Mechanisms of Chronic Metabolic Stress in Arrhythmias




Abstract
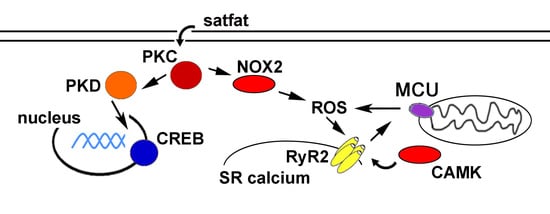
:
1. Introduction
2. Basics of Myocardial Metabolism
3. Types of Dietary Fat
4. Cardiac Abnormalities Caused by Diet-Induced Obesity (DIO)
5. De Novo Lipogenesis
6. An Overview of Arrhythmia Pathophysiology
7. Arrhythmogenic Mechanisms Caused by HFD and/or Obesity
8. Atrial Arrhythmias
9. Ventricular Arrhythmias
10. Additional Metabolic Mechanisms that Potentially Regulate Heart Rhythm
10.1. Fructose
10.2. Inflammation
10.3. Insulin Pathways
10.4. Mitochondrial DNA Damage
10.5. AMPK (Adenosine Monophosphate-Activated Protein Kinase)
10.6. Adiponectin and Other Adipokines
11. Dietary and Pharmacological Interventions
11.1. Dietary Modification
11.2. Antioxidant and Pharmacological Remedies
12. Conclusions and Future Directions
Funding
Conflicts of Interest
References
- Aggarwal, M.; Aggarwal, B.; Rao, J. Integrative Medicine for Cardiovascular Disease and Prevention. Med. Clin. 2017, 101, 895–923. [Google Scholar] [CrossRef] [PubMed]
- Schnabel, R.B.; Yin, X.; Gona, P.; Larson, M.G.; Beiser, A.S.; McManus, D.D.; Newton-Cheh, C.; Lubitz, S.A.; Magnani, J.W.; Ellinor, P.T.; et al. 50-year trends in atrial fibrillation prevalence, incidence, risk factors, and mortality in the Framingham Heart Study: A cohort study. Lancet 2015, 386, 154–162. [Google Scholar] [CrossRef] [Green Version]
- Sidney, S.; Quesenberry, C.P., Jr.; Jaffe, M.G.; Sorel, M.; Nguyen-Huynh, M.N.; Kushi, L.H.; Go, A.S.; Rana, J.S. Recent Trends in Cardiovascular Mortality in the United States and Public Health Goals. JAMA Cardiol. 2016, 1, 594–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2020 Update: A Report from the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- Andersson, C.; Vasan, R.S. Epidemiology of cardiovascular disease in young individuals. Nat. Rev. Cardiol 2018, 15, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Menke, A.; Casagrande, S.; Geiss, L.; Cowie, C.C. Prevalence of and Trends in Diabetes among Adults in the United States, 1988–2012. JAMA 2015, 314, 1021–1029. [Google Scholar] [CrossRef] [Green Version]
- Shan, Z.; Rehm, C.D.; Rogers, G.; Ruan, M.; Wang, D.D.; Hu, F.B.; Mozaffarian, D.; Zhang, F.F.; Bhupathiraju, S.N. Trends in Dietary Carbohydrate, Protein, and Fat Intake and Diet Quality Among US Adults, 1999–2016. JAMA 2019, 322, 1178–1187. [Google Scholar] [CrossRef] [Green Version]
- Filippi, A.; Sessa, E., Jr.; Mazzaglia, G.; Pecchioli, S., Jr.; Capocchi, R., Jr.; Caprari, F.; Scivales, A.C. Out of hospital sudden cardiac death in Italy: A population-based case-control study. J. Cardiovasc. Med. (Hagerstown) 2008, 9, 595–600. [Google Scholar] [CrossRef]
- Homan, E.A.; Reyes, M.V.; Hickey, K.T.; Morrow, J.P. Clinical Overview of Obesity and Diabetes Mellitus as Risk Factors for Atrial Fibrillation and Sudden Cardiac Death. Front. Physiol. 2018, 9, 1847. [Google Scholar] [CrossRef] [Green Version]
- Kolwicz, S.C., Jr.; Purohit, S.; Tian, R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ. Res. 2013, 113, 603–616. [Google Scholar] [CrossRef] [Green Version]
- Dorn, G.W., 2nd; Vega, R.B.; Kelly, D.P. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes. Dev. 2015, 29, 1981–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaper, J.; Meiser, E.; Stammler, G. Ultrastructural morphometric analysis of myocardium from dogs, rats, hamsters, mice, and from human hearts. Circ. Res. 1985, 56, 377–391. [Google Scholar] [CrossRef] [Green Version]
- Pohjoismaki, J.L.; Goffart, S. The role of mitochondria in cardiac development and protection. Free Radic. Biol. Med. 2017, 106, 345–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sikder, K.; Shukla, S.K.; Patel, N.; Singh, H.; Rafiq, K. High Fat Diet Upregulates Fatty Acid Oxidation and Ketogenesis via Intervention of PPAR-gamma. Cell. Physiol. Biochem. 2018, 48, 1317–1331. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam. Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Bjorkegren, J.; Veniant, M.; Kim, S.K.; Withycombe, S.K.; Wood, P.A.; Hellerstein, M.K.; Neese, R.A.; Young, S.G. Lipoprotein secretion and triglyceride stores in the heart. J. Biol. Chem. 2001, 276, 38511–38517. [Google Scholar] [CrossRef] [Green Version]
- Boren, J.; Veniant, M.M.; Young, S.G. Apo B100-containing lipoproteins are secreted by the heart. J. Clin. Investig. 1998, 101, 1197–1202. [Google Scholar] [CrossRef]
- Joseph, L.C.; Barca, E.; Subramanyam, P.; Komrowski, M.; Pajvani, U.; Colecraft, H.M.; Hirano, M.; Morrow, J.P. Inhibition of NADPH Oxidase 2 (NOX2) Prevents Oxidative Stress and Mitochondrial Abnormalities Caused by Saturated Fat in Cardiomyocytes. PLoS ONE 2016, 11, e0145750. [Google Scholar] [CrossRef]
- Prosser, B.L.; Ward, C.W.; Lederer, W.J. X-ROS signaling: Rapid mechano-chemo transduction in heart. Science 2011, 333, 1440–1445. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Perino, A.; Ghigo, A.; Hirsch, E.; Shah, A.M. NADPH oxidases in heart failure: Poachers or gamekeepers? Antioxid. Redox Signal. 2013, 18, 1024–1041. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Csordas, G.; Jowdy, C.; Schneider, T.G.; Csordas, N.; Wang, W.; Liu, Y.; Kohlhaas, M.; Meiser, M.; Bergem, S.; et al. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca(2+) crosstalk. Circ. Res. 2012, 111, 863–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbas, A.M.; Sakr, H.F. Simvastatin and vitamin E effects on cardiac and hepatic oxidative stress in rats fed on high fat diet. J. Physiol. Biochem. 2013, 69, 737–750. [Google Scholar] [CrossRef]
- Mayr, H.L.; Thomas, C.J.; Tierney, A.C.; Kucianski, T.; George, E.S.; Ruiz-Canela, M.; Hebert, J.R.; Shivappa, N.; Itsiopoulos, C. Randomization to 6-month Mediterranean diet compared with a low-fat diet leads to improvement in Dietary Inflammatory Index scores in patients with coronary heart disease: The AUSMED Heart Trial. Nutr. Res. 2018, 55, 94–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalliah, C.J.; Sanders, P.; Kalman, J.M. The Impact of Diet and Lifestyle on Atrial Fibrillation. Curr. Cardiol. Rep. 2018, 20, 137. [Google Scholar] [CrossRef] [PubMed]
- Sacks, F.M.; Lichtenstein, A.H.; Wu, J.H.Y.; Appel, L.J.; Creager, M.A.; Kris-Etherton, P.M.; Miller, M.; Rimm, E.B.; Rudel, L.L.; Robinson, J.G.; et al. Dietary Fats and Cardiovascular Disease: A Presidential Advisory from the American Heart Association. Circulation 2017, 136, e1–e23. [Google Scholar] [CrossRef]
- Joseph, L.C.; Avula, U.M.R.; Wan, E.Y.; Reyes, M.V.; Lakkadi, K.R.; Subramanyam, P.; Nakanishi, K.; Homma, S.; Muchir, A.; Pajvani, U.B.; et al. Dietary Saturated Fat Promotes Arrhythmia by Activating NOX2 (NADPH Oxidase 2). Circ. Arrhythm. Electrophysiol. 2019, 12, e007573. [Google Scholar] [CrossRef]
- Wanders, A.J.; Zock, P.L.; Brouwer, I.A. Trans Fat Intake and Its Dietary Sources in General Populations Worldwide: A Systematic Review. Nutrients 2017, 9, 840. [Google Scholar] [CrossRef]
- Rennison, J.H.; van Wagoner, D.R. Impact of dietary fatty acids on cardiac arrhythmogenesis. Circ. Arrhythm. Electrophysiol. 2009, 2, 460–469. [Google Scholar] [CrossRef] [Green Version]
- Casagrande, S.S.; Menke, A.; Cowie, C.C. Cardiovascular Risk Factors of Adults Age 20-49 Years in the United States, 1971-2012: A Series of Cross-Sectional Studies. PLoS ONE 2016, 11, e0161770. [Google Scholar] [CrossRef]
- Szczepaniak, L.S.; Victor, R.G.; Orci, L.; Unger, R.H. Forgotten but not gone: The rediscovery of fatty heart, the most common unrecognized disease in America. Circ. Res. 2007, 101, 759–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bello, N.A.; Cheng, S.; Claggett, B.; Shah, A.M.; Ndumele, C.E.; Roca, G.Q.; Santos, A.B.; Gupta, D.; Vardeny, O.; Aguilar, D.; et al. Association of Weight and Body Composition on Cardiac Structure and Function in the ARIC Study (Atherosclerosis Risk in Communities). Circ. Heart Fail. 2016, 9, e002978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Putte-Katier, N.; Rooman, R.P.; Haas, L.; Verhulst, S.L.; Desager, K.N.; Ramet, J.; Suys, B.E. Early cardiac abnormalities in obese children: Importance of obesity per se versus associated cardiovascular risk factors. Pediatr. Res. 2008, 64, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Franssen, W.M.A.; Beyens, M.; Hatawe, T.A.; Frederix, I.; Verboven, K.; Dendale, P.; Eijnde, B.O.; Massa, G.; Hansen, D. Cardiac function in adolescents with obesity: Cardiometabolic risk factors and impact on physical fitness. Int. J. Obes. 2019, 43, 1400–1410. [Google Scholar] [CrossRef]
- Huang, H.; Amin, V.; Gurin, M.; Wan, E.; Thorp, E.; Homma, S.; Morrow, J.P. Diet-induced obesity causes long QT and reduces transcription of voltage-gated potassium channels. J. Mol. Cell. Cardiol. 2013, 59C, 151–158. [Google Scholar] [CrossRef] [Green Version]
- Battiprolu, P.K.; Hojayev, B.; Jiang, N.; Wang, Z.V.; Luo, X.; Iglewski, M.; Shelton, J.M.; Gerard, R.D.; Rothermel, B.A.; Gillette, T.G.; et al. Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J. Clin. Investig. 2012, 122, 1109–1118. [Google Scholar] [CrossRef]
- Holzem, K.M.; Marmerstein, J.T.; Madden, E.J.; Efimov, I.R. Diet-induced obesity promotes altered remodeling and exacerbated cardiac hypertrophy following pressure overload. Physiol. Rep. 2015, 3, e12489. [Google Scholar] [CrossRef] [PubMed]
- Knowles, C.J.; Cebova, M.; Pinz, I.M. Palmitate diet-induced loss of cardiac caveolin-3: A novel mechanism for lipid-induced contractile dysfunction. PLoS ONE 2013, 8, e61369. [Google Scholar] [CrossRef] [Green Version]
- Qin, F.; Siwik, D.A.; Luptak, I.; Hou, X.; Wang, L.; Higuchi, A.; Weisbrod, R.M.; Ouchi, N.; Tu, V.H.; Calamaras, T.D.; et al. The polyphenols resveratrol and S17834 prevent the structural and functional sequelae of diet-induced metabolic heart disease in mice. Circulation 2012, 125, 1757–1764. [Google Scholar] [CrossRef] [Green Version]
- Calligaris, S.D.; Lecanda, M.; Solis, F.; Ezquer, M.; Gutierrez, J.; Brandan, E.; Leiva, A.; Sobrevia, L.; Conget, P. Mice long-term high-fat diet feeding recapitulates human cardiovascular alterations: An animal model to study the early phases of diabetic cardiomyopathy. PLoS ONE 2013, 8, e60931. [Google Scholar] [CrossRef] [Green Version]
- Sverdlov, A.L.; Elezaby, A.; Qin, F.; Behring, J.B.; Luptak, I.; Calamaras, T.D.; Siwik, D.A.; Miller, E.J.; Liesa, M.; Shirihai, O.S.; et al. Mitochondrial Reactive Oxygen Species Mediate Cardiac Structural, Functional, and Mitochondrial Consequences of Diet-Induced Metabolic Heart Disease. J. Am. Heart Assoc. 2016, 5, e002555. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, A.H.; Schildcrout, J.S.; Blakemore, D.L.; Masys, D.R.; Pulley, J.M.; Basford, M.A.; Roden, D.M.; Denny, J.C. Modulators of normal electrocardiographic intervals identified in a large electronic medical record. Heart Rhythm. 2011, 8, 271–277. [Google Scholar] [CrossRef] [Green Version]
- Messerli, F.H.; Nunez, B.D.; Ventura, H.O.; Snyder, D.W. Overweight and sudden death. Increased ventricular ectopy in cardiopathy of obesity. Arch. Intern. Med. 1987, 147, 1725–1728. [Google Scholar] [CrossRef]
- Zemva, A.; Zemva, Z. Ventricular ectopic activity, left ventricular mass, hyperinsulinemia, and intracellular magnesium in normotensive patients with obesity. Angiology 2000, 51, 101–106. [Google Scholar] [CrossRef]
- Watanabe, E.; Arakawa, T.; Uchiyama, T.; Tong, M.; Yasui, K.; Takeuchi, H.; Terasawa, T.; Kodama, I.; Hishida, H. Prognostic significance of circadian variability of RR and QT intervals and QT dynamicity in patients with chronic heart failure. Heart Rhythm. 2007, 4, 999–1005. [Google Scholar] [CrossRef]
- Okin, P.M.; Devereux, R.B.; Lee, E.T.; Galloway, J.M.; Howard, B.V. Electrocardiographic repolarization complexity and abnormality predict all-cause and cardiovascular mortality in diabetes: The strong heart study. Diabetes 2004, 53, 434–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyfeli, E.; Duru, M.; Kuvandik, G.; Kaya, H.; Yalcin, F. Effect of obesity on P-wave dispersion and QT dispersion in women. Int. J. Obes. 2006, 30, 957–961. [Google Scholar] [CrossRef] [Green Version]
- Naas, A.A.; Davidson, N.C.; Thompson, C.; Cummings, F.; Ogston, S.A.; Jung, R.T.; Newton, R.W.; Struthers, A.D. QT and QTc dispersion are accurate predictors of cardiac death in newly diagnosed non-insulin dependent diabetes: Cohort study. BMJ 1998, 316, 745–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, K.; Dorian, P.; Newman, D.; Langer, A. Association between QT dispersion and autonomic dysfunction in patients with diabetes mellitus. J. Am. Coll. Cardiol. 1995, 26, 859–863. [Google Scholar] [CrossRef] [Green Version]
- Albert, C.M.; Chae, C.U.; Grodstein, F.; Rose, L.M.; Rexrode, K.M.; Ruskin, J.N.; Stampfer, M.J.; Manson, J.E. Prospective study of sudden cardiac death among women in the United States. Circulation 2003, 107, 2096–2101. [Google Scholar] [CrossRef]
- Jouven, X.; Desnos, M.; Guerot, C.; Ducimetiere, P. Predicting sudden death in the population: The Paris Prospective Study, I. Circulation 1999, 99, 1978–1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hookana, E.; Junttila, M.J.; Puurunen, V.P.; Tikkanen, J.T.; Kaikkonen, K.S.; Kortelainen, M.L.; Myerburg, R.J.; Huikuri, H.V. Causes of nonischemic sudden cardiac death in the current era. Heart Rhythm. 2011, 8, 1570–1575. [Google Scholar]
- Mukerji, R.; Petruc, M.; Fresen, J.L.; Terry, B.E.; Govindarajan, G.; Alpert, M.A. Effect of weight loss after bariatric surgery on left ventricular mass and ventricular repolarization in normotensive morbidly obese patients. Am. J. Cardiol. 2012, 110, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Joseph, L.C.; Subramanyam, P.; Radlicz, C.; Trent, C.M.; Iyer, V.; Colecraft, H.M.; Morrow, J.P. Mitochondrial oxidative stress during cardiac lipid overload causes intracellular calcium leak and arrhythmia. Heart Rhythm. 2016, 13, 1699–1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avula, U.M.R.; Abrams, J.; Katchman, A.; Zakharov, S.; Mironov, S.; Bayne, J.; Roybal, D.; Gorti, A.; Yang, L.; Iyer, V.; et al. Heterogeneity of the action potential duration is required for sustained atrial fibrillation. JCI Insight. 2019, 5, e128765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fielitz, J.; Kim, M.S.; Shelton, J.M.; Qi, X.; Hill, J.A.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Requirement of protein kinase D1 for pathological cardiac remodeling. Proc. Natl. Acad. Sci. USA 2008, 105, 3059–3063. [Google Scholar] [CrossRef] [Green Version]
- Ozgen, N.; Guo, J.; Gertsberg, Z.; Danilo, P., Jr.; Rosen, M.R.; Steinberg, S.F. Reactive oxygen species decrease cAMP response element binding protein expression in cardiomyocytes via a protein kinase D1-dependent mechanism that does not require Ser133 phosphorylation. Mol. Pharmacol. 2009, 76, 896–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, B.M.; Bossuyt, J. Emergency Spatiotemporal Shift: The Response of Protein Kinase D to Stress Signals in the Cardiovascular System. Front. Pharmacol. 2017, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Jhun, B.S.; Jin, O.; Adaniya, S.M.; Mancini, T.J.; Cao, J.L.; King, M.E.; Landi, A.K.; Ma, H.; Shin, M.; Yang, D.; et al. Protein kinase D activation induces mitochondrial fragmentation and dysfunction in cardiomyocytes. J. Physiol. 2018, 596, 827–855. [Google Scholar] [CrossRef] [Green Version]
- Tong, M.; Saito, T.; Zhai, P.; Oka, S.I.; Mizushima, W.; Nakamura, M.; Ikeda, S.; Shirakabe, A.; Sadoshima, J. Mitophagy Is Essential for Maintaining Cardiac Function During High Fat Diet-Induced Diabetic Cardiomyopathy. Circ. Res. 2019, 124, 1360–1371. [Google Scholar] [CrossRef]
- Chen, D.; Li, X.; Zhang, L.; Zhu, M.; Gao, L. A high-fat diet impairs mitochondrial biogenesis, mitochondrial dynamics, and the respiratory chain complex in rat myocardial tissues. J. Cell. Biochem. 2018, 119, 9602. [Google Scholar] [CrossRef] [Green Version]
- Maneechote, C.; Palee, S.; Apaijai, N.; Kerdphoo, S.; Jaiwongkam, T.; Chattipakorn, S.C.; Chattipakorn, N. Mitochondrial dynamic modulation exerts cardiometabolic protection in obese insulin-resistant rats. Clin. Sci. 2019, 133, 2431–2447. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Kolwicz, S.C.; Wang, P.; Roe, N.D.; Villet, O.; Nishi, K.; Hsu, Y.A.; Flint, G.V.; Caudal, A.; Wang, W.; et al. Increasing Fatty Acid Oxidation Prevents High Fat Diet Induced Cardiomyopathy through Regulating Parkin Mediated Mitophagy. Circulation 2020, 142, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, I.J.; Reue, K.; Abumrad, N.A.; Bickel, P.E.; Cohen, S.; Fisher, E.A.; Galis, Z.S.; Granneman, J.G.; Lewandowski, E.D.; Murphy, R.; et al. Deciphering the Role of Lipid Droplets in Cardiovascular Disease: A Report from the 2017 National Heart, Lung, and Blood Institute Workshop. Circulation 2018, 138, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L. Sugar consumption, metabolic disease and obesity: The state of the controversy. Crit. Rev. Clin. Lab. Sci. 2016, 53, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.H.; Chang, G.J.; Lai, Y.J.; Chen, W.J.; Chang, S.H.; Hung, L.M.; Kuo, C.T.; Yeh, Y.H. Atrial fibrillation and its arrhythmogenesis associated with insulin resistance. Cardiovasc. Diabetol. 2019, 18, 125. [Google Scholar] [CrossRef] [Green Version]
- Federico, M.; Portiansky, E.L.; Sommese, L.; Alvarado, F.J.; Blanco, P.G.; Zanuzzi, C.N.; Dedman, J.; Kaetzel, M.; Wehrens, X.H.T.; Mattiazzi, A.; et al. Calcium-calmodulin-dependent protein kinase mediates the intracellular signalling pathways of cardiac apoptosis in mice with impaired glucose tolerance. J. Physiol. 2017, 595, 4089–4108. [Google Scholar] [CrossRef] [Green Version]
- Lanaspa, M.A.; Ishimoto, T.; Li, N.; Cicerchi, C.; Orlicky, D.J.; Ruzycki, P.; Rivard, C.; Inaba, S.; Roncal-Jimenez, C.A.; Bales, E.S.; et al. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat. Commun. 2013, 4, 2434. [Google Scholar] [CrossRef]
- Nagai, Y.; Nishio, Y.; Nakamura, T.; Maegawa, H.; Kikkawa, R.; Kashiwagi, A. Amelioration of high fructose-induced metabolic derangements by activation of PPARalpha. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E1180–E1190. [Google Scholar] [CrossRef] [Green Version]
- Stanhope, K.L.; Medici, V.; Bremer, A.A.; Lee, V.; Lam, H.; Nunez, M.V.; Chen, G.X.; Keim, N.L.; Havel, P.J. A dose-response study of consuming high-fructose corn syrup-sweetened beverages on lipid/lipoprotein risk factors for cardiovascular disease in young adults. Am. J. Clin. Nutr. 2015, 101, 1144–1154. [Google Scholar] [CrossRef] [Green Version]
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.L.; Dyachenko, A.; Zhang, W.; et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Investig. 2009, 119, 1322–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.B.; Meng, Y.H.; Chang, S.; Zhang, R.Y.; Shi, C. High fructose causes cardiac hypertrophy via mitochondrial signaling pathway. Am. J. Transl. Res. 2016, 8, 4869–4880. [Google Scholar]
- Wakili, R.; Voigt, N.; Kaab, S.; Dobrev, D.; Nattel, S. Recent advances in the molecular pathophysiology of atrial fibrillation. J. Clin. Investig. 2011, 121, 2955–2968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanner, J.T.; Georgiou, D.K.; Joshi, A.D.; Hamilton, S.L. Ryanodine receptors: Structure, expression, molecular details, and function in calcium release. Cold Spring Harb. Perspect. Biol. 2010, 2, a003996. [Google Scholar] [CrossRef] [Green Version]
- Santulli, G.; Lewis, D.; Georges, A.D.; Marks, A.R.; Frank, J. Ryanodine Receptor Structure and Function in Health and Disease. Subcell. Biochem. 2018, 87, 329–352. [Google Scholar] [PubMed] [Green Version]
- Peng, W.; Shen, H.; Wu, J.; Guo, W.; Pan, X.; Wang, R.; Chen, S.R.; Yan, N. Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science 2016, 354, aah5324. [Google Scholar] [CrossRef]
- Ai, X.; Curran, J.W.; Shannon, T.R.; Bers, D.M.; Pogwizd, S.M. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res. 2005, 97, 1314–1322. [Google Scholar] [CrossRef] [Green Version]
- Clauss, S.; Bleyer, C.; Schuttler, D.; Tomsits, P.; Renner, S.; Klymiuk, N.; Wakili, R.; Massberg, S.; Wolf, E.; Kaab, S. Animal models of arrhythmia: Classic electrophysiology to genetically modified large animals. Nat. Rev. Cardiol. 2019, 16, 457–475. [Google Scholar] [CrossRef] [PubMed]
- Pozzolini, A.; Rio, T.; Padeletti, M.; De Ponti, R.; Leonelli, F.M.; and Bagliani, G. Complex Arrhythmias Due to Reversible Causes. Card Electrophysiol. Clin. 2019, 11, 375–390. [Google Scholar] [CrossRef]
- McCauley, M.D.; Hong, L.; Sridhar, A.; Menon, A.; Perike, S.; Zhang, M.; da Silva, I.B.; Yan, J.; Bonini, M.G.; Ai, X.; et al. Ion Channel and Structural Remodeling in Obesity: Mediated Atrial Fibrillation. Circ. Arrhythm. Electrophysiol. 2020, 13, e008296. [Google Scholar] [CrossRef]
- Ashrafi, R.; Yon, M.; Pickavance, L.; Yanni Gerges, J.; Davis, G.; Wilding, J.; Jian, K.; Zhang, H.; Hart, G.; Boyett, M. Altered Left Ventricular Ion Channel Transcriptome in a High-Fat-Fed Rat Model of Obesity: Insight into Obesity-Induced Arrhythmogenesis. J. Obes. 2016, 2016, 7127898. [Google Scholar] [CrossRef] [Green Version]
- Egan Benova, T.; Viczenczova, C.; Szeiffova Bacova, B.; Knezl, V.; Dosenko, V.; Rauchova, H.; Zeman, M.; Reiter, R.J.; Tribulova, N. Obesity-associated alterations in cardiac connexin-43 and PKC signaling are attenuated by melatonin and omega-3 fatty acids in female rats. Mol. Cell. Biochem. 2019, 454, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Meng, T.; Cheng, G.; Wei, Y.; Ma, S.; Jiang, Y.; Wu, J.; Zhou, X.; Sun, C. Exposure to a chronic high-fat diet promotes atrial structure and gap junction remodeling in rats. Int. J. Mol. Med. 2017, 40, 217–225. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics-2017 Update: A Report from the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.P.; Reiffel, J.A. Drug therapy for atrial fibrillation: What will its role be in the era of increasing use of catheter ablation? Pacing Clin. Electrophysiol. PACE 2009, 32, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Pathak, R.K.; Middeldorp, M.E.; Meredith, M.; Mehta, A.B.; Mahajan, R.; Wong, C.X.; Twomey, D.; Elliott, A.D.; Kalman, J.M.; Abhayaratna, W.P.; et al. Long-Term Effect of Goal-Directed Weight Management in an Atrial Fibrillation Cohort: A Long-Term Follow-Up Study (LEGACY). J. Am. Coll. Cardiol. 2015, 65, 2159–2169. [Google Scholar] [CrossRef]
- Martinez-Gonzalez, M.A.; Toledo, E.; Aros, F.; Fiol, M.; Corella, D.; Salas-Salvado, J.; Ros, E.; Covas, M.I.; Fernandez-Crehuet, J.; Lapetra, J.; et al. Extravirgin Olive Oil Consumption Reduces Risk of Atrial Fibrillation: The PREDIMED (Prevencion con Dieta Mediterranea) Trial. Circulation 2014, 130, 18–26. [Google Scholar] [CrossRef]
- Mozaffarian, D.; Psaty, B.M.; Rimm, E.B.; Lemaitre, R.N.; Burke, G.L.; Lyles, M.F.; Lefkowitz, D.; Siscovick, D.S. Fish intake and risk of incident atrial fibrillation. Circulation 2004, 110, 368–373. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.H.; Lemaitre, R.N.; King, I.B.; Song, X.; Sacks, F.M.; Rimm, E.B.; Heckbert, S.R.; Siscovick, D.S.; Mozaffarian, D. Association of plasma phospholipid long-chain omega-3 fatty acids with incident atrial fibrillation in older adults: The cardiovascular health study. Circulation 2012, 125, 1084–1093. [Google Scholar] [CrossRef]
- Schnabel, R.B.; Sullivan, L.M.; Levy, D.; Pencina, M.J.; Massaro, J.M.; D’Agostino Sr, R.B.; Newton-Cheh, C.; Yamamoto, J.F.; Magnani, J.W.; Tadros, T.M.; et al. Development of a risk score for atrial fibrillation (Framingham Heart Study): A community-based cohort study. Lancet 2009, 373, 739–745. [Google Scholar] [CrossRef] [Green Version]
- Mayr, M.; Yusuf, S.; Weir, G.; Chung, Y.L.; Mayr, U.; Yin, X.; Ladroue, C.; Madhu, B.; Roberts, N.; De Souza, A.; et al. Combined metabolomic and proteomic analysis of human atrial fibrillation. J. Am. Coll. Cardiol. 2008, 51, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Deshmukh, A.; Barnard, J.; Sun, H.; Newton, D.; Castel, L.; Pettersson, G.; Johnston, D.; Roselli, E.; Gillinov, A.M.; McCurry, K.; et al. Left atrial transcriptional changes associated with atrial fibrillation susceptibility and persistence. Circ. Arrhythm. Electrophysiol. 2015, 8, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Herzig, S.; Long, F.; Jhala, U.S.; Hedrick, S.; Quinn, R.; Bauer, A.; Rudolph, D.; Schutz, G.; Yoon, C.; Puigserver, P.; et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 2001, 413, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Schulte, J.S.; Seidl, M.D.; Nunes, F.; Freese, C.; Schneider, M.D.; Schmitz, W.; Muller, F.U. CREB critically regulates action potential shape and duration in the adult mouse ventricle. Am. J. Physiol. Heart Circ. Physiol. 2012, 15, H1998–H2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef]
- Sano, M.; Minamino, T.; Toko, H.; Miyauchi, H.; Orimo, M.; Qin, Y.; Akazawa, H.; Tateno, K.; Kayama, Y.; Harada, M.; et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 2007, 446, 444–448. [Google Scholar] [CrossRef]
- Kim, Y.M.; Guzik, T.J.; Zhang, Y.H.; Zhang, M.H.; Kattach, H.; Ratnatunga, C.; Pillai, R.; Channon, K.M.; Casadei, B. A myocardial Nox2 containing NAD(P)H oxidase contributes to oxidative stress in human atrial fibrillation. Circ. Res. 2005, 97, 629–636. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.; Santulli, G.; Reiken, S.R.; Yuan, Q.; Osborne, B.W.; Chen, B.X.; Marks, A.R. Mitochondrial oxidative stress promotes atrial fibrillation. Sci. Rep. 2015, 5, 11427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajan, R.; Lau, D.H.; Brooks, A.G.; Shipp, N.J.; Manavis, J.; Wood, J.P.; Finnie, J.W.; Samuel, C.S.; Royce, S.G.; Twomey, D.J.; et al. Electrophysiological, Electroanatomical, and Structural Remodeling of the Atria as Consequences of Sustained Obesity. J. Am. Coll. Cardiol. 2015, 66, 1–11. [Google Scholar] [CrossRef]
- Opacic, D.; van Bragt, K.A.; Nasrallah, H.M.; Schotten, U.; Verheule, S. Atrial metabolism and tissue perfusion as determinants of electrical and structural remodelling in atrial fibrillation. Cardiovasc. Res. 2016, 109, 527–541. [Google Scholar] [CrossRef] [Green Version]
- van Bragt, K.A.; Nasrallah, H.M.; Kuiper, M.; Luiken, J.J.; Schotten, U.; Verheule, S. Atrial supply-demand balance in healthy adult pigs: Coronary blood flow, oxygen extraction, and lactate production during acute atrial fibrillation. Cardiovasc. Res. 2014, 101, 9–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrow, J.P.; Katchman, A.; Son, N.H.; Trent, C.M.; Khan, R.; Shiomi, T.; Huang, H.; Amin, V.; Lader, J.M.; Vasquez, C.; et al. Mice with Cardiac Overexpression of Peroxisome Proliferator-Activated Receptor gamma Have Impaired Repolarization and Spontaneous Fatal Ventricular Arrhythmias. Circulation 2011, 124, 2812–2821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marionneau, C.; Aimond, F.; Brunet, S.; Niwa, N.; Finck, B.; Kelly, D.P.; Nerbonne, J.M. PPARalpha-mediated remodeling of repolarizing voltage-gated K+ (Kv) channels in a mouse model of metabolic cardiomyopathy. J. Mol. Cell. Cardiol. 2008, 44, 1002–1015. [Google Scholar] [CrossRef] [Green Version]
- Chiu, H.C.; Kovacs, A.; Ford, D.A.; Hsu, F.F.; Garcia, R.; Herrero, P.; Saffitz, J.E.; Schaffer, J.E. A novel mouse model of lipotoxic cardiomyopathy. J. Clin. Investig. 2001, 107, 813–822. [Google Scholar] [CrossRef] [Green Version]
- Chiu, H.C.; Kovacs, A.; Blanton, R.M.; Han, X.; Courtois, M.; Weinheimer, C.J.; Yamada, K.A.; Brunet, S.; Xu, H.; Nerbonne, J.M.; et al. Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ. Res. 2005, 96, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Aubin, M.C.; Cardin, S.; Comtois, P.; Clement, R.; Gosselin, H.; Gillis, M.A.; Le Quang, K.; Nattel, S.; Perrault, L.P.; Calderone, A.A. High-fat diet increases risk of ventricular arrhythmia in female rats: Enhanced arrhythmic risk in the absence of obesity or hyperlipidemia. J. Appl. Physiol. 2010, 108, 933–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCully, B.H.; Hasan, W.; Streiff, C.T.; Houle, J.C.; Woodward, W.R.; Giraud, G.D.; Brooks, V.L.; Habecker, B.A. Sympathetic cardiac hyperinnervation and atrial autonomic imbalance in diet-induced obesity promote cardiac arrhythmias. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1530–H1537. [Google Scholar] [CrossRef]
- Udomkasemsab, A.; Ngamlerst, C.; Adisakwattana, P.; Aroonnual, A.; Tungtrongchitr R and Prangthip, P. Maoberry (Antidesma bunius) ameliorates oxidative stress and inflammation in cardiac t.issues of rats fed a high-fat diet. BMC Complement Altern. Med. 2018, 18, 344. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, G.; Araneda, F.; Pena, J.P.; Finkelstein, J.P.; Riquelme, J.A.; Montecinos, L.; Barrientos, G.; Llanos, P.; Pedrozo, Z.; Said, M.; et al. High-Fat-Diet-Induced Obesity Produces Spontaneous Ventricular Arrhythmias and Increases the Activity of Ryanodine Receptors in Mice. Int. J. Mol. Sci. 2018, 19, 533. [Google Scholar] [CrossRef] [Green Version]
- Dey, S.; DeMazumder, D.; Sidor, A.; Foster, D.B.; O’Rourke, B. Mitochondrial ROS Drive Sudden Cardiac Death and Chronic Proteome Remodeling in Heart Failure. Circ. Res. 2018, 123, 356–371. [Google Scholar] [CrossRef]
- Kondo, H.; Abe, I.; Gotoh, K.; Fukui, A.; Takanari, H.; Ishii, Y.; Ikebe, Y.; Kira, S.; Oniki, T.; Saito, S.; et al. Interleukin 10 Treatment Ameliorates High-Fat Diet-Induced Inflammatory Atrial Remodeling and Fibrillation. Circ. Arrhythm. Electrophysiol. 2018, 11, e006040. [Google Scholar] [CrossRef] [PubMed]
- Roncero-Ramos, I.; Rangel-Zuniga, O.A.; Lopez-Moreno, J.; Alcala-Diaz, J.F.; Perez-Martinez, P.; Jimenez-Lucena, R.; Castano, J.P.; Roche, H.M.; Delgado-Lista, J.; Ordovas, J.M.; et al. Mediterranean Diet, Glucose Homeostasis, and Inflammasome Genetic Variants: The CORDIOPREV Study. Mol. Nutr. Food Res. 2018, 62, e1700960. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Jiang, Y.P.; Wu, C.Y.; Ballou, L.M.; Liu, S.; Carpenter, E.S.; Rosen, M.R.; Cohen, I.S.; Lin, R.Z. Increased persistent sodium current due to decreased PI3K signaling contributes to QT prolongation in the diabetic heart. Diabetes 2013, 62, 4257–4265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polina, I.; Jansen, H.J.; Li, T.; Moghtadaei, M.; Bohne, L.J.; Liu, Y.; Krishnaswamy, P.; Egom, E.E.; Belke, D.D.; Rafferty, S.A.; et al. Loss of insulin signaling may contribute to atrial fibrillation and atrial electrical remodeling in type 1 diabetes. Proc. Natl. Acad. Sci. USA 2020, 117, 7990–8000. [Google Scholar] [CrossRef] [PubMed]
- Baris, O.R.; Ederer, S.; Neuhaus, J.F.; von Kleist-Retzow, J.C.; Wunderlich, C.M.; Pal, M.; Wunderlich, F.T.; Peeva, V.; Zsurka, G.; Kunz, W.S.; et al. Mosaic Deficiency in Mitochondrial Oxidative Metabolism Promotes Cardiac Arrhythmia during Aging. Cell Metab. 2015, 21, 667–677. [Google Scholar] [CrossRef] [Green Version]
- Salt, I.P.; Hardie, D.G. AMP-Activated Protein Kinase: An Ubiquitous Signaling Pathway with Key Roles in the Cardiovascular System. Circ. Res. 2017, 120, 1825–1841. [Google Scholar] [CrossRef] [Green Version]
- Harada, M.; Tadevosyan, A.; Qi, X.; Xiao, J.; Liu, T.; Voigt, N.; Karck, M.; Kamler, M.; Kodama, I.; Murohara, T.; et al. Atrial Fibrillation Activates AMP-Dependent Protein Kinase and its Regulation of Cellular Calcium Handling: Potential Role in Metabolic Adaptation and Prevention of Progression. J. Am. Coll. Cardiol. 2015, 66, 47–58. [Google Scholar] [CrossRef]
- Ozcan, C.; Battaglia, E.; Young, R.; Suzuki, G. LKB1 knockout mouse develops spontaneous atrial fibrillation and provides mechanistic insights into human disease process. J. Am. Heart Assoc. 2015, 4, e001733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, M.N.; Skibsbye, L.; Tang, C.; Petersen, F.; MacAulay, N.; Rasmussen, H.B.; Jespersen, T. PKC and AMPK regulation of Kv1.5 potassium channels. Channels 2015, 9, 121–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Light, P.E.; Wallace, C.H.; Dyck, J.R. Constitutively active adenosine monophosphate-activated protein kinase regulates voltage-gated sodium channels in ventricular myocytes. Circulation 2003, 107, 1962–1965. [Google Scholar] [CrossRef] [Green Version]
- Myers, R.W.; Guan, H.P.; Ehrhart, J.; Petrov, A.; Prahalada, S.; Tozzo, E.; Yang, X.; Kurtz, M.M.; Trujillo, M.; Gonzalez Trotter, D.; et al. Systemic pan-AMPK activator MK-8722 improves glucose homeostasis but induces cardiac hypertrophy. Science 2017, 357, 507–511. [Google Scholar] [CrossRef] [Green Version]
- Essick, E.E.; Ouchi, N.; Wilson, R.M.; Ohashi, K.; Ghobrial, J.; Shibata, R.; Pimentel, D.R.; Sam, F. Adiponectin mediates cardioprotection in oxidative stress-induced cardiac myocyte remodeling. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H984–H993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biolo, A.; Shibata, R.; Ouchi, N.; Kihara, S.; Sonoda, M.; Walsh, K.; Sam, F. Determinants of adiponectin levels in patients with chronic systolic heart failure. Am. J. Cardiol. 2010, 105, 1147–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kourliouros, A.; Karastergiou, K.; Nowell, J.; Gukop, P.; Tavakkoli Hosseini, M.; Valencia, O.; Mohamed Ali, V.; Jahangiri, M. Protective effect of epicardial adiponectin on atrial fibrillation following cardiac surgery. Eur. J. Cardiothorac. Surg. 2011, 39, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Rienstra, M.; Sun, J.X.; Lubitz, S.A.; Frankel, D.S.; Vasan, R.S.; Levy, D.; Magnani, J.W.; Sullivan, L.M.; Meigs, J.B.; Ellinor, P.T.; et al. Plasma resistin, adiponectin, and risk of incident atrial fibrillation: The Framingham Offspring Study. Am. Heart J. 2012, 163, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Liu, T.; Rose, J.L.; Stevens, R.L.; Hoyt, D.G. Sensitivity of mice to lipopolysaccharide is increased by a high saturated fat and cholesterol diet. J. Inflamm. 2007, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Yi, C.; Wang, H.; Bruce, I.C.; Xia, Q. Mitochondrial nitric oxide synthase participates in septic shock myocardial depression by nitric oxide overproduction and mitochondrial permeability transition pore opening. Shock 2012, 37, 110–115. [Google Scholar] [CrossRef]
- Otabe, S.; Yuan, X.; Fukutani, T.; Wada, N.; Hashinaga, T.; Nakayama, H.; Hirota, N.; Kojima, M.; Yamada, K. Overexpression of human adiponectin in transgenic mice results in suppression of fat accumulation and prevention of premature death by high-calorie diet. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E210–E218. [Google Scholar] [CrossRef] [Green Version]
- Park, M.; Wu, D.; Park, T.; Choi, C.S.; Li, R.K.; Cheng, K.K.; Xu, A.; Sweeney, G. APPL1 transgenic mice are protected from high-fat diet-induced cardiac dysfunction. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E795–E804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, X.; Palanivel, R.; Cresser, J.; Schram, K.; Ganguly, R.; Thong, F.S.; Tuinei, J.; Xu, A.; Abel, E.D.; Sweeney, G. An APPL1-AMPK signaling axis mediates beneficial metabolic effects of adiponectin in the heart. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E721–E729. [Google Scholar] [CrossRef] [Green Version]
- Awazawa, M.; Ueki, K.; Inabe, K.; Yamauchi, T.; Kaneko, K.; Okazaki, Y.; Bardeesy, N.; Ohnishi, S.; Nagai, R.; Kadowaki, T. Adiponectin suppresses hepatic SREBP1c expression in an AdipoR1/LKB1/AMPK dependent pathway. Biochem. Biophys. Res. Commun. 2009, 382, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Shibata, R.; Sato, K.; Pimentel, D.R.; Takemura, Y.; Kihara, S.; Ohashi, K.; Funahashi, T.; Ouchi, N.; Walsh, K. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat. Med. 2005, 11, 1096–1103. [Google Scholar] [CrossRef]
- Komatsu, M.; Ohfusa, H.; Sato, Y.; Yajima, H.; Yamauchi, K.; Aizawa, T.; Hashizume, K. Strong inverse correlation between serum adiponectin level and heart rate-corrected QT interval in an apparently healthy population: A suggestion for a direct antiatherogenic effect of adiponectin. Diabetes Care 2004, 27, 1237–1238. [Google Scholar] [CrossRef] [Green Version]
- Hoyda, T.D.; Ferguson, A.V. Adiponectin modulates excitability of rat paraventricular nucleus neurons by differential modulation of potassium currents. Endocrinology 2010, 151, 3154–3162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, W.; Zhang, F.; Zhang, R.; Zhang, X.; Wang, Y.; Zhou, F.; Xia, Y.; Liu, P.; Gao, C.; Wang, H.; et al. Adiponectin regulates SR Ca cycling following ischemia/reperfusion via sphingosine 1-phosphate-CaMKII signaling in mice. J. Mol. Cell. Cardiol. 2014, 74C, 183–192. [Google Scholar] [CrossRef]
- Fukui, A.; Ikebe-Ebata, Y.; Kondo, H.; Saito, S.; Aoki, K.; Fukunaga, N.; Shinohara, T.; Masaki, T.; Teshima, Y.; Takahashi, N. Hyperleptinemia Exacerbates High-Fat Diet-Mediated Atrial Fibrosis and Fibrillation. J. Cardiovasc. Electrophysiol. 2017, 28, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Wu, L.; Terrar, D.A.; Huang, C.L. Modernized Classification of Cardiac Antiarrhythmic Drugs. Circulation 2018, 138, 1879–1896. [Google Scholar] [CrossRef]
- Harling, L.; Rasoli, S.; Vecht, J.A.; Ashrafian, H.; Kourliouros, A.; Athanasiou, T. Do antioxidant vitamins have an anti-arrhythmic effect following cardiac surgery? A meta-analysis of randomised controlled trials. Heart 2011, 97, 1636–1642. [Google Scholar] [CrossRef]
- Rodrigo, R.; Korantzopoulos, P.; Cereceda, M.; Asenjo, R.; Zamorano, J.; Villalabeitia, E.; Baeza, C.; Aguayo, R.; Castillo, R.; Carrasco, R.; et al. A Randomized Controlled Trial to Prevent Postoperative Atrial Fibrillation by Antioxidant Reinforcement. J. Am. Coll. Cardiol. 2013, 62, 1457–1465. [Google Scholar] [CrossRef] [Green Version]
- Ni, R.; Cao, T.; Xiong, S.; Ma, J.; Fan, G.C.; Lacefield, J.C.; Lu, Y.; Le Tissier, S.; Peng, T. Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic. Biol. Med. 2016, 90, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Souza, D.S.; Menezes-Filho, J.E.R.; Santos-Miranda, A.; Jesus, I.C.G.; Silva Neto, J.A.; Guatimosim, S.; Cruz, J.S.; Vasconcelos, C.M.L. Calcium overload-induced arrhythmia is suppressed by farnesol in rat heart. Eur. J. Pharmacol. 2019, 859, 172488. [Google Scholar] [CrossRef] [PubMed]
- Szucs, G.; Murlasits, Z.; Torok, S.; Kocsis, G.F.; Paloczi, J.; Gorbe, A.; Csont, T.; Csonka, C.; Ferdinandy, P. Cardioprotection by farnesol: Role of the mevalonate pathway. Cardiovasc. Drugs Ther. 2013, 27, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhao, W.; Thomson, J.K.; Gao, X.; DeMarco, D.M.; Carrillo, E.; Chen, B.; Wu, X.; Ginsburg, K.S.; Bakhos, M.; et al. Stress Signaling JNK2 Crosstalk with CaMKII Underlies Enhanced Atrial Arrhythmogenesis. Circ. Res. 2018, 122, 821–835. [Google Scholar] [CrossRef] [PubMed]
- van Petegem, F. Ryanodine receptors: Structure and function. J. Biol. Chem. 2012, 287, 31624–31632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwong, J.Q.; Lu, X.; Correll, R.N.; Schwanekamp, J.A.; Vagnozzi, R.J.; Sargent, M.A.; York, A.J.; Zhang, J.; Bers, D.M.; Molkentin, J.D. The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell Rep. 2015, 12, 15–22. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Dey et al. | Joseph et al. ‘16 | Joseph et al. ‘19 | McCauley et al. | Sanchez et al. | |
|---|---|---|---|---|---|
| Species | Guinea pig | Mouse | Mouse | Mouse | Mouse |
| Disease Model | Heart failure | Transgenic-induced cardiac lipid overload | DIO | DIO | DIO |
| HFD | N/A | N/A | 60% fat, palm or olive oil | 60% fat, lard | 60% fat, lard |
| Diet Duration | N/A | N/A | 4 weeks | ~5 months | 8 weeks |
| Antioxidant | mitoTEMPO | mitoTEMPO | Apocynin | mitoTEMPO | Apocynin |
| Results | Prevented HF, decreased vent. arrhythmia | Decreased PVCs | Prevented PVCs | Reduced AF, restored Nav1.5, Kv1.5 expression | Prevented vent. arrhythmia |
| Reference | 90 | 51 | 24 | 72 | 89 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gowen, B.H.; Reyes, M.V.; Joseph, L.C.; Morrow, J.P. Mechanisms of Chronic Metabolic Stress in Arrhythmias. Antioxidants 2020, 9, 1012. https://doi.org/10.3390/antiox9101012
Gowen BH, Reyes MV, Joseph LC, Morrow JP. Mechanisms of Chronic Metabolic Stress in Arrhythmias. Antioxidants. 2020; 9(10):1012. https://doi.org/10.3390/antiox9101012
Chicago/Turabian StyleGowen, Blake H., Michael V. Reyes, Leroy C. Joseph, and John P. Morrow. 2020. "Mechanisms of Chronic Metabolic Stress in Arrhythmias" Antioxidants 9, no. 10: 1012. https://doi.org/10.3390/antiox9101012