ca2+-dependent activation of Rho and Rho kinase in membrane depolarization-induced and receptor stimulation-induced vascular smooth muscle contraction. Circ. Res. 2003, 93, 548–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukata, Y.; Kaibuchi, K.; Amano, M. Rho–Rho-kinase pathway in smooth muscle contraction and cytoskeletal reorganization of non-muscle cells. Trends Pharmacol. Sci. 2001, 22, 32–39. [Google Scholar] [CrossRef]
- Turcotte, S.; Desrosiers, R.R.; Béliveau, R. HIF-1alpha mRNA and protein upregulation involves Rho GTPase expression during hypoxia in renal cell carcinoma. J. Cell Sci. 2003, 116 Pt 11, 2247–2260. [Google Scholar] [CrossRef] [Green Version]
- Wennerberg, K.; Der, C.J. Rho-family GTPases: It’s not only Rac and Rho (and I like it). J. Cell Sci. 2004, 117 Pt 8, 1301–1312. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.; Campbell, S.L. Mechanism of Redox-mediated Guanine Nucleotide Exchange on Redox-active Rho GTPases. J. Biol. Chem. 2005, 280, 31003–31010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aghajanian, A.; Wittchen, E.S.; Campbell, S.L.; Burridge, K. Direct Activation of RhoA by Reactive Oxygen Species Requires a Redox-Sensitive Motif. PLoS ONE 2009, 4, e8045. [Google Scholar] [CrossRef] [Green Version]
- Chaumais, M.-C.; Ranchoux, B.; Montani, D.; Dorfmüller, P.; Tu, L.; Lecerf, F.; Raymond, N.; Guignabert, C.; Price, L.; Simonneau, G.; et al. N-acetylcysteine improves established monocrotaline-induced pulmonary hypertension in rats. Respir. Res. 2014, 15, 65. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Zhang, Y.; Kang, B.; Xiao, J.; Xie, B.; Wang, Z. Protection of oral hydrogen water as an antioxidant on pulmonary hypertension. Mol. Biol. Rep. 2013, 40, 5513–5521. [Google Scholar] [CrossRef] [Green Version]
- Jefferson, J.A.; Simoni, J.M.; Escudero, E.; Hurtado, M.-E.; Swenson, E.R.; E Wesson, D.; Schreiner, G.F.; Schoene, R.B.; Johnson, R.J.; Hurtado, A. Increased Oxidative Stress Following Acute and Chronic High Altitude Exposure. High Alt. Med. Biol. 2004, 5, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Cracowski, J.-L.; Cracowski, C.; Bessard, G.; Pépin, J.-L.; Bessard, J.; Schwebel, C.; Stanke-Labesque, F.; Pison, C. Increased Lipid Peroxidation in Patients with Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2001, 164, 1038–1042. [Google Scholar] [CrossRef] [PubMed]
- Smukowska-Gorynia, A.D.; Rzymski, P.; Marcinkowska, J.; Poniedziałek, B.; Komosa, A.; Cieslewicz, A.; Slawek-Szmyt, S.; Janus, M.; Araszkiewicz, A.; Jankiewicz, S.; et al. Prognostic Value of Oxidative Stress Markers in Patients with Pulmonary Arterial or Chronic Thromboembolic Pulmonary Hypertension. Oxid. Med. Cell. Longev. 2019, 2019, 3795320. [Google Scholar] [CrossRef] [PubMed]
- Robbins, I.M.; Morrow, J.D.; Christman, B.W. Oxidant stress but not thromboxane decreases with epoprostenol therapy. Free Radic. Biol. Med. 2005, 38, 568–574. [Google Scholar] [CrossRef]
- Hemnes, A.R.; Rathinasabapathy, A.; Austin, E.A.; Brittain, E.L.; Carrier, E.J.; Chen, X.; Fessel, J.P.; Fike, C.D.; Fong, P.; Fortune, N.; et al. A potential therapeutic role for angiotensin-converting enzyme 2 in human pulmonary arterial hypertension. Eur. Respir. J. 2018, 51, 1702638. [Google Scholar] [CrossRef]
- Westerhof, B.E.; Saouti, N.; Van Der Laarse, W.J.; Westerhof, N.; Noordegraaf, A.V. Treatment strategies for the right heart in pulmonary hypertension. Cardiovasc. Res. 2017, 113, 1465–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, J.; Farha, S.; Park, M.M.; Comhair, S.A.; Lundgrin, E.L.; Tang, W.W.; Bongard, R.D.; Merker, M.P.; Erzurum, S.C. Coenzyme Q supplementation in pulmonary arterial hypertension. Redox Biol. 2014, 2, 884–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]






| NOX Isoform | Expression in PAEC | Expression in PASMC | ROS Generated |
|---|---|---|---|
| NOX1 | Human [81,82,83], rat [84] | Human [85], rat [86,87], mouse [85,88] | O2.− [83,85,86,87,88] |
| NOX2 | Human [81,82,89,90], rat [84], mouse [89,91] | Rat [92] | O2.− [81,89] |
| NOX3 | Human [82] | N/A | O2.− [93] |
| NOX4 | Human [81,82,94], rat [84], mouse [95] | Human [90,96], rat [86,92], mouse [90] | O2.− [82,97], H2O2 [82,95,96] |
| Channel/Molecule | Alteration by CH | Functions |
|---|---|---|
| L-type VGCC | Increased current density [195], channel upregulation (Cav1.2) [196] | Positive: Mediate CH-induced enhanced pulmonary vascular tone [195] and PA vasoconstriction to KCl [195,196] and to L-type VGCC activator [195] |
| Negative: 1. Without effects on basal [Ca2+]i in PASMCs or basal PA tension [180] 2. Responsible for 30–40% of basal [Ca2+]i in cultured PASMCs but not affect basal PA tone [178] 3. Do not contribute to increase PA wall basal [Ca2+]i or elevated PA constriction to UTP following CH [17] 4. Do not contribute to CH-induced augmentation of PA myogenic tone [21] 5. CH-induced PH is not acutely alleviated by L-type VGCC inhibition in SD rats [197] or COPD patients [198,199] | ||
| T-type VGCC | Channel upregulation (Cav3.2) [196] | Positive: Mediate CH-induced augmented PA constriction to K+ and U-46619 [196] |
| Negative: 1. Do not contribute to increase PA wall basal [Ca2+]i following CH [17] 2. Do not contribute to CH-induced augmentation of PA myogenic tone [21] | ||
| TRPC1 | Channel upregulation [177,178,200] | CH-induced PH [201,202]; SOCE in PASMC [178,200]; CH-induced augmented basal tone and vasoconstriction to 5-HT [202] |
| TRPC6 | Channel upregulation [177,178,203] | CH-induced PH [202,203]; ROCE in PASMC [178]; augmented SOCE in PASMCs following CH [203]; basal tone under normoxia [202]; CH-induced augmented vasoconstriction to 5-HT [202] |
| TRPV4 | Channel upregulation in PASMCs [204,205], increased channel activities in PASMCs [204,205] | 1. CH-induced PH development [204,206] 2. CH-induced enhanced myogenic tone [204] and augmented vasoconstriction to serotonin [206] and TRPV4 agonist [205] but not to U46619 [204], PE [206] or ET-1 [206] in endothelium-disrupted PAs 3. Ca2+-induced Ca2+ release in PASMCs [205] |
| ASIC1 | Unaltered expression [207] | Contribute to augmented SOCE and SOCE-induced vasoconstriction in PAs following CH [17]; CH-induced PH [207] |
| Orai1 | Upregulation [14,200,203,208] | CH-induced increases in basal Ca2+ [14] and SOCE [14,200] in PASMCs |
| Orai2 | Upregulation [14,203,208] | CH-induced increases in basal Ca2+ and SOCE in PASMCs [14] |
| Orai3 | Unaltered expression [14] | CH-induced increases in basal Ca2+ and SOCE in PASMCs [14] |
| STIM1 | Upregulation [200,208], unaltered expression [14] | CH-induced increases in basal Ca2+ [14] and SOCE [14,200] in PASMCs |
| STIM2 | Upregulation [203,208] | Enhanced SOCE in PASMCs from PH patients [209] |
| MSC | Increased channel activities [176] | CH-induced augmentation of PA myogenic tone [19,21,176] |
| Type | Channel | Function | Ref. |
|---|---|---|---|
| Kir | KATP | Gain of function protects against CH-induced PH indices including mPAP, RV hypertrophy and PA remodeling | [256] |
| KCa | Large conductance KCa (BKCa) | Gain of function protects against monocrotaline-induced PH, reduces PDGF-induced PASMC proliferation | [257] |
| Loss of function does not affect PH development following CH | [258] | ||
| K2P | TREK-1 (K2P2.1) | Gain of function leads to PAEC hyperpolarization and PA relaxation | [259] |
| K2P | TWIK-2 (K2P6.1) | Gain of function leads to PAEC hyperpolarization and PA relaxation | [259] |
| K2P | TWIK-2 (KCNK6) | Loss of function results in increased RVSP, PA thickening, greater PA vasoconstrictor to U46619 | [260] |
| Loss of function causes PASMC depolarization, enhanced [Ca2+]i and PA constriction to U46619 | [261] | ||
| K2P | TASK-1 (KCNK3) | Loss of function favors proliferation of (PAEC, PASMC and fibroblast) and enhanced basal tone | [262] |
| Gain of function protects against monocrotaline-induced PH | [262] | ||
| Loss of function is without effects on CH-induced PH | [263] |
| Outcome | ROS | ||
|---|---|---|---|
| O2.− | H2O2 | ONOO− | |
| KATP activation | Mesenteric artery SMC [264] | Mesenteric arteries [265] Cerebral arteries [266] Retinal microvessels [267] Cardiomyocytes [268] | Cerebral arteries [266] Internal carotid arteries [269] |
| KATP inhibition | Cerebral arteries [270] | A10 cell line [271] | N/A |
| KCa activation | Cerebral arteries [266] | Coronary arteries [272,273,274] Cerebral arteries [266,275] | Arteriolar SMC [276] |
| KCa inhibition | Coronary arteries [277] Cerebral arteries [270] | Renal arteries [278] | Coronary artery SMC [279] Gracilis arteries [280] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, S.; Resta, T.C.; Jernigan, N.L. Vasoconstrictor Mechanisms in Chronic Hypoxia-Induced Pulmonary Hypertension: Role of Oxidant Signaling. Antioxidants 2020, 9, 999. https://doi.org/10.3390/antiox9100999
Yan S, Resta TC, Jernigan NL. Vasoconstrictor Mechanisms in Chronic Hypoxia-Induced Pulmonary Hypertension: Role of Oxidant Signaling. Antioxidants. 2020; 9(10):999. https://doi.org/10.3390/antiox9100999
Chicago/Turabian StyleYan, Simin, Thomas C. Resta, and Nikki L. Jernigan. 2020. "Vasoconstrictor Mechanisms in Chronic Hypoxia-Induced Pulmonary Hypertension: Role of Oxidant Signaling" Antioxidants 9, no. 10: 999. https://doi.org/10.3390/antiox9100999