A Mechanistic Evaluation of Antioxidant Nutraceuticals on Their Potential against Age-Associated Neurodegenerative Diseases



Abstract
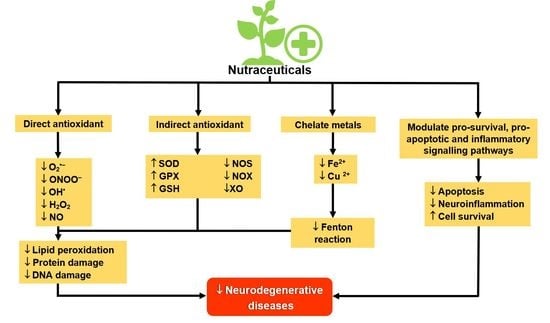
:
1. Introduction
1.1. Alzheimer and Parkinson Disease
2. Free Radicals and Oxidative Damage to Biomolecules
2.1. Free Radical Formation
2.2. Brain Susceptibility to Oxidative Damage
2.3. Nutraceuticals as Free Radical Scavenger and Prevents Damage to Biomolecules
2.3.1. Resveratrol
2.3.2. Grape Seed Extract
2.3.3. Ginkgo biloba Extract
2.3.4. Green Tea Polyphenols
2.3.5. Curcumin
2.3.6. Xanthorrhizol
2.3.7. Magnolol
2.3.8. Pycnogenol®
2.3.9. Guarana Seed Extract
2.3.10. Vitamin E
3. Metal Ions and Neurodegeneration
3.1. Pathophysiology of Neurodegenerative Disease Involving Transition Metals
3.2. Metal Chelation in the Treatment of Neurodegenerative Diseases
3.3. Nutraceutical that Acts as Metal Chelators
3.3.1. Epigallocatechin Gallate (EGCG)
3.3.2. Curcumin
4. Dysregulation of Antioxidant and Pro-Oxidant Enzymes
4.1. Internal Antioxidation Enzymes
4.2. Gene Expression for the Antioxidant Enzymes
4.3. Pro-Oxidant Enzymes
4.4. Modulation of Antioxidant and Pro-Oxidant Enzymes by Nutraceuticals
4.4.1. Resveratrol
4.4.2. Curcumin
4.4.3. Green Tea Polyphenols
4.4.4. Ginger Extract
4.4.5. Vitamin E
5. Oxidative Stress and Intracellular Signaling Pathway
5.1. Inflammatory Pathway
5.1.1. Microglia and Astrocyte Activation
5.1.2. MAPK Pathway
5.1.3. NF-κB Pathway
5.2. Cell Survival and Apoptotic Pathways
5.2.1. PI3K/AKT Pathway
5.2.2. Extrinsic Apoptotic Pathway
5.2.3. Intrinsic Apoptotic Pathway
5.3. Modulation of Inflammatory, Survival, and Apoptotic Signaling Pathways by Nutraceuticals
5.3.1. Curcumin
5.3.2. Green Tea Polyphenols
5.3.3. Berberine
5.3.4. Magnolol
5.3.5. Vitamin E
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Dementia: Fact Sheets. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 21 September 2020).
- Alzheimer’s Association Report. 2020 Alzheimer’s disease facts and figures. Alzheimers Dement. 2020, 16, 391–460. [Google Scholar] [CrossRef] [PubMed]
- GBD 2016 Parkinson’s Disease Collaborators. Global, regional, and national burden of Parkinson’s disease, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet. Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Chen, M. The maze of APP processing in Alzheimer’s disease: Where did we go wrong in reasoning? Front. Cell. Neurosci. 2015, 9, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naini, S.M.A.; Soussi-Yanicostas, N. Tau hyperphosphorylation and oxidative stress, a critical vicious circle in neurodegenerative tauopathies? Oxid. Med. Cell. Longev. 2015, 2015, 151979. [Google Scholar]
- Kametani, F.; Hasegawa, M. Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer’s disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [Green Version]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R.J.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.S.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jack, C.R.J.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Pratico, D. Oxidative stress hypothesis in Alzheimer’s disease: A reappraisal. Trends Pharmacol. Sci. 2008, 29, 609–615. [Google Scholar] [CrossRef]
- Izuo, N.; Kasahara, C.; Murakami, K.; Kume, T.; Maeda, M.; Irie, K.; Yokote, K.; Shimizu, T. A Toxic conformer of Aβ42 with a turn at 22–23 is a novel therapeutic target for Alzheimer’s disease. Sci. Rep. 2017, 7, 11811. [Google Scholar] [CrossRef]
- Salahuddin, P.; Fatima, M.T.; Abdelhameed, A.S.; Nusrat, S.; Khan, R.H. Structure of amyloid oligomers and their mechanisms of toxicities: Targeting amyloid oligomers using novel therapeutic approaches. Eur. J. Med. Chem. 2016, 114, 41–58. [Google Scholar] [CrossRef] [PubMed]
- McLellan, M.E.; Kajdasz, S.T.; Hyman, B.T.; Bacskai, B.J. In vivo imaging of reactive oxygen species specifically associated with thioflavine S-positive amyloid plaques by multiphoton microscopy. J. Neurosci. 2003, 23, 2212–2217. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Massano, J.; Bhatia, K.P. Clinical approach to Parkinson’s disease: Features, diagnosis, and principles of management. Cold Spring Harb. Perspect. Med. 2012, 2, a008870. [Google Scholar] [CrossRef]
- Michel, P.P.; Hirsch, E.C.; Hunot, S. Understanding dopaminergic cell death pathways in Parkinson disease. Neuron 2016, 90, 675–691. [Google Scholar] [CrossRef] [Green Version]
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. Alpha-synuclein structure and Parkinson’s disease–lessons and emerging principles. Mol. Neurodegener. 2019, 14, 29. [Google Scholar] [CrossRef] [Green Version]
- Stefanis, L. α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [Green Version]
- Hwang, O. Role of oxidative stress in Parkinson’s disease. Exp. Neurobiol. 2013, 22, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Bosco, D.A.; Fowler, D.M.; Zhang, Q.; Nieva, J.; Powers, E.T.; Wentworth, P.J.; Lerner, R.A.; Kelly, J.W. Elevated levels of oxidized cholesterol metabolites in Lewy body disease brains accelerate alpha-synuclein fibrilization. Nat. Chem. Biol. 2006, 2, 249–253. [Google Scholar] [CrossRef]
- Nakabeppu, Y.; Tsuchimoto, D.; Yamaguchi, H.; Sakumi, K. Oxidative damage in nucleic acids and Parkinson’s disease. J. Neurosci. Res. 2007, 85, 919–934. [Google Scholar] [CrossRef]
- Miyazaki, I.; Asanuma, M. Dopaminergic neuron-specific oxidative stress caused by dopamine itself. Acta Med. Okayama 2008, 62, 141–150. [Google Scholar]
- Varzakas, T.; Kandylis, P.; Dimitrellou, D.; Salamoura, C.; Zakynthinos, G.; Proestos, C. Innovative and fortified food: Probiotics, prebiotics, GMOs, and superfood. In Preparation and Processing of Religious and Cultural Foods; Ali, M.E., Nizar, N.N.A., Eds.; Woodhead Publishing: Sawston, UK, 2018; pp. 67–129. [Google Scholar]
- Kelsey, N.A.; Wilkins, H.M.; Linseman, D.A. Nutraceutical antioxidants as novel neuroprotective agents. Molecules 2010, 15, 7792–7814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittler, R. ROS Are Good. Trends Plant Sci. 2017, 22, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Caruso, G.; Spampinato, S.F.; Cardaci, V.; Caraci, F.; Sortino, M.A.; Merlo, S. β-amyloid and oxidative stress: Perspectives in drug development. Curr. Pharm. Des. 2019, 25, 4771–4781. [Google Scholar] [CrossRef]
- Smith, J.V.; Luo, Y. Elevation of oxidative free radicals in Alzheimer’s disease models can be attenuated by Ginkgo biloba extract EGb 761. J. Alzheimers Dis. 2003, 5, 287–300. [Google Scholar] [CrossRef]
- Massaccesi, L.; Galliera, E.; Galimberti, D.; Fenoglio, C.; Arcaro, M.; Goi, G.; Barassi, A.; Corsi Romanelli, M.M. Lag-time in Alzheimer’s disease patients: A potential plasmatic oxidative stress marker associated with ApoE4 isoform. Immun. Ageing 2019, 16, 7. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Treberg, J.R.; Brand, M.D. Mechanisms of mitochondrial free radical production and their relationship to the aging process. In Handbooks of Aging, 7th ed.; Masoro, E.J., Austad, S.N., Eds.; Academic Press: San Diego, CA, USA, 2011; pp. 47–61. [Google Scholar]
- Tönnies, E.; Trushina, E. Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J. Alzheimers Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [Green Version]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Mailloux, R.J. Mitochondrial antioxidants and the maintenance of cellular hydrogen peroxide levels. Oxid. Med. Cell. Longev. 2018, 2018, 7857251. [Google Scholar] [CrossRef]
- Handy, D.E.; Loscalzo, J. Redox regulation of mitochondrial function. Antioxid. Redox Signal. 2012, 16, 123–139. [Google Scholar] [CrossRef]
- Sheu, S.-S.; Nauduri, D.; Anders, M.W. Targeting antioxidants to mitochondria: A new therapeutic direction. Biochim. Biophys. Acta Mol. Basis Dis. 2006, 1762, 256–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanti Das, T.; Wati, M.R.; Fatima-Shad, K. Oxidative stress gated by fenton and haber weiss reactions and its association with Alzheimer’s disease. Arch. Neurosci. 2015, 2, e60038. [Google Scholar] [CrossRef] [Green Version]
- Watts, M.E.; Pocock, R.; Claudianos, C. Brain energy and oxygen metabolism: Emerging role in normal function and disease. Front. Mol. Neurosci. 2018, 11, 216. [Google Scholar] [CrossRef]
- Pizzimenti, S.; Ciamporcero, E.; Daga, M.; Pettazzoni, P.; Arcaro, A.; Cetrangolo, G.; Minelli, R.; Dianzani, C.; Lepore, A.; Gentile, F.; et al. Interaction of aldehydes derived from lipid peroxidation and membrane proteins. Front. Physiol. 2013, 4, 242. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.J.; Green, P.; Mann, J.J.; Rapoport, S.I.; Sublette, M.E. Pathways of polyunsaturated fatty acid utilization: Implications for brain function in neuropsychiatric health and disease. Brain Res. 2015, 1597, 220–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative stress in neurodegenerative diseases: From a mitochondrial point of view. Oxid. Med. Cell. Longev. 2019, 2019, 2105607. [Google Scholar] [CrossRef] [Green Version]
- Shichiri, M. The role of lipid peroxidation in neurological disorders. J. Clin. Biochem. Nutr. 2014, 54, 151–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taso, O.V.; Philippou, A.; Moustogiannis, A.; Zevolis, E.; Koutsilieris, M. Lipid peroxidation products and their role in neurodegenerative diseases. Ann. Res. Hosp. 2019, 3, 2. [Google Scholar] [CrossRef]
- Boonruamkaew, P.; Chonpathompikunlert, P.; Vong, L.B.; Sakaue, S.; Tomidokoro, Y.; Ishii, K.; Tamaoka, A.; Nagasaki, Y. Chronic treatment with a smart antioxidative nanoparticle for inhibition of amyloid plaque propagation in Tg2576 mouse model of Alzheimer’s disease. Sci. Rep. 2017, 7, 3785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, E.; Morel, A.; Saso, L.; Saluk, J. Isoprostanes and neuroprostanes as biomarkers of oxidative stress in neurodegenerative diseases. Oxid. Med. Cell. Longev. 2014, 2014, 572491. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Roles of the lipid peroxidation product 4-hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Exp. Gerontol. 2009, 44, 625–633. [Google Scholar] [CrossRef] [Green Version]
- Greenough, M.A.; Camakaris, J.; Bush, A.I. Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem. Int. 2013, 62, 540–555. [Google Scholar] [CrossRef]
- Contestabile, A. Oxidative stress in neurodegeneration: Mechanisms and therapeutic perspectives. Curr. Top. Med. Chem. 2001, 1, 553–568. [Google Scholar] [CrossRef]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z. Iron and oxidizing species in oxidative stress and Alzheimer’s disease. Aging Med. 2019, 2, 82–87. [Google Scholar] [CrossRef]
- Sharma, P.; Jha, A.B.; Dubey, R.S.; Pessarakli, M. Reactive oxygen species, oxidative damage, and antioxidative defense mechanism in plants under stressful conditions. J. Bot. 2012, 2012, 217037. [Google Scholar] [CrossRef] [Green Version]
- Aksenov, M.Y.; Aksenova, M.V.; Butterfield, D.A.; Geddes, J.W.; Markesbery, W.R. Protein oxidation in the brain in Alzheimer’s disease. Neuroscience 2001, 103, 373–383. [Google Scholar] [CrossRef]
- Grimm, S.; Hoehn, A.; Davies, K.J.; Grune, T. Protein oxidative modifications in the ageing brain: Consequence for the onset of neurodegenerative disease. Free Radic. Res. 2011, 45, 73–88. [Google Scholar] [PubMed] [Green Version]
- Lovell, M.A.; Markesbery, W.R. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acids Res. 2007, 35, 7497–7504. [Google Scholar] [CrossRef] [Green Version]
- Salminen, L.E.; Paul, R.H. Oxidative stress and genetic markers of suboptimal antioxidant defense in the aging brain: A theoretical review. Rev. Neurosci. 2014, 25, 805–819. [Google Scholar]
- Patel, M. Targeting oxidative stress in central nervous system disorders. Trends Pharmacol. Sci. 2016, 37, 768–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, K.; Luo, X.; Xu, K.; Ven Murthy, M.R. Role of oxidative stress in neurodegeneration: Recent developments in assay methods for oxidative stress and nutraceutical antioxidants. Prog. Neuropsychopharmacol. Biol. Psychiatry 2004, 28, 771–799. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, D.; Megha, K.; Mishra, R.; Mandal, P.K. Glutathione in brain: Overview of its conformations, functions, biochemical characteristics, quantitation and potential therapeutic role in brain disorders. Neurochem. Res. 2020, 45, 1461–1480. [Google Scholar]
- Sinet, P.M.; Heikkila, R.E.; Cohen, G. Hydrogen peroxide production by rat brain in vivo. J. Neurochem. 1980, 34, 1421–1428. [Google Scholar] [CrossRef]
- Ren, X.; Zou, L.; Zhang, X.; Branco, V.; Wang, J.; Carvalho, C.; Holmgren, A.; Lu, J. redox signaling mediated by thioredoxin and glutathione systems in the central nervous system. Antioxid. Redox Signal. 2017, 27, 989–1010. [Google Scholar] [CrossRef]
- Fishel, M.L.; Vasko, M.R.; Kelley, M.R. DNA repair in neurons: So if they don’t divide what’s to repair? Mutat. Res. 2007, 614, 24–36. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative stress: A key modulator in neurodegenerative diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramovič, H.; Grobin, B.; Poklar Ulrih, N.; Cigić, B. Relevance and standardization of in vitro antioxidant assays: ABTS, DPPH, and Folin–Ciocalteu. J. Chem. 2018, 2018, 4608405. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.-K.; Shibamoto, T. Antioxidant assays for plant and food components. J. Agric. Food Chem. 2009, 57, 1655–1666. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.; Yokota, T.; Ashihara, H.; Lean, M.E.J.; Crozier, A. Plant foods and herbal sources of resveratrol. J. Agric. Food Chem. 2002, 50, 3337–3340. [Google Scholar] [CrossRef]
- Tian, B.; Liu, J. Resveratrol: A review of plant sources, synthesis, stability, modification and food application. J. Sci. Food Agric. 2020, 100, 1392–1404. [Google Scholar] [CrossRef]
- Lin, Y.-T.; Wu, Y.-C.; Sun, G.-C.; Ho, C.-Y.; Wong, T.-Y.; Lin, C.-H.; Chen, H.-H.; Yeh, T.-C.; Li, C.-J.; Tseng, C.-J.; et al. Effect of resveratrol on reactive oxygen species-induced cognitive impairment in rats with angiotensin II-induced early Alzheimer’s disease. J. Clin. Med. 2018, 7, 329. [Google Scholar] [CrossRef] [Green Version]
- Bao, D.; Wang, J.; Pang, X.; Liu, H. Protective effect of quercetin against oxidative stress-induced cytotoxicity in rat pheochromocytoma (PC-12) cells. Molecules 2017, 22, 1122. [Google Scholar] [CrossRef]
- Kim, H.; Lee, K.; Lee, H.J. Protective effects of piceatannol against beta-amyloid–induced neuronal cell death. Ann. N. Y. Acad. Sci. 2007, 1095, 473–482. [Google Scholar] [CrossRef]
- Sharma, M.; Gupta, Y.K. Chronic treatment with trans resveratrol prevents intracerebroventricular streptozotocin induced cognitive impairment and oxidative stress in rats. Life Sci. 2002, 71, 2489–2498. [Google Scholar] [CrossRef]
- Petralia, S.; Spatafora, C.; Tringali, C.; Foti, M.C.; Sortino, S. Hydrogen atom abstraction from resveratrol and two lipophilic derivatives by tert-butoxyl radicals. A laser flash photolysis study. New J. Chem. 2004, 28, 1484–1487. [Google Scholar] [CrossRef]
- Iuga, C.; Alvarez-Idaboy, J.R.; Russo, N. Antioxidant activity of trans-resveratrol toward hydroxyl and hydroperoxyl radicals: A quantum chemical and computational kinetics study. J. Org. Chem. 2012, 77, 3868–3877. [Google Scholar] [CrossRef] [PubMed]
- El-Beshbishy, H.A.; Mohamadin, A.M.; Abdel-Naim, A.B. In vitro evaluation of the antioxidant activities of grape seed (vitis vinifera) extract, blackseed (nigella sativa) extract and curcumin. J. Taibah Univ. Med. Sci. 2009, 4, 23–35. [Google Scholar]
- Balu, M.; Sangeetha, P.; Murali, G.; Panneerselvam, C. Modulatory role of grape seed extract on age-related oxidative DNA damage in central nervous system of rats. Brain Res. Bull. 2006, 68, 469–473. [Google Scholar] [CrossRef]
- Balu, M.; Sangeetha, P.; Murali, G.; Panneerselvam, C. Age-related oxidative protein damages in central nervous system of rats: Modulatory role of grape seed extract. Int. J. Dev. Neurosci. 2005, 23, 501–507. [Google Scholar] [CrossRef]
- Martínez-Solís, I.; Acero, N.; Bosch-Morell, F.; Castillo, E.; González-Rosende, M.E.; Muñoz-Mingarro, D.; Ortega, T.; Sanahuja, M.A.; Villagrasa, V. Neuroprotective potential of Ginkgo biloba in retinal diseases. Planta Med. 2019, 85, 1292–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silberstein, R.B.; Pipingas, A.; Song, J.; Camfield, D.A.; Nathan, P.J.; Stough, C. Examining brain-cognition effects of ginkgo biloba extract: Brain activation in the left temporal and left prefrontal cortex in an object working memory task. Evid. Based Complement. Alternat. Med. 2011, 2011, 164139. [Google Scholar] [CrossRef] [PubMed]
- Kanowski, S.; Hoerr, R. Ginkgo biloba extract EGb 761 in dementia: Intent-to-treat analyses of a 24-week, multi-center, double-blind, placebo-controlled, randomized trial. Pharmacopsychiatry 2003, 36, 297–303. [Google Scholar]
- Legeay, S.; Rodier, M.; Fillon, L.; Faure, S.; Clere, N. Epigallocatechin gallate: A review of its beneficial properties to prevent metabolic syndrome. Nutrients 2015, 7, 5443–5468. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.A.; Mandal, A.K.A.; Khan, Z.A. Potential neuroprotective properties of epigallocatechin-3-gallate (EGCG). Nutr. J. 2016, 15, 60. [Google Scholar] [CrossRef] [Green Version]
- Arab, H.; Mahjoub, S.; Hajian-Tilaki, K.; Moghadasi, M. The effect of green tea consumption on oxidative stress markers and cognitive function in patients with Alzheimer’s disease: A prospective intervention study. Casp. J. Intern. Med. 2016, 7, 188–194. [Google Scholar]
- Frautschy, S.A.; Hu, W.; Kim, P.; Miller, S.A.; Chu, T.; Harris-White, M.E.; Cole, G.M. Phenolic anti-inflammatory antioxidant reversal of Abeta-induced cognitive deficits and neuropathology. Neurobiol. Aging 2001, 22, 993–1005. [Google Scholar] [CrossRef]
- Seo, J.-S.; Leem, Y.-H.; Lee, K.-W.; Kim, S.-W.; Lee, J.-K.; Han, P.-L. Severe motor neuron degeneration in the spinal cord of the Tg2576 mouse model of Alzheimer disease. J. Alzheimers Dis. 2010, 21, 263–276. [Google Scholar] [PubMed] [Green Version]
- Chen, W.-F.; Deng, S.-L.; Zhou, B.; Yang, L.; Liu, Z.-L. Curcumin and its analogues as potent inhibitors of low density lipoprotein oxidation: H-atom abstraction from the phenolic groups and possible involvement of the 4-hydroxy-3-methoxyphenyl groups. Free Radic. Biol. Med. 2006, 40, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Priyadarsini, K.I.; Maity, D.K.; Naik, G.H.; Kumar, M.S.; Unnikrishnan, M.K.; Satav, J.G.; Mohan, H. Role of phenolic O–H and methylene hydrogen on the free radical reactions and antioxidant activity of curcumin. Free Radic. Biol. Med. 2003, 35, 475–484. [Google Scholar] [CrossRef]
- Dei Cas, M.; Ghidoni, R. Dietary Curcumin: Correlation between bioavailability and health potential. Nutrients 2019, 11, 2147. [Google Scholar]
- Shelat, D.Y.; Acharya, S.R. CUR-CA-THIONE: A novel curcumin concoction with enhanced water solubility and brain bio-availability. Int. J. Pharm. Pharm. Sci. 2016, 8, 265–270. [Google Scholar] [CrossRef] [Green Version]
- Yallapu, M.M.; Jaggi, M.; Chauhan, S.C. Curcumin nanoformulations: A future nanomedicine for cancer. Drug Discov. Today 2012, 17, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Shelat, D.Y.; Acharya, S.R. Neuroprotective activity of novel CUR-CA-THIONE and its oxidative stress study. Int. J. Pharm. Pharm. Sci. 2016, 8, 167–173. [Google Scholar] [CrossRef] [Green Version]
- Ramkumar, M.; Rajasankar, S.; Gobi, V.V.; Dhanalakshmi, C.; Manivasagam, T.; Justin Thenmozhi, A.; Essa, M.M.; Kalandar, A.; Chidambaram, R. Neuroprotective effect of Demethoxycurcumin, a natural derivative of Curcumin on rotenone induced neurotoxicity in SH-SY 5Y Neuroblastoma cells. BMC Complement. Altern. Med. 2017, 17, 217. [Google Scholar]
- Lim, C.S.; Jin, D.Q.; Mok, H.; Oh, S.J.; Lee, J.U.; Hwang, J.K.; Ha, I.; Han, J.S. Antioxidant and antiinflammatory activities of xanthorrhizol in hippocampal neurons and primary cultured microglia. J. Neurosci. Res. 2005, 82, 831–838. [Google Scholar]
- Muroyama, A.; Fujita, A.; Lv, C.; Kobayashi, S.; Fukuyama, Y.; Mitsumoto, Y. Magnolol protects against MPTP/MPP(+)-induced toxicity via inhibition of oxidative stress in in vivo and in vitro models of Parkinson’s disease. Parkinsons Dis. 2012, 2012, 985157. [Google Scholar] [PubMed] [Green Version]
- Ogata, M.; Hoshi, M.; Shimotohno, K.; Urano, S.; Endo, T. Antioxidant activity of magnolol, honokiol, and related phenolic compounds. J. Am. Oil Chem. Soc. 1997, 74, 557–562. [Google Scholar] [CrossRef]
- Dong, L.; Zhou, S.; Yang, X.; Chen, Q.; He, Y.; Huang, W. Magnolol protects against oxidative stress-mediated neural cell damage by modulating mitochondrial dysfunction and PI3K/Akt signaling. J. Mol. Neurosci. 2013, 50, 469–481. [Google Scholar] [CrossRef]
- Rohdewald, P.J. Pycnogenol, French maritime pine bark extract. In Encyclopedia of Dietary Supplements, 1st ed.; Coates, P.M., Paul, M.C., Blackman, M., Blackman, M.R., Cragg, G.M., Levine, M., White, J.D., Moss, J., Eds.; CRC Press: Boca Raton, FL, USA, 2004; pp. 545–553. [Google Scholar]
- Packer, L.; Rimbach, G.; Virgili, F. Antioxidant activity and biologic properties of a procyanidin-rich extract from pine (Pinus maritima) bark, pycnogenol. Free Radic. Biol. Med. 1999, 27, 704–724. [Google Scholar] [CrossRef]
- Ansari, M.A.; Keller, J.N.; Scheff, S.W. Protective effect of pycnogenol in human neuroblastoma SH-SY5Y cells following acrolein-induced cytotoxicity. Free Radic. Biol. Med. 2008, 45, 1510–1519. [Google Scholar] [CrossRef] [Green Version]
- Ishrat, T.; Parveen, K.; Hoda, M.N.; Khan, M.B.; Yousuf, S.; Ansari, M.A.; Saleem, S.; Islam, F. Effects of pycnogenol and vitamin E on cognitive deficits and oxidative damage induced by intracerebroventricular streptozotocin in rats. Behav. Pharmacol. 2009, 20, 567–575. [Google Scholar] [CrossRef]
- Devaraj, S.; Vega-López, S.; Kaul, N.; Schönlau, F.; Rohdewald, P.; Jialal, I. Supplementation with a pine bark extract rich in polyphenols increases plasma antioxidant capacity and alters the plasma lipoprotein profile. Lipids 2002, 37, 931–934. [Google Scholar] [CrossRef]
- Belcaro, G.; Hu, S.; Cesarone, M.R.; Dugall, M. A controlled study shows daily intake of 50 mg of French pine bark extract (Pycnogenol®) lowers plasma reactive oxygen metabolites in healthy smokers. Minerva Med. 2013, 104, 439–446. [Google Scholar]
- Dalonso, N.; de Oliveira Petkowicz, C.L. Guarana powder polysaccharides: Characterisation and evaluation of the antioxidant activity of a pectic fraction. Food Chem. 2012, 134, 1804–1812. [Google Scholar] [CrossRef] [Green Version]
- Sereia, A.L.; de Oliveira, M.T.; Baranoski, A.; Marques, L.L.M.; Ribeiro, F.M.; Isolani, R.G.; de Medeiros, D.C.; Chierrito, D.; Lazarin-Bidóia, D.; Zielinski, A.A.F.; et al. In vitro evaluation of the protective effects of plant extracts against amyloid-beta peptide-induced toxicity in human neuroblastoma SH-SY5Y cells. PLoS ONE 2019, 14, e0212089. [Google Scholar] [CrossRef]
- De Oliveira, D.M.; Barreto, G.; Galeano, P.; Romero, J.I.; Holubiec, M.I.; Badorrey, M.S.; Capani, F.; Alvarez, L.D.G. Paullinia cupana Mart. var. Sorbilis protects human dopaminergic neuroblastoma SH-SY5Y cell line against rotenone-induced cytotoxicity. Hum. Exp. Toxicol. 2011, 30, 1382–1391. [Google Scholar] [CrossRef]
- Peixoto, H.; Roxo, M.; Röhrig, T.; Richling, E.; Wang, X.; Wink, M. Anti-aging and antioxidant potential of paullinia cupana var. sorbilis: Findings in caenorhabditis elegans indicate a new utilization for roasted seeds of guarana. Medicines 2017, 4, 61. [Google Scholar] [CrossRef] [PubMed]
- Boasquívis, P.F.; Silva, G.M.M.; Paiva, F.A.; Cavalcanti, R.M.; Nunez, C.V.; de Paula Oliveira, R. Guarana (Paullinia cupana) extract protects Caenorhabditis elegans models for Alzheimer disease and Huntington disease through activation of antioxidant and protein degradation pathways. Oxid. Med. Cell. Longev. 2018, 2018, 9241308. [Google Scholar] [CrossRef] [Green Version]
- Da Silva Bittencourt, L.; Zeidán-Chuliá, F.; Yatsu, F.K.J.; Schnorr, C.E.; Moresco, K.S.; Kolling, E.A.; Gelain, D.P.; Bassani, V.L.; Moreira, J.C.F. Guarana (Paullinia cupana Mart.) prevents β-amyloid aggregation, generation of advanced glycation-end products (AGEs), and acrolein-induced cytotoxicity on human neuronal-like cells. Phytother. Res. 2014, 28, 1615–1624. [Google Scholar] [CrossRef]
- Lee, G.Y.; Han, S.N. The role of vitamin E in immunity. Nutrients 2018, 10, 1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermizi, H.; Faizah, O.; Ima-Nirwana, S.; Nazrun, S.A.; Norazlina, M. Beneficial effects of tocotrienol and tocopherol on bone histomorphometric parameters in sprague-dawley male rats after nicotine cessation. Calcif. Tissue Int. 2009, 84, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Zarkasi, K.A.; Jen-Kit, T.; Jubri, Z. Molecular understanding of the cardiomodulation in myocardial infarction and the mechanism of vitamin E protections. Mini Rev. Med. Chem. 2019, 19, 1407–1426. [Google Scholar] [CrossRef]
- Sekikawa, T.; Kizawa, Y.; Li, Y.; Takara, T. Cognitive function improvement with astaxanthin and tocotrienol intake: A randomized, double-blind, placebo-controlled study. J. Clin. Biochem. Nutr. 2020. [Google Scholar] [CrossRef]
- Taridi, N.M.; Rani, N.A.; Latiff, A.A.; Ngah, W.Z.W.; Mazlan, M. Tocotrienol rich fraction reverses age-related deficits in spatial learning and memory in aged rats. Lipids 2014, 49, 855–869. [Google Scholar] [CrossRef]
- Gulcin, İ. Antioxidants and antioxidant methods: An updated overview. Arch. Toxicol. 2020, 94, 651–715. [Google Scholar] [CrossRef] [Green Version]
- Goon, J.A.; Nor Azman, N.H.E.; Abdul Ghani, S.M.; Hamid, Z.; Wan Ngah, W.Z. Comparing palm oil tocotrienol rich fraction with α-tocopherol supplementation on oxidative stress in healthy older adults. Clin. Nutr. ESPEN 2017, 21, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Taridi, N.M.; Yahaya, M.F.; Teoh, S.L.; Latiff, A.A.; Ngah, W.Z.W.; Das, S.; Mazlan, M. Tocotrienol rich fraction (TRF) supplementation protects against oxidative DNA damage and improves cognitive functions in Wistar rats. Clin. Ter. 2011, 162, 93–98. [Google Scholar] [PubMed]
- Damanhuri, H.A.; Rahim, N.I.A.; Nasri, W.N.W.; Tan, J.K.; Makpol, S.; Mazlan, M.; Tooyama, I.; Ngah, W.Z.W. Tocotrienol-rich fraction supplementation modulates antioxidant enzymes activity and reduces DNA damage in APPswe/PS1dE9 Alzheimer’s disease mouse model. Sains Malays. 2016, 45, 1363–1370. [Google Scholar]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet. Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef] [Green Version]
- Farina, M.; Avila, D.S.; da Rocha, J.B.T.; Aschner, M. Metals, oxidative stress and neurodegeneration: A focus on iron, manganese and mercury. Neurochem. Int. 2013, 62, 575–594. [Google Scholar] [CrossRef] [Green Version]
- Maynard, C.J.; Bush, A.I.; Masters, C.L.; Cappai, R.; Li, Q.-X. Metals and amyloid-beta in Alzheimer’s disease. Int. J. Exp. Pathol. 2005, 86, 147–159. [Google Scholar] [CrossRef]
- Cristóvão, J.S.; Santos, R.; Gomes, C.M. Metals and neuronal metal binding proteins implicated in Alzheimer’s disease. Oxid. Med. Cell. Longev. 2016, 2016, 9812178. [Google Scholar] [CrossRef] [Green Version]
- Berg, D.; Youdim, M.B.H. Role of iron in neurodegenerative disorders. Top. Magn. Reson. Imaging 2006, 17, 5–17. [Google Scholar] [CrossRef]
- Faller, P. Copper and zinc binding to amyloid-beta: Coordination, dynamics, aggregation, reactivity and metal-ion transfer. ChemBioChem 2009, 10, 2837–2845. [Google Scholar] [CrossRef]
- Cheignon, C.; Faller, P.; Testemale, D.; Hureau, C.; Collin, F. Metal-catalyzed oxidation of Aβ and the resulting reorganization of Cu binding sites promote ROS production. Metallomics 2016, 8, 1081–1089. [Google Scholar] [CrossRef] [Green Version]
- Šileikytė, J.; Forte, M. The mitochondrial permeability transition in mitochondrial disorders. Oxid. Med. Cell. Longev. 2019, 2019, 3403075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hare, D.J.; Double, K.L. Iron and dopamine: A toxic couple. Brain 2016, 139, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Bolton, J.L.; Dunlap, T. Formation and biological targets of quinones: Cytotoxic versus cytoprotective effects. Chem. Res. Toxicol. 2017, 30, 13–37. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Lee, J.; Darley-Usmar, V.M.; Zhang, J. Distinct effects of rotenone, 1-methyl-4-phenylpyridinium and 6-hydroxydopamine on cellular bioenergetics and cell death. PLoS ONE 2012, 7, e44610. [Google Scholar] [CrossRef] [PubMed]
- Varešlija, D.; Tipton, K.F.; Davey, G.P.; McDonald, A.G. 6-Hydroxydopamine: A far from simple neurotoxin. J. Neural Transm. 2020, 127, 213–230. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Garrett, M.R.; Men, P.; Zhu, X.; Perry, G.; Smith, M.A. Nanoparticle and other metal chelation therapeutics in Alzheimer disease. Biochim. Biophys. Acta-Mol. Basis Dis. 2005, 1741, 246–252. [Google Scholar] [CrossRef] [Green Version]
- Fine, J.M.; Forsberg, A.C.; Renner, D.B.; Faltesek, K.A.; Mohan, K.G.; Wong, J.C.; Arneson, L.C.; Crow, J.M.; Frey, W.H.; Hanson, L.R. Intranasally-administered deferoxamine mitigates toxicity of 6-OHDA in a rat model of Parkinson׳s disease. Brain Res. 2014, 1574, 96–104. [Google Scholar] [CrossRef]
- Sangchot, P.; Sharma, S.; Chetsawang, B.; Porter, J.; Govitrapong, P.; Ebadi, M. Deferoxamine attenuates iron-induced oxidative stress and prevents mitochondrial aggregation and alpha-synuclein translocation in SK-N-SH cells in culture. Dev. Neurosci. 2002, 24, 143–153. [Google Scholar] [CrossRef]
- Devos, D.; Moreau, C.; Devedjian, J.C.; Kluza, J.; Petrault, M.; Laloux, C.; Jonneaux, A.; Ryckewaert, G.; Garçon, G.; Rouaix, N.; et al. Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid. Redox Signal. 2014, 21, 195–210. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.-H.; Liang, D.-C.; Lin, H.-C.; Cheng, S.-Y.; Chen, L.-J.; Liu, H.-C. Auditory and visual toxicity during deferoxamine therapy in transfusion-dependent patients. J. Pediatr. Hematol. Oncol. 2005, 27, 651–653. [Google Scholar] [CrossRef]
- Hegde, M.L.; Bharathi, P.; Suram, A.; Venugopal, C.; Jagannathan, R.; Poddar, P.; Srinivas, P.; Sambamurti, K.; Rao, K.J.; Scancar, J.; et al. Challenges associated with metal chelation therapy in Alzheimer’s disease. J. Alzheimers Dis. 2009, 17, 457–468. [Google Scholar] [CrossRef] [Green Version]
- Mandel, S.; Maor, G.; Youdim, M.B.H. Iron and alpha-synuclein in the substantia nigra of MPTP-treated mice: Effect of neuroprotective drugs R-apomorphine and green tea polyphenol (−)-epigallocatechin-3-gallate. J. Mol. Neurosci. 2004, 24, 401–416. [Google Scholar] [CrossRef]
- Zhao, J.; Xu, L.; Liang, Q.; Sun, Q.; Chen, C.; Zhang, Y.; Ding, Y.; Zhou, P. Metal chelator EGCG attenuates Fe(III)-induced conformational transition of α-synuclein and protects AS-PC12 cells against Fe(III)-induced death. J. Neurochem. 2017, 143, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Ryan, P.; Hynes, M.J. The kinetics and mechanisms of the complex formation and antioxidant behaviour of the polyphenols EGCg and ECG with iron(III). J. Inorg. Biochem. 2007, 101, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Zhao, J.; Ding, L.; Ding, Y.; Zhou, P. Complex of EGCG with Cu(II) suppresses amyloid aggregation and Cu(II)-induced cytotoxicity of α-Synuclein. Molecules 2019, 24, 2940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiss, J.E. Laboratory and genetic assessment of iron deficiency in blood donors. Clin. Lab. Med. 2015, 35, 73–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reznichenko, L.; Amit, T.; Zheng, H.; Avramovich-Tirosh, Y.; Youdim, M.B.H.; Weinreb, O.; Mandel, S. Reduction of iron-regulated amyloid precursor protein and beta-amyloid peptide by (−)-epigallocatechin-3-gallate in cell cultures: Implications for iron chelation in Alzheimer’s disease. J. Neurochem. 2006, 97, 527–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, Y.; Wilkinson, J., 4th; Pietsch, E.C.; Buss, J.L.; Wang, W.; Planalp, R.; Torti, F.M.; Torti, S.V. Iron chelation in the biological activity of curcumin. Free Radic. Biol. Med. 2006, 40, 1152–1160. [Google Scholar] [CrossRef]
- Du, X.-X.; Xu, H.-M.; Jiang, H.; Song, N.; Wang, J.; Xie, J.-X. Curcumin protects nigral dopaminergic neurons by iron-chelation in the 6-hydroxydopamine rat model of Parkinson’s disease. Neurosci. Bull. 2012, 28, 253–258. [Google Scholar] [CrossRef] [Green Version]
- Dairam, A.; Fogel, R.; Daya, S.; Limson, J.L. Antioxidant and iron-binding properties of curcumin, capsaicin, and S-allylcysteine reduce oxidative stress in rat brain homogenate. J. Agric. Food Chem. 2008, 56, 3350–3356. [Google Scholar] [CrossRef]
- Yan, F.-S.; Sun, J.-L.; Xie, W.-H.; Shen, L.; Ji, H.-F. Neuroprotective effects and mechanisms of curcumin-Cu(II) and -Zn(II) complexes systems and their pharmacological implications. Nutrients 2017, 10, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mary, C.P.V.; Vijayakumar, S.; Shankar, R. Metal chelating ability and antioxidant properties of curcumin-metal complexes–A DFT approach. J. Mol. Graph. Model. 2018, 79, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rainey, N.E.; Moustapha, A.; Saric, A.; Nicolas, G.; Sureau, F.; Petit, P.X. Iron chelation by curcumin suppresses both curcumin-induced autophagy and cell death together with iron overload neoplastic transformation. Cell Death Discov. 2019, 5, 150. [Google Scholar] [CrossRef] [PubMed]
- Riezzo, I.; Cerretani, D.; Fiore, C.; Bello, S.; Centini, F.; D’Errico, S.; Fiaschi, A.I.; Giorgi, G.; Neri, M.; Pomara, C.; et al. Enzymatic-nonenzymatic cellular antioxidant defense systems response and immunohistochemical detection of MDMA, VMAT2, HSP70, and apoptosis as biomarkers for MDMA (Ecstasy) neurotoxicity. J. Neurosci. Res. 2010, 88, 905–916. [Google Scholar] [CrossRef]
- Shim, S.-Y.; Kim, H.-S. Oxidative stress and the antioxidant enzyme system in the developing brain. Korean J. Pediatr. 2013, 56, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, P.; Jaleel, C.A.; Salem, M.A.; Nabi, G.; Sharma, S. Roles of enzymatic and nonenzymatic antioxidants in plants during abiotic stress. Crit. Rev. Biotechnol. 2010, 30, 161–175. [Google Scholar] [CrossRef]
- Koyama, H.; Nojiri, H.; Kawakami, S.; Sunagawa, T.; Shirasawa, T.; Shimizu, T. Antioxidants improve the phenotypes of dilated cardiomyopathy and muscle fatigue in mitochondrial superoxide dismutase-deficient mice. Molecules 2013, 18, 1383–1393. [Google Scholar] [CrossRef]
- Youssef, P.; Chami, B.; Lim, J.; Middleton, T.; Sutherland, G.T.; Witting, P.K. Evidence supporting oxidative stress in a moderately affected area of the brain in Alzheimer’s disease. Sci. Rep. 2018, 8, 11553. [Google Scholar] [CrossRef] [Green Version]
- Power, J.H.T.; Blumbergs, P.C. Cellular glutathione peroxidase in human brain: Cellular distribution, and its potential role in the degradation of Lewy bodies in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol. 2009, 117, 63–73. [Google Scholar] [CrossRef]
- Dringen, R.; Hirrlinger, J. Glutathione pathways in the brain. Biol. Chem. 2003, 384, 505–516. [Google Scholar] [CrossRef]
- Das, K.; Roychoudhury, A. Reactive oxygen species (ROS) and response of antioxidants as ROS-scavengers during environmental stress in plants. Front. Environ. Sci. 2014, 2, 53. [Google Scholar] [CrossRef] [Green Version]
- Shungu, D.; Mao, X.; Weiduschat, N.; Hanineva, A.; Zhao, Y.; Mangat, H.; Kang, G.; Henchcliffe, C. Nigrostriatal glutathione deficit in Parkinson’s disease measured in vivo with MRS supports oxidative stress in disease pathophysiology. J. Neurol. Sci. 2017, 381, 737. [Google Scholar] [CrossRef]
- Smeyne, M.; Smeyne, R.J. Glutathione metabolism and Parkinson’s disease. Free Radic. Biol. Med. 2013, 62, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bermejo, P.; Martín-Aragón, S.; Benedí, J.; Susín, C.; Felici, E.; Gil, P.; Ribera, J.M.; Villar, A.M. Peripheral levels of glutathione and protein oxidation as markers in the development of Alzheimer’s disease from Mild Cognitive Impairment. Free Radic. Res. 2008, 42, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of Nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Robledinos-Antón, N.; Fernández-Ginés, R.; Manda, G.; Cuadrado, A. Activators and inhibitors of Nrf2: A review of their potential for clinical development. Oxid. Med. Cell. Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Kazantsev, A.G. The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J. 2018, 285, 3576–3590. [Google Scholar] [CrossRef] [Green Version]
- Rojo, A.I.; Pajares, M.; Rada, P.; Nuñez, A.; Nevado-Holgado, A.J.; Killik, R.; van Leuven, F.; Ribe, E.; Lovestone, S.; Yamamoto, M.; et al. Nrf2 deficiency replicates transcriptomic changes in Alzheimer’s patients and worsens APP and TAU pathology. Redox Biol. 2017, 13, 444–451. [Google Scholar] [CrossRef]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [Green Version]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Z.; Zhu, H.; Li, Y.; Misra, H.P. Potent inhibition of peroxynitrite-induced DNA strand breakage and hydroxyl radical formation by dimethyl sulfoxide at very low concentrations. Exp. Biol. Med. 2010, 235, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Kostić, D.A.; Dimitrijević, D.S.; Stojanović, G.S.; Palić, I.R.; Đorđević, A.S.; Ickovski, J.D. Xanthine oxidase: Isolation, assays of activity, and inhibition. J. Chem. 2015, 2015, 294858. [Google Scholar] [CrossRef] [Green Version]
- Mokni, M.; Elkahoui, S.; Limam, F.; Amri, M.; Aouani, E. Effect of resveratrol on antioxidant enzyme activities in the brain of healthy rat. Neurochem. Res. 2007, 32, 981–987. [Google Scholar] [CrossRef]
- Cosín-Tomàs, M.; Senserrich, J.; Arumí-Planas, M.; Alquézar, C.; Pallàs, M.; Martín-Requero, Á.; Suñol, C.; Kaliman, P.; Sanfeliu, C. Role of resveratrol and selenium on oxidative stress and expression of antioxidant and anti-aging genes in immortalized lymphocytes from Alzheimer’s disease patients. Nutrients 2019, 11, 1764. [Google Scholar] [CrossRef] [Green Version]
- Franklin, C.C.; Backos, D.S.; Mohar, I.; White, C.C.; Forman, H.J.; Kavanagh, T.J. Structure, function, and post-translational regulation of the catalytic and modifier subunits of glutamate cysteine ligase. Mol. Aspects Med. 2009, 30, 86–98. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-Y.; Jang, J.-H.; Li, M.-H.; Surh, Y.-J. Resveratrol upregulates heme oxygenase-1 expression via activation of NF-E2-related factor 2 in PC12 cells. Biochem. Biophys. Res. Commun. 2005, 331, 993–1000. [Google Scholar] [CrossRef]
- Chen, J.; Tang, X.Q.; Zhi, J.L.; Cui, Y.; Yu, H.M.; Tang, E.H.; Sun, S.N.; Feng, J.Q.; Chen, P.X. Curcumin protects PC12 cells against 1-methyl-4-phenylpyridinium ion-induced apoptosis by bcl-2-mitochondria-ROS-iNOS pathway. Apoptosis 2006, 11, 943–953. [Google Scholar] [CrossRef]
- Zhang, M.; An, C.; Gao, Y.; Leak, R.K.; Chen, J.; Zhang, F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog. Neurobiol. 2013, 100, 30–47. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Tian, X.; Guo, Y.; Duan, W.; Bu, H.; Li, C. Activation of nuclear factor erythroid 2-related factor 2 cytoprotective signaling by curcumin protect primary spinal cord astrocytes against oxidative toxicity. Biol. Pharm. Bull. 2011, 34, 1194–1197. [Google Scholar] [CrossRef] [Green Version]
- Cui, Q.; Li, X.; Zhu, H. Curcumin ameliorates dopaminergic neuronal oxidative damage via activation of the Akt/Nrf2 pathway. Mol. Med. Rep. 2016, 13, 1381–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes-Fermín, L.M.; González-Reyes, S.; Tarco-Álvarez, N.G.; Hernández-Nava, M.; Orozco-Ibarra, M.; Pedraza-Chaverri, J. Neuroprotective effect of α-mangostin and curcumin against iodoacetate-induced cell death. Nutr. Neurosci. 2012, 15, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Panchal, H.D.; Vranizan, K.; Lee, C.Y.; Ho, J.; Ngai, J.; Timiras, P.S. Early anti-oxidative and anti-proliferative curcumin effects on neuroglioma cells suggest therapeutic targets. Neurochem. Res. 2008, 33, 1701–1710. [Google Scholar] [CrossRef] [PubMed]
- Levites, Y.; Weinreb, O.; Maor, G.; Youdim, M.B.; Mandel, S. Green tea polyphenol (-)-epigallocatechin-3-gallate prevents N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced dopaminergic neurodegeneration. J. Neurochem. 2001, 78, 1073–1082. [Google Scholar] [CrossRef]
- Kim, J.S.; Kim, J.-M.; Jeong-Ja, O.; Jeon, B.S. Inhibition of inducible nitric oxide synthase expression and cell death by (−)-epigallocatechin-3-gallate, a green tea catechin, in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2010, 17, 1165–1168. [Google Scholar] [CrossRef]
- Li, R.; Huang, Y.-G.; Fang, D.; Le, W.-D. (−)-Epigallocatechin gallate inhibits lipopolysaccharide-induced microglial activation and protects against inflammation-mediated dopaminergic neuronal injury. J. Neurosci. Res. 2004, 78, 723–731. [Google Scholar] [CrossRef]
- Biasibetti, R.; Tramontina, A.C.; Costa, A.P.; Dutra, M.F.; Quincozes-Santos, A.; Nardin, P.; Bernardi, C.L.; Wartchow, K.M.; Lunardi, P.S.; Gonçalves, C.-A. Green tea (-)epigallocatechin-3-gallate reverses oxidative stress and reduces acetylcholinesterase activity in a streptozotocin-induced model of dementia. Behav. Brain Res. 2013, 236, 186–193. [Google Scholar] [CrossRef]
- Liu, Z.-M.; Tse, L.A.; Chen, B.; Wu, S.; Chan, D.; Kowk, T.; Woo, J.; Xiang, Y.-T.; Wong, S.Y.-S. Dietary acrylamide exposure was associated with mild cognition decline among non-smoking Chinese elderly men. Sci. Rep. 2017, 7, 6395. [Google Scholar] [CrossRef] [Green Version]
- Esmaeelpanah, E.; Razavi, B.M.; Hasani, F.V.; Hosseinzadeh, H. Evaluation of epigallocatechin gallate and epicatechin gallate effects on acrylamide-induced neurotoxicity in rats and cytotoxicity in PC 12 cells. Drug Chem. Toxicol. 2018, 41, 441–448. [Google Scholar] [CrossRef]
- Singh, J.C.H.; Alagarsamy, V.; Kumar, S.S.; Reddy, Y.N. Neurotransmitter metabolic enzymes and antioxidant status on Alzheimer’s disease induced mice treated with Alpinia galanga (L.) Willd. Phytother. Res. 2011, 25, 1061–1067. [Google Scholar] [CrossRef]
- Peng, S.; Yao, J.; Liu, Y.; Duan, D.; Zhang, X.; Fang, J. Activation of Nrf2 target enzymes conferring protection against oxidative stress in PC12 cells by ginger principal constituent 6-shogaol. Food Funct. 2015, 6, 2813–2823. [Google Scholar] [CrossRef] [PubMed]
- Magesh, S.; Chen, Y.; Hu, L. Small molecule modulators of Keap1-Nrf2-ARE pathway as potential preventive and therapeutic agents. Med. Res. Rev. 2012, 32, 687–726. [Google Scholar] [CrossRef] [Green Version]
- Andriollo-Sanchez, M.; Hininger-Favier, I.; Meunier, N.; Venneria, E.; O’Connor, J.M.; Maiani, G.; Coudray, C.; Roussel, A.M. Age-related oxidative stress and antioxidant parameters in middle-aged and older European subjects: The ZENITH study. Eur. J. Clin. Nutr. 2005, 59, S58–S62. [Google Scholar] [CrossRef] [PubMed]
- Azman, N.H.E.N.; Goon, J.A.; Ghani, S.M.A.; Hamid, Z.; Ngah, W.Z.W. Comparing palm oil, tocotrienol-rich fraction and α-tocopherol supplementation on the antioxidant levels of older adults. Antioxidants 2018, 7, 74. [Google Scholar] [CrossRef] [Green Version]
- Zarkasi, K.A.; Zainalabidin, S.; Jen-Kit, T.; Hakimi, N.H.; Ramli, N.Z.; Jubri, Z. Tocotrienol-rich fraction modulates cardiac metabolic profile changes in isoprenaline-induced myocardial infarction rats. Sains Malays. 2020, 49, 357–373. [Google Scholar] [CrossRef]
- Ozkan, A.; Fiskin, K.; Ayhan, A.G. Effect of vitamin E and selenium on antioxidant enzymes in brain, kidney and liver of cigarette smoke-exposed mice. Biologia 2007, 62, 360–364. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-Y.; Wen, L.-L.; Huang, Y.-N.; Chen, Y.-T.; Ku, M.-C. Dual effects of antioxidants in neurodegeneration: Direct neuroprotection against oxidative stress and indirect protection via suppression of glia-mediated inflammation. Curr. Pharm. Des. 2006, 12, 3521–3533. [Google Scholar] [CrossRef]
- Costantini, E.; D’Angelo, C.; Reale, M. The Role of immunosenescence in neurodegenerative diseases. Mediat. Inflamm. 2018, 2018, 6039171. [Google Scholar] [CrossRef]
- Son, Y.; Cheong, Y.-K.; Kim, N.-H.; Chung, H.-T.; Kang, D.G.; Pae, H.-O. Mitogen-activated protein kinases and reactive oxygen species: How can ROS activate MAPK pathways? J. Signal Transduct. 2011, 2011, 792639. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [Green Version]
- Thalhamer, T.; McGrath, M.A.; Harnett, M.M. MAPKs and their relevance to arthritis and inflammation. Rheumatology 2008, 47, 409–414. [Google Scholar] [CrossRef] [Green Version]
- Yue, J.; López, J.M. Understanding MAPK Signaling Pathways in Apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- León-Buitimea, A.; Rodríguez-Fragoso, L.; Lauer, F.T.; Bowles, H.; Thompson, T.A.; Burchiel, S.W. Ethanol-induced oxidative stress is associated with EGF receptor phosphorylation in MCF-10A cells overexpressing CYP2E1. Toxicol. Lett. 2012, 209, 161–165. [Google Scholar] [CrossRef] [Green Version]
- Matsuzawa, A.; Ichijo, H. Redox control of cell fate by MAP kinase: Physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochim. Biophys. Acta 2008, 1780, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Huang, J.-Z.; Chen, L.; Chen, Y.; Li, X. In vivo and in vitro studies on the roles of p38 mitogen-activated protein kinase and NADPH-cytochrome P450 reductase in Alzheimer’s disease. Exp. Ther. Med. 2017, 14, 4755–4760. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.; Anand, V.S.; Ludwig, B.; Nawoschik, S.; Dunlop, J.; Braithwaite, S.P. The JNK pathway amplifies and drives subcellular changes in tau phosphorylation. Neuropharmacology 2009, 57, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Wang, M.; Du, Y.; Zhang, W.; Bai, M.; Zhang, Z.; Li, Z.; Miao, J. Inhibition of c-Jun N-terminal kinase activation reverses Alzheimer disease phenotypes in APPswe/PS1dE9 mice. Ann. Neurol. 2015, 77, 637–654. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Dresselhaus, E.C.; Meffert, M.K. Cellular specificity of NF-κB function in the nervous system. Front. Immunol. 2019, 10, 1043. [Google Scholar] [CrossRef]
- Chen, C.-H.; Zhou, W.; Liu, S.; Deng, Y.; Cai, F.; Tone, M.; Tone, Y.; Tong, Y.; Song, W. Increased NF-κB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2012, 15, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Chao, Y.; Wong, S.C.; Tan, E.K. Evidence of inflammatory system involvement in Parkinson’s disease. BioMed Res. Int. 2014, 2014, 308654. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Orosz, Z.; Labinskyy, N.; Rivera, A.; Xiangmin, Z.; Smith, K.; Csiszar, A. Increased mitochondrial H2O2 production promotes endothelial NF-κB activation in aged rat arteries. Am. J. Physiol. Circ. Physiol. 2007, 293, H37–H47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riquelme, D.; Alvarez, A.; Leal, N.; Adasme, T.; Espinoza, I.; Valdés, J.A.; Troncoso, N.; Hartel, S.; Hidalgo, J.; Hidalgo, C.; et al. High-frequency field stimulation of primary neurons enhances ryanodine receptor-mediated Ca2+ release and generates hydrogen peroxide, which jointly stimulate NF-κB activity. Antioxid. Redox Signal. 2011, 14, 1245–1259. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, M.; Lahiri, D.K. Significancenof NF-κB as a pivotal therapeutic target in the neurodegenerative pathologies of Alzheimer’s disease and multiple sclerosis. Expert Opin. Ther. Targets 2015, 19, 471–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valerio, A.; Boroni, F.; Benarese, M.; Sarnico, I.; Ghisi, V.; Bresciani, L.G.; Ferrario, M.; Borsani, G.; Spano, P.; Pizzi, M. NF-κB pathway: A target for preventing β-amyloid (Aβ)-induced neuronal damage and Aβ42 production. Eur. J. Neurosci. 2006, 23, 1711–1720. [Google Scholar] [CrossRef]
- Singh, S.S.; Rai, S.N.; Birla, H.; Zahra, W.; Rathore, A.S.; Singh, S.P. NF-κB-mediated neuroinflammation in Parkinson’s disease and potential therapeutic effect of polyphenols. Neurotox. Res. 2020, 37, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef] [Green Version]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.-J.; Koh, S.-H. The role of PI3K/AKT pathway and its therapeutic possibility in Alzheimer’s disease. Hanyang Med. Rev. 2017, 37, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Timmons, S.; Coakley, M.F.; Moloney, A.M.; O’Neill, C. Akt signal transduction dysfunction in Parkinson’s disease. Neurosci. Lett. 2009, 467, 30–35. [Google Scholar] [CrossRef]
- Cao, J.; Xu, D.; Wang, D.; Wu, R.; Zhang, L.; Zhu, H.; He, Q.; Yang, B. ROS-driven Akt dephosphorylation at Ser-473 is involved in 4-HPR-mediated apoptosis in NB4 cells. Free Radic. Biol. Med. 2009, 47, 536–547. [Google Scholar] [CrossRef]
- Caulfield, A.J.; Lathem, W.W. Disruption of fas-fas ligand signaling, apoptosis, and innate immunity by bacterial pathogens. PLoS Pathog. 2014, 10, e1004252. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.-W.; Seo, J.; Jeong, M.; Lee, S.; Song, J. The roles of FADD in extrinsic apoptosis and necroptosis. BMB Rep. 2012, 45, 496–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Che, X.; Zheng, Q.; Wu, A.; Pan, K.; Shao, A.; Wu, Q.; Zhang, J.; Hong, Y. Caspases: A molecular switch node in the crosstalk between autophagy and apoptosis. Int. J. Biol. Sci. 2014, 10, 1072–1083. [Google Scholar] [CrossRef]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Zhou, L.; Zhao, T.; Liu, X.; Zhang, P.; Liu, Y.; Zheng, X.; Li, Q. Caspase-9: Structure, mechanisms and clinical application. Oncotarget 2017, 8, 23996–24008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Miao, X.; Wang, H.; Xu, Z.; Li, B. The tissue dependent interactions between p53 and Bcl-2 in vivo. Oncotarget 2015, 6, 35699–35709. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.S.; Muhammad, T.; Ikram, M.; Kim, M.O. Dietary supplementation of the antioxidant curcumin halts systemic lps-induced neuroinflammation-associated neurodegeneration and memory/synaptic impairment via the JNK/NF-κB/Akt signaling pathway in adult rats. Oxid. Med. Cell. Longev. 2019, 2019, 7860650. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Jin, M.L.; Kim, Y.H.; Kim, Y.; Lee, S.J. Anti-inflammatory effects of aromatic-turmerone through blocking of NF-κB, JNK, and p38 MAPK signaling pathways in amyloid β-stimulated microglia. Int. Immunopharmacol. 2012, 14, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-J.; Li, Z.-H.; Liu, L.; Tang, W.-X.; Wang, Y.; Dong, M.-R.; Xiao, C. Curcumin attenuates beta-amyloid-induced neuroinflammation via activation of peroxisome proliferator-activated receptor-gamma function in a rat model of Alzheimer’s disease. Front. Pharmacol. 2016, 7, 261. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Sun, P.; Zhang, J.-C.; Zhang, Q.; Yao, S.-L. Proinflammatory effects of S100A8/A9 via TLR4 and RAGE signaling pathways in BV-2 microglial cells. Int. J. Mol. Med. 2017, 40, 31–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Othman, I.; Aamir, K.; Shaikh, M.F. Impact of HMGB1, RAGE, and TLR4 in Alzheimer’s Disease (AD): From Risk Factors to Therapeutic Targeting. Cells 2020, 9, 383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikram, M.; Saeed, K.; Khan, A.; Muhammad, T.; Khan, M.S.; Jo, M.G.; Rehman, S.U.; Kim, M.O. Natural dietary supplementation of curcumin protects mice brains against ethanol-induced oxidative stress-mediated neurodegeneration and memory impairment via Nrf2/TLR4/RAGE signaling. Nutrients 2019, 11, 1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.-F.; Zhang, Y.-J.; Zhou, H.-Y.; Wang, H.-M.; Tian, L.-P.; Liu, J.; Ding, J.-Q.; Chen, S.-D. Curcumin ameliorates the neurodegenerative pathology in A53T α-synuclein cell model of Parkinson’s disease through the downregulation of mTOR/p70S6K signaling and the recovery of macroautophagy. J. Neuroimmune Pharmacol. 2013, 8, 356–369. [Google Scholar] [CrossRef]
- Griner, E.M.; Kazanietz, M.G. Protein kinase C and other diacylglycerol effectors in cancer. Nat. Rev. Cancer 2007, 7, 281–294. [Google Scholar] [CrossRef]
- Levites, Y.; Amit, T.; Youdim, M.B.H.; Mandel, S. Involvement of protein kinase C activation and cell survival/cell cycle genes in green tea polyphenol (−)-epigallocatechin 3-gallate neuroprotective action. J. Biol. Chem. 2002, 277, 30574–30580. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-López, L.; Márquez-Valadez, B.; Gómez-Sánchez, A.; Silva-Lucero, M.D.C.; Torres-Pérez, M.; Téllez-Ballesteros, R.I.; Ichwan, M.; Meraz-Ríos, M.A.; Kempermann, G.; Ramírez-Rodríguez, G.B. Green tea compound epigallo-catechin-3-gallate (EGCG) increases neuronal survival in adult hippocampal neurogenesis in vivo and in vitro. Neuroscience 2016, 322, 208–220. [Google Scholar] [CrossRef]
- Cong, L.; Cao, C.; Cheng, Y.; Qin, X.-Y. Green tea polyphenols attenuated glutamate excitotoxicity via antioxidative and antiapoptotic pathway in the primary cultured cortical neurons. Oxid. Med. Cell. Longev. 2016, 2016, 2050435. [Google Scholar] [CrossRef]
- Imenshahidi, M.; Hosseinzadeh, H. Berberis Vulgaris and Berberine: An update review. Phytother. Res. 2016, 30, 1745–1764. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Sharma, B. Toxicological effects of berberine and sanguinarine. Front. Mol. Biosci. 2018, 5, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, L.; Liu, J.; Song, Z.; Pan, X.; Chen, L.; Cui, X.; Wang, M. Berberine suppresses amyloid-beta-induced inflammatory response in microglia by inhibiting nuclear factor-kappaB and mitogen-activated protein kinase signalling pathways. J. Pharm. Pharmacol. 2012, 64, 1510–1521. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Zhao, J.; Wang, H.; Jiang, Y.; Yang, Q.; Fu, Y.; Zeng, H.; Hölscher, C.; Xu, J.; Zhang, Z. Magnolol alleviates Alzheimer’s disease-like pathology in transgenic C. elegans by promoting microglia phagocytosis and the degradation of beta-amyloid through activation of PPAR-γ. Biomed. Pharmacother. 2020, 124, 109886. [Google Scholar] [CrossRef]
- Nasri, W.N.W.; Makpol, S.; Mazlan, M.; Tooyama, I.; Ngah, W.Z.W.; Damanhuri, H.A. Tocotrienol rich fraction supplementation modulate brain hippocampal gene expression in APPswe/PS1dE9 Alzheimer’s disease mouse model. J. Alzheimers Dis. 2019, 70, S239–S254. [Google Scholar] [CrossRef] [Green Version]
- Kalani, K.; Yan, S.F.; Yan, S.S. Mitochondrial permeability transition pore: A potential drug target for neurodegeneration. Drug Discov. Today 2018, 23, 1983–1989. [Google Scholar] [CrossRef]


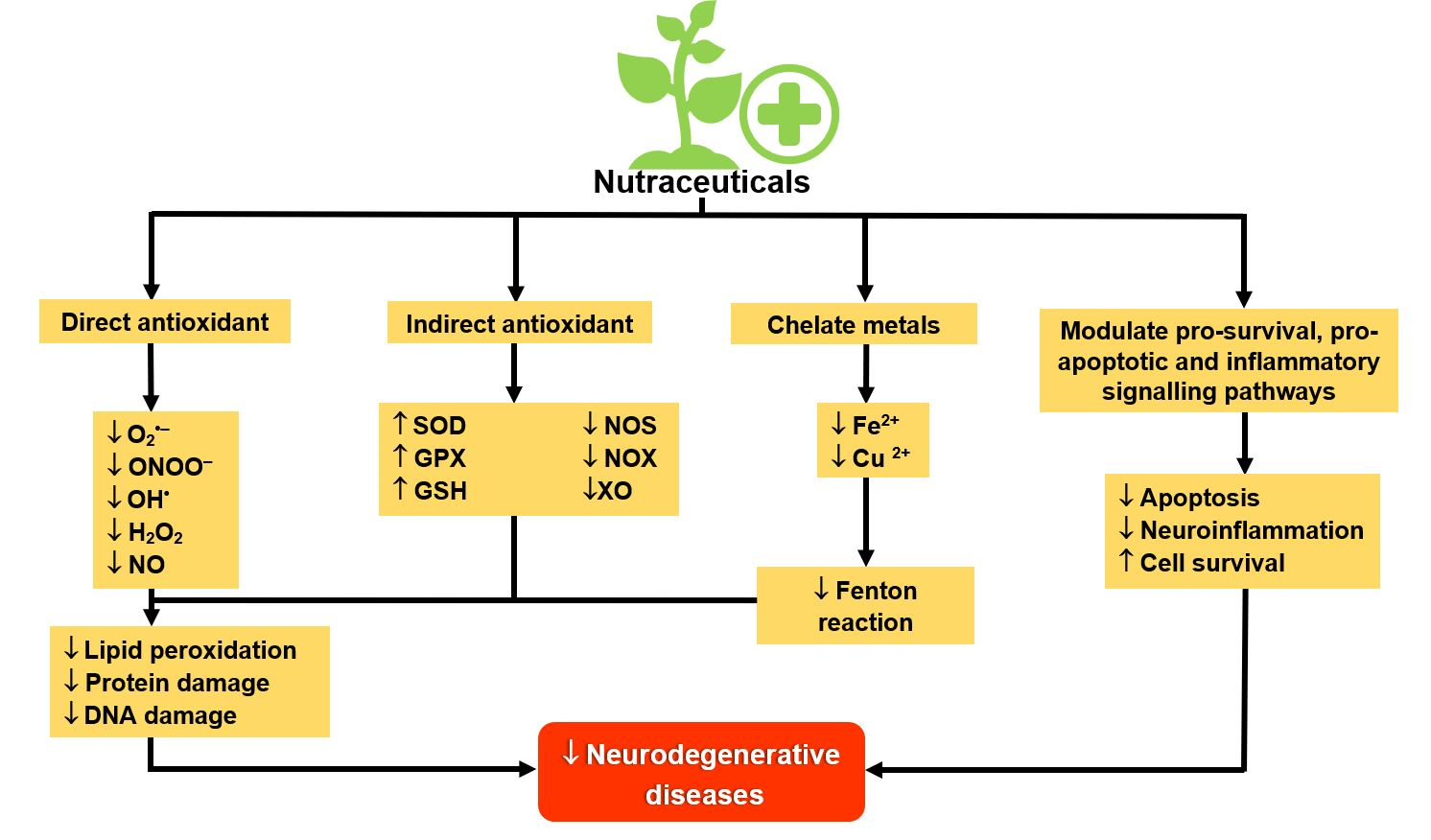{kind=link}
{kind=link}
| Nutraceutical and Its Active Compounds | Dosage | Experimental Models | Direct Antioxidant Effects | References |
|---|---|---|---|---|
| Resveratrol | 10 mg/kg BW/day for two weeks | Angiotensin II-induced early AD rats | ↓ superoxide levels in the nucleus tractus solitarius and hippocampus. | [69] |
| 10 and 20 mg/kg BW for 21 days | ICV-STZ infused rats | ↓ MDA levels in the brain. | [72] | |
| Piceatannol | 10 and 20 µM | Aβ-induced cytotoxicity PC 12 cells | ↓ the intracellular ROS generation. | [71] |
| Grape seed extract. Active compound: Proanthocyanidins | 100 mg/kg BW/day for 30 days | Aged rats | ↓ 8-OHdG and DNA protein cross-links levels in the rat brains and spinal cords. | [76] |
| ↓ ROS and PC levels in the rat brains and spinal cords. | [77] | |||
| EGb 761 extract. Active compounds: 24% flavonols (kaempferol, quercetin and isorhamnetin), 6% terpene lactones (ginkgolide-A, -B, -C -M, and -J and bilobalide B) | Pre-treatment of EGb 761 (100 µg/mL) for 48 h | Aβ-secreting mutant cells | ↓ intracellular ROS levels. | [29] |
| Green tea polyphenols | 2 g/day of green tea pills. Each pill contained 50 mg of total polyphenols (EGCG, EC, and ECG). | Severe AD patients | Levels of MDA, 8-OHdG and PC ↓ significantly as compared to baseline values. | [83] |
| ↑ FRAP assay. | ||||
| Curcumin | 2000 parts per million (ppm) of curcumin mixed in the diet | Aβ-induced cognitive deficits and neuropathology rats | ↓ IsoPs levels in rat brains. | [84] |
| 500 ppm of curcumin mixed in the diet | APP transgenic mouse | ↓ LPO in the spinal cord. | [85] | |
| CUR-CA-THIONE | 500 mg/kg BW/day for fifteen days | Aluminum chloride-induced AD rats | Inhibits LPO in the rat brains. | [91] |
| Demethoxycurcumin | 50 nM | Rotenone induced neurotoxicity in neuroblastoma cells | ↓ the intracellular ROS generation. | [92] |
| Xanthorrhizol | 0.5, 1, 5, and 10 µM | H2O2-induced lipid peroxidation in rat brain homogenate | ↓ LPO in the rat brains. | [93] |
| 2 µM | Glutamate-induced cytotoxicity in hippocampal neurons | ↓ intracellular ROS generation in the hippocampal neurons. | ||
| Magnolol | 30 mg/kg BW, given once after MPTP injection | MPTP-induced Parkinson in mice | ↓ lipid peroxidation in the striatum. | [94] |
| 16 and 32 µM | Acrolein-induced oxidative damage in neuroblastoma cells | ↓ PC, HNE, and superoxide levels. | [96] | |
| Pycnogenol®. Active compounds: Procyanidins, phenolic acids, and polyphenols (catechin, epicatechin, and taxifolin) | 50 and 100 μg/mL | Acrolein-induced oxidative damage in neuroblastoma cells | ↓ ROS, superoxide, PC, and 4-HNE levels. | [99] |
| Pre-treatment of Pycnogenol®, 10 mg/kg BW for three weeks | ICV-STZ induces AD in rats | ↓ PC and LPO in the hippocampus and cerebral cortex. | [100] | |
| 150 mg/day for six weeks | Healthy subjects | ↑ antioxidant capacity of plasma compared to baseline. | [101] | |
| 50 mg/day for eight weeks | Human subjects (smokers) | ↓ plasma reactive oxygen metabolites and ↑ antioxidant potential compared to smokers taking placebo. | [102] | |
| Guarana seed extract. Active compound: Polyphenols (catechins and epicatechins) | 100 and 1000 μg/mL | Acrolein-induced cytotoxicity on neuronal cells | ↓ intracellular ROS production. | [108] |
| 10 and 50 mg/mL of GHE | C. elegans models of AD | [107] | ||
| Tocotrienol rich fraction (TRF). Active compounds: α-tocopherol (23.40%) and α-, β-, γ-, and δ-tocotrienol (27.30, 3.34, 35.51, and 10.45%, respectively) | 150 mg/day for six months | Older adults aged 50–55 years old | ↓ plasma MDA and DNA damage. | [115] |
| 200 mg/kg BW for eight months | APPswe/PS1dE9 transgenic mouse model of AD | ↓ DNA strand breaks in the blood. | [117] |
| Nutraceutical and Its Active Compounds | Dosage | Experimental Models | Metal Chelators Effect | References |
|---|---|---|---|---|
| EGCG | 20 µM | Fe3+ induced fibrillation of α-Syn and Fe3+ induced cytotoxicity in α-Syn-PC 12 cells | Molecular: Slowed the formation of α-Syn fibrillation and β-sheet conformers, inhibited the conformational transition of α-Syn in the nucleation period, remodel amyloid fibrils into soluble amorphous aggregates. | [137] |
| Cellular: ↓ ROS generation and ↑ cell viability. | ||||
| 20 µM | Cu2+ induced fibrillation of α-Syn and Cu2+ induced cytotoxicity in α-Syn-PC 12 cells | Molecular: inhibited the formation of Cu2+-induced amyloid fibrillation of α-Syn, inhibited β-sheet conformation, restructure amyloid fibrils into soluble amorphous aggregates, forming EGCG/Cu2+ complex and binds to Tyr in α-Syn. | [139] | |
| Cellular: ↓ expression and aggregation of α-Syn, cell apoptotic percentage, ↓ ROS generation. | ||||
| 5 and 10µM | SH-SY5Y cells | ↑ the TfR and TfR mRNA expressions as well as ↓ APP expression. | [141] | |
| Curcumin | In vitro: 12.5, 25 and 50 µM | Liver cell line BNL CL.2 and mice fed with 2% of dietary curcumin | ↓ H and L ferritin protein expression. | [142] |
| In vivo: 2.0% dietary curcumin for twelve weeks | ||||
| 200 mg/kg BW/twice a day for twenty-four days | 6-OHDA-induced PD in Wistar rats | ↓ the number of iron positive cell count in substantia nigra. | [143] | |
| 25 µM | Wistar brain homogenate | ↓ Fe2+ and Quinolic acid-induced lipid peroxidation. | [144] | |
| 2:1 and 1:1 curcumin-Cu2+ complexes; 2:1 and 1:1 curcumin-Zn2+ complexes | H2O2-induced injury in PC 12 cells | ↑ antioxidant enzymes, CAT, SOD and GPX levels. | [145] | |
| ↓ ROS and MDA levels and ↑ cell viability. |
| Nutraceutical and Its Active Compounds | Dosage | Experimental Models | Indirect Antioxidant Effects | References |
|---|---|---|---|---|
| Resveratrol | 12.5 mg/kg BW/day for seven days | Healthy Wistar rats | ↑ CAT, SOD, and peroxidase in rat brains. | [168] |
| 50 µM | Cultured lymphoblastoid cell lines of AD patients | ↑ CAT, SOD2, and NFE2L2 gene expressions. | [169] | |
| 15 µM | H2O2 induced cytotoxicity on PC 12 cells | ↑ HO-1 and GCLC gene expression and stimulated Nrf2 nuclear translocation. | [171] | |
| Curcumin | 20 µmol/L | MPP+ induced cytotoxicity on PC 12 cells | ↓ iNOS overexpression, and attenuated MPP+ induced apoptosis. | [172] |
| 5, 10, and 15 µM | Nrf2+/+ primary astrocytes with H2O2 insult | ↑ Nrf2 target genes (HO-1, NQO1, GCLC) in Nrf2+/+ primary astrocytes. | [174] | |
| 100 mg/kg BW/twice a day for 50 days | Rotenone-induced PD rats | ↑ GSH, HO-1 and NQO1 protein through the Akt/Nrf2 pathway. | [175] | |
| 15, 20, and 30 µM | Iodoacetate induced cytotoxicity on primary rat cerebellar granule neurons | ↑ HO-1 expression. | [176] | |
| 5 µM | C6 rat glioma cells | ↑ genes involved in antioxidant response including HO-1, NQO1, CAT, TrxR, and GCLC. | [177] | |
| EGCG | 2 mg/kg BW/day for 10 days | MPTP-induced parkinsonism in male C57/BL mice | Prevented the ↑ of SOD and CAT in the striatum induced by MPTP. | [178] |
| 10 and 50 mg/kg BW/day for 14 days | MPTP-induced parkinsonism mice | ↓ iNOS expression in substantia nigra and striatum. | [179] | |
| 1, 5, and 10 µM | Lipopolysaccharide-induced microglial activation | ↓ iNOS in activated microglia. | [180] | |
| 10 mg/kg BW/day for four weeks | ICV-STZ infusion in rats | ↑ GPX and maintained GSH as well as, ↓ ROS and NO levels in the hippocampus. | [181] | |
| 20 mg/kg BW/day for 14 days | Acrylamide-induced neurotoxicity in rats and cytotoxicity in PC 12 cells | ↑ GSH level in the cerebral cortex. | [183] | |
| Ginger extract | 200 and 400 mg/kg BW/day for 14 days | Aβ induced AD mice | ↑ SOD, GPX, CAT, and non-enzymatic antioxidant vitamin C in the brain. Inhibited the MAO A and B activity. | [184] |
| 6-shogaols | 20 µM | H2O2- and 6-OHDA-induced cytotoxicity in PC 12 cells | ↑ phase two enzymes genes expression and the respective enzyme’s activity; HO-1, NQO1, TrxR1, GCLC, and GCLM via activation of the NrF2/ARE pathway. | [185] |
| ↑ translocation of Nrf2 from the cytosol to nucleus | ||||
| Tocotrienol rich fraction (TRF). Active compounds: α-tocopherol (23.40%) and α, β, γ, and δ-tocotrienol (27.30, 3.34, 35.51, and 10.45%, respectively) | 150 mg/day for six months | Healthy older adults aged 50 to 55 years | ↑ SOD, GPX, and GSH/GSSG ratio in the blood compared to baseline | [188] |
| 200 mg/kg BW/day for three months | Aged Wistar rats (21 months old) | ↑ SOD, CAT, and GPX in the blood. Higher concentrations of total vitamin E in the brain. | [113] |
| Nutraceutical and Its Active Compounds | Dosage | Experimental Models | Signaling Pathways Affected | References |
|---|---|---|---|---|
| Curcumin | 300 mg/kg BW/day for 14 days | Lipopolysaccharide-induced neurotoxicity in rats and microglial cells | ↓ p-JNK, p-NF-κB, TNF-α, IL-1β, GFAP, and Iba-1 in the hippocampus. | [224] |
| ↓ apoptotic markers (Bax, caspase-3, Cyt c, and PARP-1), while anti-apoptotic protein Bcl-2 was ↑. | ||||
| Inhibited p-JNK activation similar to the JNK inhibitor, SP600125, in vitro. | ||||
| Ar-turmerone | 20 μM | Aβ-stimulated microglial cells | ↓ MCP1, TNF-α, IL-1β, and IL-6 by inhibiting MAPK (p-JNK and p38) and NF-κB signaling pathways. | [225] |
| Curcumin | 150 mg/kg BW/day for four consecutive weeks | Transgenic APP/PS1 mice and mixed neuronal/glial cultures | ↓ microglial activation as well as inflammatory mediators, including TNF-α, IL-1β, COX-2, and NO. | [226] |
| Inhibited IκBα degradation and NF-κB p65 translocation. | ||||
| ↑ in PPARγ transcriptional activity and PPARγ protein expression. | ||||
| 50 mg/kg BW daily for six weeks | Ethanol-induced oxidative stress in mice brains | ↓ TLR4, RAGE, GFAP, and Iba-1 expressions. | [229] | |
| ↓ p-JNK, p-NF-κB, COX-2, IL-1β, and TNF-α. | ||||
| 6 µM | A53T α-syn cell model of Parkinsonism | Recovered autophagy via inhibition of the mTOR/p70S6K signaling pathway. | [230] | |
| ↑ clearance of A53T α-syn accumulation. | ||||
| Demethoxycurcumin | 50 nM | Rotenone induced neurotoxicity in SH-SY5Y neuroblastoma cells | ↑ in the anti-apoptotic protein expression (Bcl-2 and Bcl-xL). | [92] |
| ↓ pro-apoptotic protein expressions (Bad, caspase-3, caspase-6, caspase-8, caspase-9). | ||||
| Epigallocatechin gallate | 1 μM | 6-OHDA-induced cytotoxicity in neuroblastoma cells | ↑ PKC and ERK1/2 activities. | [232] |
| ↓ mRNAs expression of Bax, Bad, and Mdm2 while ↑ in Bcl-2, Bcl-w, and Bcl-xL. | ||||
| 2.5 mg/kg BW daily for two weeks | Balb/C mice | ↑ neurogenesis in the hippocampus via the PI3K/Akt pathway. | [233] | |
| Green tea polyphenols | 10 μM | Glutamate excitotoxicity in primary cultured cortical neurons | ↓ the expression of proapoptotic proteins Bax and caspase-3. | [234] |
| ↑ the antiapoptotic protein Bcl-2. | ||||
| Berberine | 5 μM | Aβ-stimulated microglial cells | ↓ MCP1 and IL-6 and their mRNA expressions. | [237] |
| Inhibited NF-κB nuclear translocation by suppressing phosphorylation of IκB. | ||||
| Inhibited phosphorylation of Akt, ERK1/2, and p38. | ||||
| Magnolol | 2.5 µM | BV2 cells exposed to oligomeric Aβ42 | ↓ NF-κB target gene expressions including iNOS, TNF-α, and IL-1β. | [238] |
| 16 µM | Acrolein-induced apoptosis in neuroblastoma SH-SY5Y cells | ↓ apoptotic cell via the PI3K/MEK/ERK and PI3K/Akt/FoxO1 signaling pathways. | [96] | |
| Tocotrienol rich fraction (TRF). Active compounds: α-tocopherol (23.40%) and α, β, γ, and δ-tocotrienol (27.30, 3.34, 35.51, and 10.45%, respectively) | 200 mg/kg BW/day for six months | APPswe/PS1dE9 AD mouse model | ↓ Hdac2 and Iqgap2 genes involved in the NF-κB signaling pathway. | [239] |
| ↓ expression of pro-apoptotic genes (Casp7, Casp8, Casp12, and Bid). | ||||
| Regulated the voltage-dependent calcium channel genes (Cacna1c and Cacna1d) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramli, N.Z.; Yahaya, M.F.; Tooyama, I.; Damanhuri, H.A. A Mechanistic Evaluation of Antioxidant Nutraceuticals on Their Potential against Age-Associated Neurodegenerative Diseases. Antioxidants 2020, 9, 1019. https://doi.org/10.3390/antiox9101019
Ramli NZ, Yahaya MF, Tooyama I, Damanhuri HA. A Mechanistic Evaluation of Antioxidant Nutraceuticals on Their Potential against Age-Associated Neurodegenerative Diseases. Antioxidants. 2020; 9(10):1019. https://doi.org/10.3390/antiox9101019
Chicago/Turabian StyleRamli, Nur Zuliani, Mohamad Fairuz Yahaya, Ikuo Tooyama, and Hanafi Ahmad Damanhuri. 2020. "A Mechanistic Evaluation of Antioxidant Nutraceuticals on Their Potential against Age-Associated Neurodegenerative Diseases" Antioxidants 9, no. 10: 1019. https://doi.org/10.3390/antiox9101019