Peroxiredoxin6 in Endothelial Signaling


Institute for Environmental Medicine and Department of Physiology, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA 19104, USA
*
Author to whom correspondence should be addressed.
Antioxidants 2019, 8(3), 63; https://doi.org/10.3390/antiox8030063
Submission received: 25 January 2019
/
Revised: 21 February 2019
/
Accepted: 5 March 2019
/
Published: 13 March 2019
(This article belongs to the Special Issue Peroxiredoxin 6 as a Unique Member of the Peroxiredoxin Family)
Abstract
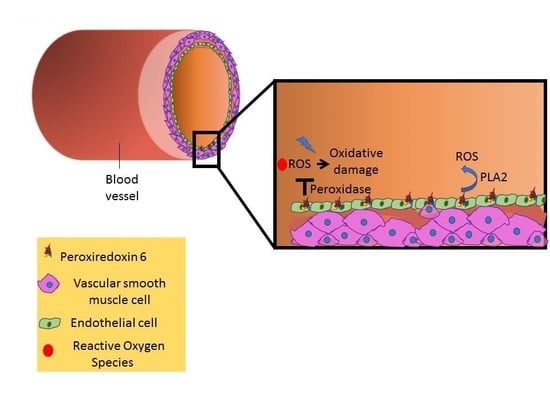
:Peroxiredoxins (Prdx) are a ubiquitous family of highly conserved antioxidant enzymes with a cysteine residue that participate in the reduction of peroxides. This family comprises members Prdx1–6, of which Peroxiredoxin 6 (Prdx6) is unique in that it is multifunctional with the ability to neutralize peroxides (peroxidase activity) and to produce reactive oxygen species (ROS) via its phospholipase (PLA2) activity that drives assembly of NADPH oxidase (NOX2). From the crystal structure, a C47 residue is responsible for peroxidase activity while a catalytic triad (S32, H26, and D140) has been identified as the active site for its PLA2 activity. This paradox of being an antioxidant as well as an oxidant generator implies that Prdx6 is a regulator of cellular redox equilibrium (graphical abstract). It also indicates that a fine-tuned regulation of Prdx6 expression and activity is crucial to cellular homeostasis. This is specifically important in the endothelium, where ROS production and signaling are critical players in inflammation, injury, and repair, that collectively signal the onset of vascular diseases. Here we review the role of Prdx6 as a regulator of redox signaling, specifically in the endothelium and in mediating various pathologies.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Peroxiredoxins (Prdx) are a family of enzymes that primarily function as antioxidants to scavenge peroxide in biological systems. The six isoforms of mammalian Prdxs (Prdx1 to 6) are distributed across various cellular sites of reactive oxygen species (ROS) production, such as the cytosol, mitochondria, and peroxisomes [1,2]. Prdxs are divided into two subgroups: Those that have one (Prdx6) cysteine (Cys) residue to participate in the redox cycle and those that have two (Prdx1–5). The enzymes Prdx1–5 use thioredoxin as an electron donor in their redox cycle. However, Prdx6 does not use thioredoxin and uses glutathione (GSH) as the reductant instead [3]. Prdxs are small proteins with sizes varying between 22 and 27 KDa; of these, Prdx6 in its native form is a 26 KDa protein and consists of 224 amino acids. The human Prdx6 gene comprises 11,542 base pairs and is located on Chromosome 1. The protein encoded by this gene is a member of the thiol-specific antioxidant protein family. Prdx6 is reported to be expressed in almost all cell types with high expression levels noted in lung endothelial and epithelial cells, lens epithelial cells, hepatocytes, leukocytes, neutrophils, etc. [4,5].
Prdx6 has long been established as a bifunctional enzyme. The antioxidant property, i.e., the peroxidase activity, is dependent on the catalytic Cys at position 47 [6], which is reduced by GSH S-transferase-bound GSH to complete the catalytic cycle [7], while the other enzymatic function of Prdx6 (a calcium-independent phospholipase (PLA2) activity) is dependent on a catalytic triad: Ser32, His26, and Asp140 [8], which catalyze the hydrolysis of the acyl group of phospholipids. The PLA2 activity of Prdx6 has been reported to play a major role in the metabolism of the phospholipids of lung surfactant [9] and to activate NADPH oxidase [10,11]. Prdx6 is a cytosolic enzyme, but upon stimulation of cells (with inflammatory stimuli), it is phosphorylated and the phosphorylated form translocates to the plasma membrane, where it supports NADPH oxidase activity [5].
Thus Prdx6 is unique in that it possesses both glutathione peroxidase (GPx) and calcium independent phospholipase A2 (PLA2) activities [9]. Prdx6 is able to act as an antioxidant by limiting oxidative stress by reducing short-chain hydroperoxides through its peroxidase activity. However, the PLA2 activity specific to Prdx6 leads to the generation of oxidants [11]. Thus, while the peroxidase activity is crucial in protecting against oxidative stress, the PLA2 activity plays an important role in the production of reactive oxygen species (ROS). These two paradoxical abilities of Prdx6 seem to point to a regulatory role of this enzyme in oxidative stress. It is conceivable that Prdx6 acts as a regulator or rheostat to fine tune ROS levels via the two activities, so as to enable optimal ROS levels for maintaining vascular homeostasis.
The lungs are one of the major organs exposed to the environment. The airways are in direct contact with a wide range of chemical and biological components in the atmosphere. Environmental agents also affect the lung vasculature via the alveolar–capillary structure, where pulmonary gas exchange occurs. The lung is also highly vascularized, with the endothelium that lines the lung vessels comprising >30% of the lungs [12]. Moreover, the endothelium, specifically the pulmonary endothelium, is the converging site of inflammation, whereby polymorphonuclear neutrophils (PMN) adhere to the vessel wall, followed by their transmigration into tissue. Reports have established that endothelial redox signaling facilitates lung inflammation via upregulation of adherence and transmigration of PMN, macrophages, and other immune cells [12,13,14].
Endothelial oxidative stress arises as a consequence of an imbalance between the production of ROS and the antioxidant defenses. The increase in ROS (either external or generated by the cell) leads to alteration of cellular proteins and organelles and is a major cause of cell death. Among the antioxidant defenses is Prdx6, which is highly expressed in the lung; indeed both endothelial and non-endothelial cells of the lung express high amounts of Prdx6 [11,15]. In addition to acting as an antioxidant, we reported that Prdx6 also participates in the generation of ROS, specifically in endothelial cells [11]. This review will focus on how these contradictory roles of Prdx6 facilitate the regulation vascular homeostasis and disease.
2. Peroxiredoxin 6 in the Endothelium
The endothelium is a critical component of vascular function by virtue of the signaling pathways that are activated in response to both physical forces, associated with blood flow (endothelial mechanotransduction) [16,17] and chemical stimuli (endothelial chemotransduction) [12,18] from chemical toxins, bacterial endotoxins, etc. Prdx6 expression in the endothelium has been reported, and it has been observed showing a steep increase with oxidative pathologies [19]. Examination of the aortic wall of subjects with aortic aneurysm showed a high expression of Prdx6 in the atherosclerotic plaques [20]. Lack of Prdx6 in the endothelium has been noted to increase sensitivity to oxidative stress. Conversely, it also leads to decreased ROS production in response to various stimuli [21]. Studies evaluating the relative roles of the peroxidase (GPx) and PLA2 activities of Prdx6 in pulmonary microvascular cells exposed directly to the oxidant (tert-butyl hydroperoxide) revealed that both activities participated in protecting cells from oxidative stress [22].
3. Peroxiredoxin 6 in Endothelial Mechanotransduction
The endothelium by virtue of its location “senses” the alteration of blood flow, as would occur with ischemia–reperfusion (I/R). Our work in the past decade has shown that the stop and restart of blood flow to the lung activates “flow-sensitive” machinery on lung endothelium comprising a KATP channel and NADPH oxidase 2 (NOX2) that leads to the production of ROS [23,24]. Our work has shown that the deletion of NOX2 (null mice) leads to a complete abrogation of ROS production with lung ischemia and reperfusion [25,26]. The deletion of Prdx6 (Prdx6-null mice) also led to diminution of ROS with lung ischemia [27]. This seemed paradoxical, as a lack of the antioxidant would be expected to increase ROS production. Our investigations revealed that Prdx6 was required for NOX2 activation. Indeed, we showed that in pulmonary microvascular endothelial cells, the phospholipase activity (PLA2) of Prdx6 was a key event in NOX2 activation [11,28].
However, Prdx6 does act as an antioxidant in other models of I/R that involve systemic organs [29], for instance, in liver I/R, where ROS production is believed to be predominantly via mitochondrial ROS, deletion of Prdx6, and increased injury. Isolation of mitochondria after ischemia or I/R demonstrated that a lack of mitochondrial Prdx6 led to mitochondrial dysfunction via disruption of mitochondrial respiration [29]. In a model of intestinal I/R (achieved by occlusion of the superior mesenteric artery), Prdx6 was found to play a protective role as delivery of exogenous Prdx6 significantly reduced I/R injury. However the administration of mutant forms of Prx6 (Prx6C47S) that do not possess the peroxidase activity had no protective effect [30]. Elsewhere, a hind limb model of I/R was found to show reduced injury upon exogenous administration of Prdx6 [31]. In a cerebral ischemia–reperfusion model, where non-endothelial cells were also evaluated, blocking Prdx6–PLA2 (either by siRNA or by a PLA2 inhibitor MJ33) reduced pro-inflammatory cytokines, and increased neuronal survival was compared to the wild type. In this study, both in vitro (oxygen–glucose deprivation/recovery) and in vivo (middle cerebral artery occlusion) models were used and I/R represented hypoxia–reoxygenation [32]. Thus, in several systemic models of I/R, Prdx6, either in endogenous form or administered exogenously, neutralizes oxidative stress, thus reducing the extent of tissue injury destruction with I/R. Overall, in the models of systemic I/R, the antioxidant effect of Prdx6 was significant, while lung I/R showed Prdx6–PLA2 activity. This discrepancy arises because systemic I/R models differ greatly from lung I/R—in the lung, the stop of blood flow does not compromise oxygen supply as the lung parenchyma obtains oxygen from the alveolus, but the systemic organs are dependent on blood flow for oxygen supply [33]. Lung I/R is thus a mechanotransduction event, where endothelial signaling with alteration of flow occurs due to loss of the mechanical component of flow and is independent of the partial oxygen pressures. In contrast, systemic I/R represents both altered flow stimulus and the effects of anoxia or hypoxia, followed by reoxygenation. The ROS generated by lung I/R has been reported to be via NADPH oxidase 2, whose assembly is dependent on the PLA2 activity of Prx6 [11,27]. Thus, loss of Prdx6 compromises ROS production with mechanotransduction. In systemic I/R, ROS is produced via either xanthine oxidase or the disrupted mitochondrial electron system (complex I–complex III) [34,35]. These pathways do not seem to involve Prdx6. Thus, in metabolically active organs, lack of Prdx6 leads to enhanced I/R injury, while in lungs where normoxia is maintained throughout I/R, and where the ROS production occurs via Prdx6–PLA2 activity, lack of Prdx6 leads to compromised ROS production [27].
4. Peroxiredoxin 6 in Endothelial Chemotransduction
Pulmonary endothelial cells are exposed to inhaled agonists via the alveolar capillary interface or agonists in the systemic circulation. Exposure to paraquat (PQ), a toxic herbicide, results in cell death of lung endothelial cells [36,37,38]. PQ, a prototypical redox cycling agent, is rapidly taken up and accumulates in endothelial cells. It produces ROS, leading to oxidative stress and apoptosis by a mitochondrial dependent pathway [39]. Pulmonary endothelial cells, when exposed to PQ, show a significant increase in oxidative damage and cell death [40]. In Prdx6-null mice, PQ administration led to increased lung injury. Although the effect on the pulmonary endothelium was not evaluated per se, vascular leakiness, an index of endothelial injury, showed a significant increase in Prdx6-null mice treated with PQ [36]. This indicates that Prdx6 is pivotal in protection, presumably via its peroxidase activity, against PQ.
Chemotransduction, associated with the endotoxin LPS, involves a role for Prdx6–PLA2 activity [41]. LPS is a component of the outer membrane in Gram-negative bacteria and is involved in the pathogenesis of sepsis [42,43]. We reported that LPS exposure on endothelial cells leads to ROS production via the NOX2 pathway [41]. This requires PLA2 activation of Prdx6. Indeed, we previously demonstrated that Prdx6–PLA2 generates lysophosphatidylcholine (LPC), which is converted to lysophosphatidic acid (LPA) by the lysophospholipase D activity of autotaxin. The binding of LPA to its receptor (LPAR) leads to an assembly of NOX2 components on the cell membrane [28]. Thus Prdx6–PLA2 activity facilitates (via LPA production) ROS generation by endothelial cells in response to agonists such as LPS (Figure 1). However, it is not clear if the PLA2 signaling pathway is effective in NOX2 activation only within the same cell. Studies elsewhere have shown that PLA2 products, such as eicosanoids and platelet-activating factor engage receptors in a paracrine fashion [44]. Other cells, such as PMN, also show Prdx6–PLA2 activity and, therefore, it is possible that PLA2 products secreted from non-endothelial and endothelial cells can activate NOX2 in a paracrine manner. Investigations into intercellular cross talk via PLA2 products have not been reported as yet. Our studies monitored PLA2 and NOX2 activation in entire endothelial monolayers; such methods are insufficient to detect the local and paracrine effects of PLA2 products, therefore, the NOX2 activation within each cell type of our ROS measurements, and thus, to an extent, the PLA2 cleavage. The methods we used showed a conclusive link between Prdx6–PLA2 activity; indeed we demonstrated that a lack of Prdx6–PLA2 activity, as achieved by mutations in Asp140, led to reduction in ROS [28].
In vivo exposures to LPS either via the systemic circulation or intratracheal instillation also resulted in ROS generation by the lung endothelium via the NOX2 pathway. Deleting Prdx6 by the use of Prdx6-null mice or by the inhibitor MJ33 resulted in a decrease in endothelial ROS production [41,45]. However, in other models of LPS-induced systemic injury, such as kidney damage, that involve an inflammatory response, Prdx6 seemed to be participating in a protective role. Prdx6-overexpressing mice showed decreased mortality and renal injury following the LPS challenge, compared to wild type (WT) mice. The inflammatory response of the kidney to LPS in the form of infiltration of macrophages, T-cells, and neutrophils, were also lower in Prdx6-overexpressing mice, as compared to wild-type mice [46]. This indicated that LPS-induced chemotransduction involved ROS generation (Prdx6–PLA2 activity) in certain models of injury, while in others, it was the antioxidant activity of Prdx6 (peroxidase activity) that was observed in response to ROS generation that presumably occurred via non NOX2 pathways.
Angiotensin II (Ang II) is an endogenous agonist that raises blood pressure primarily through vasoconstriction by acting on the endothelium and producing ROS [47]. We and others showed that Ang II activates the NOX2 pathway on endothelial cells in vitro and on the lung endothelium in vivo, via Prdx6–PLA2 activation [11,28]. Blocking PLA2 activation can potentially provide a pharmacological approach to Ang II-induced vasocontriction.
A unique feature of Prdx6 is the regulation of its antioxidant action by partner protein glutathione S-transferase Pi (GSTπ). GSTπ is a member of the phase II detoxification enzyme family that catalyzes the formation of thioether bonds between GSH and electrophilic centers on small proteins. One such protein is Prdx6 and GST-π is well established to catalyze the conjugation of the antioxidant GSH to Prdx6 [7,48]. GSH is not able to access the catalytic cysteine residue on Prdx6. Upon conversion to its anionic form, GS- (by GSTπ), it can bind to Prdx6 (Figure 2). The glutathionylation of the oxidized cysteine in Prdx6 is followed by a spontaneous reduction of the mixed disulfide and restoration of enzymatic activity of Prdx6 (Figure 2B) [7]. GSTπ has been reported to be highly expressed in endothelial cells, specifically with pathologies such as neurological disorders [49], and protects against endothelial permeability and damage in response to inflammatory stimuli [50]. Studies using several oxidant and inflammation stimuli have demonstrated that GSTπ acts as a negative regulator of endothelial dysfunction [51]. These studies did not investigate the role of Prdx6 per se; however, based on the fact that GST-π can exert its protective function via activation of the oxidized Prdx6 [48], it is presumable that Prdx6 plays a role in protection against endothelial dysfunction in oxidant-induced pathologies.
5. Peroxiredoxin 6 in Inflammatory Response
ROS-induced signaling has been well established as playing a role in the onset and amplification of inflammation. The endothelium is a converging site of inflammation, where PMN, macrophages, and other immune cells adhere to and extravasate from blood to tissue. An enhanced ROS generation on the endothelium is thus a first step in inflammation [52]. We showed that ROS production by the endothelium in response to either chemotransduction or mechanotransduction is an inflammatory stimulus [53]. Endothelial ROS leads to an increase in cellular adhesion molecules and pro-inflammatory cytokines that recruit PMN and other immune cells to the vascular wall. ROS produced by PMN in addition to the endothelial ROS, leads to oxidative stress in the vasculature and leads to opening of inter-endothelial cell–cell junctions that in turn promotes the migration of PMN, etc., across the vascular wall [52]. Thus, enzymes with roles in redox balance, i.e., with both antioxidants as well as ROS-generating capacity, play a critical role in inflammatory and immune responses. Prdx6 possesses both of these activities and, therefore, it is reasonable to assume that this enzyme would regulate inflammation in vivo.
Upregulation of Prdx6 has been observed under conditions of increased ROS generation in various models of injury, implying that oxidative stress leads to increased Prdx6 transcription. This also implies that Prdx6 expression can be indicative of oxidative stress pathologies. Studies in patients with peripheral arterial disease show that circulating levels of PRDX1, 2, 4, and 6 are markedly raised [54]. In patients with abdominal aortic aneurysms, there are increased levels of Prdx6, both in the plasma and in the tissue from the aneurysm [20]. Lack of Prdx6 (PRDX6-null mice) showed significantly higher aortic lesions in response to oxidative stress [55]. Prdx6 is also high in serum of patients with osteoarthritis and those with femoral neck fracture and there are strong correlations between the levels of these molecules in the serum and severity of these conditions [56]. It is not clear in all of these models of inflammation whether Prdx6 participates in redox imbalance through protective antioxidant functions or by redox signaling via activation of NOX2. In an oxidative stress environment, hyperoxidation of Prdx6 is reported to upregulate its PLA2 activity [57]. Conversely, environments with a high inflammation and oxidative stress load, such as aortic lesions or aneurysms, can cause post-translation modification of Prdx6, such that it loses its antioxidant activity [58]. The major pro-inflammatory transcription factor NFκB has been observed to be regulated by Prdx6 [19]. The Prdx6 promoter (−1139 bp) containing κB binding sites, showed reduced promoter activity in Prdx6 null cells [59]. Besides the peroxidase activity that facilitates protection from oxidative stress, Prdx6 also protects by downregulating NFκB [60].
6. Prdx6 in Wound Repair
Wound repair is based on the balance between oxidative stress and regeneration. Thus, most of the injury- and repair-regulated genes encode for antioxidant enzymes, which scavenge on ROS [61,62]. Among the wound-regulated genes that have been identified, one is Prdx6 [61]. In mice, skin wounds were found to have an overexpression of Prdx6 in the epidermis [63]. The epidermis of psoriatic patients and cells of the wound granulation tissue also showed high expression of Prdx6 [61,63].
Prdx6 seems to be protective against dermal injury and to facilitate wound repair. Indeed, mice overexpressing Prdx6 in the epidermis were protected from UVA- and UVB-induced skin damage [61,63]. These mice also showed accelerated wound closure [64]. Similarly, lack of Prdx6, i.e., Prdx6-null mice, increased keratinocyte apoptosis and endothelial damage. In another study using an excision model of injury, the role of the Prdx6 in the endothelium was found to be crucial in the repair of endothelial cells damaged by oxidative stress [21]. Prdx6-null mice showed the appearance of granulated tissue associated with hemorrhage due to endothelial damage. Using chimeric mice (wild-type and Prdx6-null mice with WT- and Prdx6-null bone marrow cells) and ultrastructural analysis of tissue, it was observed that Prdx6 expression on the endothelium correlated with the formation of new blood vessels, suggesting that Prdx6 is required for endothelial cell integrity and viability in the wound tissue [21]. The onset of hemorrhage correlating with the formation of novel blood vessels suggests the importance of Prdx6 for endothelial cell maintenance, structure, and viability in wounded tissue. However, lack of Prdx6 affected the endothelial cell integrity, structure, and function, only during oxidative stress and not under normal conditions, implying that the protective role was crucial only post-injury and did not affect the endothelium under basal conditions. Overall, these studies seem to reveal the role for the peroxidase activity of Prdx6 in protecting the epidermis from oxidative damage. However, it is not clear if peroxidase and/or PLA2 activities might participate in wound repair. This is because transcription factors, such as NFκB, that are crucial for skin wound repair processes are regulated both by ROS and Prdx6 [65] and there is some evidence for the role of Prdx6–PLA2 activity in NFκB regulation [59].
7. Prdx6 in the Pathogenesis of Diabetes
Increased oxidative stress appears to be a major factor leading to insulin resistance and pathogenesis of diabetes [66]. Pancreatic β-cells that produce insulin express very low levels of antioxidant enzymes and are thus susceptible to oxidative stress [67]. Although β-cells express Prdx6 [68], studies using insulin-producing RINm5F cells have shown that Prdx6 expression (both mRNA and protein) is downregulated in response to proinflammatory cytokines [69]. This reduces the antioxidant capacity of Prdx6. Furthermore, lack of Prdx6 (in mice) causes the development of a phenotype similar to early-stage Type II diabetes in terms of both reduced glucose-dependent insulin secretion and increased insulin resistance [70].
Dysfunction of the endothelium is similarly observed in diabetes [71]. Under conditions of oxidative stress, the endothelial repair mechanisms are hindered. This is because endothelial repair occurs via endothelial progenitor cells (EPC) [72]. Under basal conditions, EPCs are immature cells and differentiate into mature endothelial cells. With vascular injury, EPC mobilization from the peripheral circulation (aided via growth factors and cytokines) to sites of damage is a crucial component of repair and angiogenesis. Both type 1 and type 2 diabetics have less circulating EPCs than matched healthy subjects. Diabetic EPCs also show reduced proliferation, adhesion, migration, and impaired development of tubules for the formation of a vascular network [71,73].
As oxidative stress and insulin resistance are associated with a proinflammatory state, inflammation often accompanies diabetes [71]. The link between diabetes, inflammation, and Prdx6 is not clear. On one hand, inflammation can often lower expression of Prdx6, while on the other, it is overexpressed in some inflammatory pathologies, such as inflammatory bowel disease (IBD) [74]. Indeed, human colonic biopsies of IBD patients showed high expression of Prdx6. Mice that lack Prdx6 show a marked increase in proinflammatory cytokines (IL-1β and TNF-α) and in matrix metalloproteases (MMP-2 and -9) in response to an inflammatory stimulus [75]. Elsewhere, these mice showed an insulin resistance phenotype [70]. Insulin resistance-associated inflammation could be partly regulated by the Prdx6, although the role of its PLA2 versus peroxidase activities in this regulation has not been clearly defined.
A recent report seems to point to a role for Prdx6 in glucose homeostasis, primarily via its antioxidant role [69]. Reports elsewhere indicate that the pathophysiological impairments associated with diabetes, such as sarcopenia (loss of muscle mass), muscle atrophy, and diabetic myopathy can potentially be rescued by Prdx6 [76]. Gene expression of MyoD and myogenin, that participate in the differentiation of muscle cells, was observed to be significantly lower in Prdx6-null mice as compared to wild type controls [76]. This points to a role for Prdx6 in the maintenance of muscle mass and points to a possible use of this enzyme for therapy in diabetic myopathy and sarcopenia. Thus, Prdx6 could potentially represent diabetes susceptibility in humans. Moreover, associations between Prdx6 expression and inflammatory endothelial phenotype are possible factors in endothelial dysfunction with diabetes.
8. Conclusions
The family of peroxiredoxins has been recognized as major scavenging enzymes in mammalian systems. Of these, Prdx6 is unique in that its peroxidase activity facilities oxidant scavenging, while Prdx6–PLA2 activity is crucial in oxidant generation (Figure 3). The dual function of this enzyme presumably enables it to act as a “regulatory body”, whereby its protective role and its oxidant-generating action is not exceeded, so as to perturb vascular homeostasis. This optimal regulation of Prdx6 expression and activity is crucial in fine tuning cellular ROS levels and seems to occur via complex signaling cascades that involve NFκB and oxidants. While upregulation of Prdx6 is protective, it is also associated with several pathologies ranging from aortic lesions to diabetes susceptibility. Low levels or lack of Prdx6 are also associated with inflammatory diseases. These observations and reports indicate that regulation of the peroxidase and PLA2 activities is complex and intersects with several other transcriptional pathways. Further studies using Prdx6 mutants and null systems on diverse models of inflammation and injury are needed to strengthen our knowledge regarding the relationship between Prdx6 regulation and oxidant and antioxidant signaling and the final physiological and pathophysiological effects.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rhee, S.G.; Chae, H.Z.; Kim, K. Peroxiredoxins: A historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic. Biol. Med. 2005, 38, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Kang, S.W.; Chang, T.S.; Jeong, W.; Kim, K. Peroxiredoxin, a novel family of peroxidases. IUBMB Life 2001, 52, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Sorokina, E.M.; Harper, S.; Li, H.; Ralat, L.; Dodia, C.; Speicher, D.W.; Feinstein, S.I.; Fisher, A.B. Peroxiredoxin 6 homodimerization and heterodimerization with glutathione S-transferase pi are required for its peroxidase but not phospholipase A2 activity. Free Radic. Biol. Med. 2016, 94, 145–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolussi, A.; D’Inzeo, S.; Capalbo, C.; Giannini, G.; Coppa, A. The role of peroxiredoxins in cancer. Mol. Clin. Oncol. 2017, 6, 139–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambruso, D.R.; Ellison, M.A.; Thurman, G.W.; Leto, T.L. Peroxiredoxin 6 translocates to the plasma membrane during neutrophil activation and is required for optimal NADPH oxidase activity. Biochim. Biophys. Acta. 2012, 1823, 306–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.W.; Dodia, C.; Feinstein, S.I.; Jain, M.K.; Fisher, A.B. 1-Cys peroxiredoxin, a bifunctional enzyme with glutathione peroxidase and phospholipase A2 activities. J. Biol. Chem. 2000, 275, 28421–28427. [Google Scholar] [CrossRef] [PubMed]
- Manevich, Y.; Feinstein, S.I.; Fisher, A.B. Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with pi GST. Proc. Natl. Acad. Sci. USA 2004, 101, 3780–3785. [Google Scholar] [CrossRef]
- Manevich, Y.; Reddy, K.S.; Shuvaeva, T.; Feinstein, S.I.; Fisher, A.B. Structure and phospholipase function of peroxiredoxin 6: Identification of the catalytic triad and its role in phospholipid substrate binding. J. Lipid Res. 2007, 48, 2306–2318. [Google Scholar] [CrossRef]
- Manevich, Y.; Fisher, A.B. Peroxiredoxin 6, a 1-Cys peroxiredoxin, functions in antioxidant defense and lung phospholipid metabolism. Free Radic. Biol. Med. 2005, 38, 1422–1432. [Google Scholar] [CrossRef]
- Fisher, A.B. The phospholipase A2 activity of peroxiredoxin 6. J. Lipid Res. 2018, 59, 1132–1147. [Google Scholar] [CrossRef]
- Chatterjee, S.; Feinstein, S.I.; Dodia, C.; Sorokina, E.; Lien, Y.C.; Nguyen, S.; Debolt, K.; Speicher, D.; Fisher, A.B. Peroxiredoxin 6 phosphorylation and subsequent phospholipase A2 activity are required for agonist-mediated activation of NADPH oxidase in mouse pulmonary microvascular endothelium and alveolar macrophages. J. Biol. Chem. 2011, 286, 11696–11706. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S. Endothelial Mechanotransduction, Redox Signaling and the Regulation of Vascular Inflammatory Pathways. Front. Physiol. 2018, 9, 524. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Fisher, A.B. Endothelium: A Comprehensive Reference; Cambridge University Press: Cambridge, MA, USA, 2007. [Google Scholar]
- Sarantos, M.R.; Zhang, H.; Schaff, U.Y.; Dixit, N.; Hayenga, H.N.; Lowell, C.A.; Simon, S.I. Transmigration of neutrophils across inflamed endothelium is signaled through LFA-1 and Src family kinase. J. Immunol. 2008, 181, 8660–8669. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Pak, J.H.; Gonzales, L.W.; Feinstein, S.I.; Fisher, A.B. Regulation of 1-cys peroxiredoxin expression in lung epithelial cells. Am. J. Respir. Cell Mol. Biol. 2002, 27, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Browning, E.A.; Chatterjee, S.; Fisher, A.B. Stop the flow: A paradigm for cell signaling mediated by reactive oxygen species in the pulmonary endothelium. Annu. Rev. Physiol. 2012, 74, 403–424. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Fisher, A.B. Mechanotransduction: Forces, sensors, and redox signaling. Antioxid. Redox Signal. 2014, 20, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Fisher, A.B. Mechanotransduction in the endothelium: role of membrane proteins and reactive oxygen species in sensing, transduction, and transmission of the signal with altered blood flow. Antioxid. Redox Signal. 2014, 20, 899–913. [Google Scholar] [CrossRef] [PubMed]
- Chhunchha, B.; Fatma, N.; Bhargavan, B.; Kubo, E.; Kumar, A.; Singh, D.P. Specificity protein, Sp1-mediated increased expression of Prdx6 as a curcumin-induced antioxidant defense in lens epithelial cells against oxidative stress. Cell Death Dis. 2011, 2, e234. [Google Scholar] [CrossRef] [PubMed]
- Burillo, E.; Jorge, I.; Martinez-Lopez, D.; Camafeita, E.; Blanco-Colio, L.M.; Trevisan-Herraz, M.; Ezkurdia, I.; Egido, J.; Michel, J.B.; Meilhac, O.; et al. Quantitative HDL Proteomics Identifies Peroxiredoxin-6 as a Biomarker of Human Abdominal Aortic Aneurysm. Sci. Rep. 2016, 6, 38477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumin, A.; Schafer, M.; Epp, N.; Bugnon, P.; Born-Berclaz, C.; Oxenius, A.; Klippel, A.; Bloch, W.; Werner, S. Peroxiredoxin 6 is required for blood vessel integrity in wounded skin. J. Cell Biol. 2007, 179, 747–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lien, Y.C.; Feinstein, S.I.; Dodia, C.; Fisher, A.B. The roles of peroxidase and phospholipase A2 activities of peroxiredoxin 6 in protecting pulmonary microvascular endothelial cells against peroxidative stress. Antioxid. Redox Signal. 2012, 16, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Levitan, I.; Wei, Z.; Fisher, A.B. KATP channels are an important component of the shear-sensing mechanism in the pulmonary microvasculature. Microcirculation 2006, 13, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Browning, E.A.; Hong, N.; DeBolt, K.; Sorokina, E.M.; Liu, W.; Birnbaum, M.J.; Fisher, A.B. Membrane depolarization is the trigger for PI3K/Akt activation and leads to the generation of ROS. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H105–H114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Matsuzaki, I.; Chatterjee, S.; Fisher, A.B. Activation of endothelial NADPH oxidase during normoxic lung ischemia is KATP channel dependent. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 289, L954–L961. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chatterjee, S.; Wei, Z.; Liu, W.D.; Fisher, A.B. Rac and PI3 kinase mediate endothelial cell-reactive oxygen species generation during normoxic lung ischemia. Antioxid. Redox Signal. 2008, 10, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Feinstein, S.; Hong, N.K.; Debolt, K. Paradoxical response of ROS production in peroxiredoxin 6 null mice to ischemia. FASEB J. 2007, 21, A1201. [Google Scholar]
- Vazquez-Medina, J.P.; Dodia, C.; Weng, L.; Mesaros, C.; Blair, I.A.; Feinstein, S.I.; Chatterjee, S.; Fisher, A.B. The phospholipase A2 activity of peroxiredoxin 6 modulates NADPH oxidase 2 activation via lysophosphatidic acid receptor signaling in the pulmonary endothelium and alveolar macrophages. FASEB J. 2016, 30, 2885–2898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eismann, T.; Huber, N.; Shin, T.; Kuboki, S.; Galloway, E.; Wyder, M.; Edwards, M.J.; Greis, K.D.; Shertzer, H.G.; Fisher, A.B.; et al. Peroxiredoxin-6 protects against mitochondrial dysfunction and liver injury during ischemia-reperfusion in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G266–G274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordeeva, A.E.; Temnov, A.A.; Charnagalov, A.A.; Sharapov, M.G.; Fesenko, E.E.; Novoselov, V.I. Protective Effect of Peroxiredoxin 6 in Ischemia/Reperfusion-Induced Damage of Small Intestine. Dig. Dis. Sci. 2015, 60, 3610–3619. [Google Scholar] [CrossRef] [PubMed]
- Kubyshkin, A.V.; Novosyolov, S.V.; Fomochkina, I.I.; Kharchenko, V.Z.; Pisarev, A.A.; Gordeeva, A.E.; Beketov, A.A.; Kochkina, A.V.; Fedosov, M.I.; Anisimova, L.V.; et al. Expression of caspase-3 and the cytokine level in experimental reperfusion syndrome upon treatment with peroxiredoxin 6. Biophysics 2017, 62, 848. [Google Scholar] [CrossRef]
- Shanshan, Y.; Beibei, J.; Li, T.; Minna, G.; Shipeng, L.; Li, P.; Yong, Z. Phospholipase A2 of Peroxiredoxin 6 Plays a Critical Role in Cerebral Ischemia/Reperfusion Inflammatory Injury. Front. Cell. Neurosci. 2017, 11, 99. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Chapman, K.E.; Fisher, A.B. Lung ischemia: A model for endothelial mechanotransduction. Cell Biochem. Biophys. 2008, 52, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef]
- Wu, M.Y.; Yiang, G.T.; Liao, W.T.; Tsai, A.P.; Cheng, Y.L.; Cheng, P.W.; Li, C.Y.; Li, C.J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef]
- Wang, Y.; Feinstein, S.I.; Manevich, Y.; Ho, Y.S.; Fisher, A.B. Peroxiredoxin 6 gene-targeted mice show increased lung injury with paraquat-induced oxidative stress. Antioxid. Redox Signal. 2006, 8, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Feinstein, S.I.; Wang, Y.; Dodia, C.; Fisher, D.; Yu, K.; Ho, Y.S.; Fisher, A.B. Comparison of glutathione peroxidase 1 and peroxiredoxin 6 in protection against oxidative stress in the mouse lung. Free Radic. Biol. Med. 2010, 49, 1172–1181. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.; Heath, D. The pathology of the lung in paraquat poisoning. J. Clin. Pathol. Suppl. (R. Coll. Pathol.) 1975, 9, 81–93. [Google Scholar] [CrossRef]
- Day, B.J.; Patel, M.; Calavetta, L.; Chang, L.Y.; Stamler, J.S. A mechanism of paraquat toxicity involving nitric oxide synthase. Proc. Natl. Acad. Sci. USA 1999, 96, 12760–12765. [Google Scholar] [CrossRef] [Green Version]
- Tsukamoto, M.; Tampo, Y.; Sawada, M.; Yonaha, M. Paraquat-induced oxidative stress and dysfunction of the glutathione redox cycle in pulmonary microvascular endothelial cells. Toxicol. Appl. Pharmacol. 2002, 178, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Dodia, C.; Chatterjee, S.; Feinstein, S.I.; Fisher, A.B. Protection against LPS-induced acute lung injury by a mechanism based inhibitor of NADPH-oxidase (Type 2). Am. J. Physiol. Lung Cell Mol. Physiol. 2014. [Google Scholar] [CrossRef]
- Parrillo, J.E.; Parker, M.M.; Natanson, C.; Suffredini, A.F.; Danner, R.L.; Cunnion, R.E.; Ognibene, F.P. Septic shock in humans. Advances in the understanding of pathogenesis, cardiovascular dysfunction, and therapy. Ann. Intern. Med. 1990, 113, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B.; Rietschel, E.T. Innate immune sensing and its roots: The story of endotoxin. Nat. Rev. Immunol. 2003, 3, 169–176. [Google Scholar] [CrossRef]
- Lagarde, M.; Gualde, N.; Rigaud, M. Metabolic interactions between eicosanoids in blood and vascular cells. Biochem. J. 1989, 257, 313–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hood, E.D.; Greineder, C.F.; Dodia, C.; Han, J.; Mesaros, C.; Shuvaev, V.V.; Blair, I.A.; Fisher, A.B.; Muzykantov, V.R. Antioxidant protection by PECAM-targeted delivery of a novel NADPH-oxidase inhibitor to the endothelium in vitro and in vivo. J. Controll. Release off. J. Controll. Release Soc. 2012, 163, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.H.; Park, J.H.; Han, S.B.; Yoon, D.Y.; Jung, Y.Y.; Hong, J.T. Peroxiredoxin 6 overexpression attenuates lipopolysaccharide-induced acute kidney injury. Oncotarget 2017, 8, 51096–51107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, T.; Kajikuri, J.; Tada, T.; Suzuki, Y.; Mabuchi, Y. Angiotensin II-induced modulation of endothelium-dependent relaxation in rabbit mesenteric resistance arteries. J. Physiol. 2003, 548, 893–906. [Google Scholar] [CrossRef]
- Ralat, L.A.; Manevich, Y.; Fisher, A.B.; Colman, R.F. Direct evidence for the formation of a complex between 1-cysteine peroxiredoxin and glutathione S-transferase π with activity changes in both enzymes. Biochemistry 2006, 45, 360–372. [Google Scholar] [CrossRef]
- Shang, W.; Liu, W.H.; Zhao, X.H.; Sun, Q.J.; Bi, J.Z.; Chi, Z.F. Expressions of glutathione S-transferase alpha, mu, and pi in brains of medically intractable epileptic patients. BMC Neurosci. 2008, 9, 67. [Google Scholar] [CrossRef]
- Yang, Y.; Yin, F.; Hang, Q.; Dong, X.; Chen, J.; Li, L.; Cao, P.; Yin, Z.; Luo, L. Regulation of Endothelial Permeability by Glutathione S-Transferase Pi Against Actin Polymerization. Cell. Physiol. Biochem. 2018, 45, 406–418. [Google Scholar] [CrossRef]
- Conklin, D.J.; Haberzettl, P.; Prough, R.A.; Bhatnagar, A. Glutathione-S-transferase P protects against endothelial dysfunction induced by exposure to tobacco smoke. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1586–H1597. [Google Scholar] [CrossRef]
- Walzog, B.; Gaehtgens, P. Adhesion Molecules: The Path to a New Understanding of Acute Inflammation. News Physiol. Sci. 2000, 15, 107–113. [Google Scholar] [CrossRef]
- Browning, E.; Wang, H.; Hong, N.; Yu, K.; Buerk, D.G.; DeBolt, K.; Gonder, D.; Sorokina, E.M.; Patel, P.; De Leon, D.D.; et al. Mechanotransduction drives post ischemic revascularization through K(ATP) channel closure and production of reactive oxygen species. Antioxid. Redox Signal. 2014, 20, 872–886. [Google Scholar] [CrossRef]
- El Eter, E.; Al Masri, A.; Habib, S.; Al Zamil, H.; Al Hersi, A.; Al Hussein, F.; Al Omran, M. Novel links among peroxiredoxins, endothelial dysfunction, and severity of atherosclerosis in type 2 diabetic patients with peripheral atherosclerotic disease. Cell Stress Chaperones 2014, 19, 173–181. [Google Scholar] [CrossRef]
- Wang, X.; Phelan, S.A.; Forsman-Semb, K.; Taylor, E.F.; Petros, C.; Brown, A.; Lerner, C.P.; Paigen, B. Mice with targeted mutation of peroxiredoxin 6 develop normally but are susceptible to oxidative stress. J. Biol. Chem. 2003, 278, 25179–25190. [Google Scholar] [CrossRef] [PubMed]
- Kuroiwa, T. New biomarkers of osteoarthritis. Osteoarthr. Cartil. 2016, 24, S89. [Google Scholar] [CrossRef]
- Kim, S.Y.; Jo, H.Y.; Kim, M.H.; Cha, Y.Y.; Choi, S.W.; Shim, J.H.; Kim, T.J.; Lee, K.Y. H2O2-dependent hyperoxidation of peroxiredoxin 6 (Prdx6) plays a role in cellular toxicity via up-regulation of iPLA2 activity. J. Biol. Chem. 2008, 283, 33563–33568. [Google Scholar] [CrossRef] [PubMed]
- Chhunchha, B.; Fatma, N.; Kubo, E.; Singh, D.P. Aberrant sumoylation signaling evoked by reactive oxygen species impairs protective function of Prdx6 by destabilization and repression of its transcription. FEBS J. 2014, 281, 3357–3381. [Google Scholar] [CrossRef] [PubMed]
- Fatma, N.; Kubo, E.; Takamura, Y.; Ishihara, K.; Garcia, C.; Beebe, D.C.; Singh, D.P. Loss of NF-kappaB control and repression of Prdx6 gene transcription by reactive oxygen species-driven SMAD3-mediated transforming growth factor beta signaling. J. Biol. Chem. 2009, 284, 22758–22772. [Google Scholar] [CrossRef] [PubMed]
- Tu, Q.; Xiong, Y.; Fan, L.; Qiao, B.; Xia, Z.; Hu, L.; Wang, Y.; Peng, G.; Ye, Q. Peroxiredoxin 6 attenuates ischemia and hypoxiainduced liver damage of braindead donors. Mol. Med. Rep. 2016, 13, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Munz, B.; Frank, S.; Hubner, G.; Olsen, E.; Werner, S. A novel type of glutathione peroxidase: Expression and regulation during wound repair. Biochem. J. 1997, 326 Pt 2, 579–585. [Google Scholar] [CrossRef]
- auf dem Keller, U.; Kumin, A.; Braun, S.; Werner, S. Reactive oxygen species and their detoxification in healing skin wounds. J. Investig. Dermatol. Symp. Proc. 2006, 11, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.; Munz, B.; Werner, S. The human homologue of a bovine non-selenium glutathione peroxidase is a novel keratinocyte growth factor-regulated gene. Oncogene 1997, 14, 915–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumin, A.; Huber, C.; Rulicke, T.; Wolf, E.; Werner, S. Peroxiredoxin 6 is a potent cytoprotective enzyme in the epidermis. Am. J. Pathol. 2006, 169, 1194–1205. [Google Scholar] [CrossRef] [PubMed]
- Ambrozova, N.; Ulrichova, J.; Galandakova, A. Models for the Study of Skin Wound Healing. The Role of Nrf2 and NF-kappaB; Biomedical Papers of the Medical Faculty of Palacky University in Olomouc; Palacky University in Olomouc: Olomouc, Czech Republic, 2017; Volume 161, pp. 1–13. [Google Scholar]
- Hurrle, S.; Hsu, W.H. The etiology of oxidative stress in insulin resistance. Biomed. J. 2017, 40, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Kajimoto, Y.; Kaneto, H. Role of oxidative stress in pancreatic beta-cell dysfunction. Ann. N. Y. Acad. Sci. 2004, 1011, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Wood, Z.A.; Schroder, E.; Robin Harris, J.; Poole, L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003, 28, 32–40. [Google Scholar] [CrossRef]
- Paula, F.M.; Ferreira, S.M.; Boschero, A.C.; Souza, K.L. Modulation of the peroxiredoxin system by cytokines in insulin-producing RINm5F cells: down-regulation of PRDX6 increases susceptibility of beta cells to oxidative stress. Mol. Cell. Endocrinol. 2013, 374, 56–64. [Google Scholar] [CrossRef]
- Pacifici, F.; Arriga, R.; Sorice, G.P.; Capuani, B.; Scioli, M.G.; Pastore, D.; Donadel, G.; Bellia, A.; Caratelli, S.; Coppola, A.; et al. Peroxiredoxin 6, a novel player in the pathogenesis of diabetes. Diabetes 2014, 63, 3210–3220. [Google Scholar] [CrossRef]
- Avogaro, A.; Albiero, M.; Menegazzo, L.; de Kreutzenberg, S.; Fadini, G.P. Endothelial dysfunction in diabetes: The role of reparatory mechanisms. Diabetes Care 2011, 34 (Suppl. 2), S285–S290. [Google Scholar] [CrossRef]
- Altabas, V. Diabetes, Endothelial Dysfunction, and Vascular Repair: What Should a Diabetologist Keep His Eye on? Int. J. Endocrinol. 2015, 2015, 848272. [Google Scholar] [CrossRef]
- Fadini, G.P.; Sartore, S.; Albiero, M.; Baesso, I.; Murphy, E.; Menegolo, M.; Grego, F.; Vigili de Kreutzenberg, S.; Tiengo, A.; Agostini, C.; et al. Number and function of endothelial progenitor cells as a marker of severity for diabetic vasculopathy. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2140–2146. [Google Scholar] [CrossRef] [PubMed]
- Melhem, H.; Spalinger, M.R.; Cosin-Roger, J.; Atrott, K.; Lang, S.; Wojtal, K.A.; Vavricka, S.R.; Rogler, G.; Frey-Wagner, I. Prdx6 Deficiency Ameliorates DSS Colitis: Relevance of Compensatory Antioxidant Mechanisms. J. Crohns Colitis 2017, 11, 871–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Song, Y.; Wang, X.; Sun, J.; Ben, Y.; An, X.; Tong, L.; Bi, J.; Bai, C. Deletion of peroxiredoxin 6 potentiates lipopolysaccharide-induced acute lung injury in mice. Crit. Care Med. 2011, 39, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Pacifici, F.; Capuani, B.; Piermarini, F.; Pastore, D.; Arriga, R.; Coppola, A.; Rea, S.; Donadel, G.; Bellia, A.; Della-morte, D.; et al. Prdx6 Prevents Diabetic Myopathy by Improving Skeletal Muscle Cell Differentiation; American Diabetes Association: Arlington, VA, USA, 2018; Volume 67. [Google Scholar]
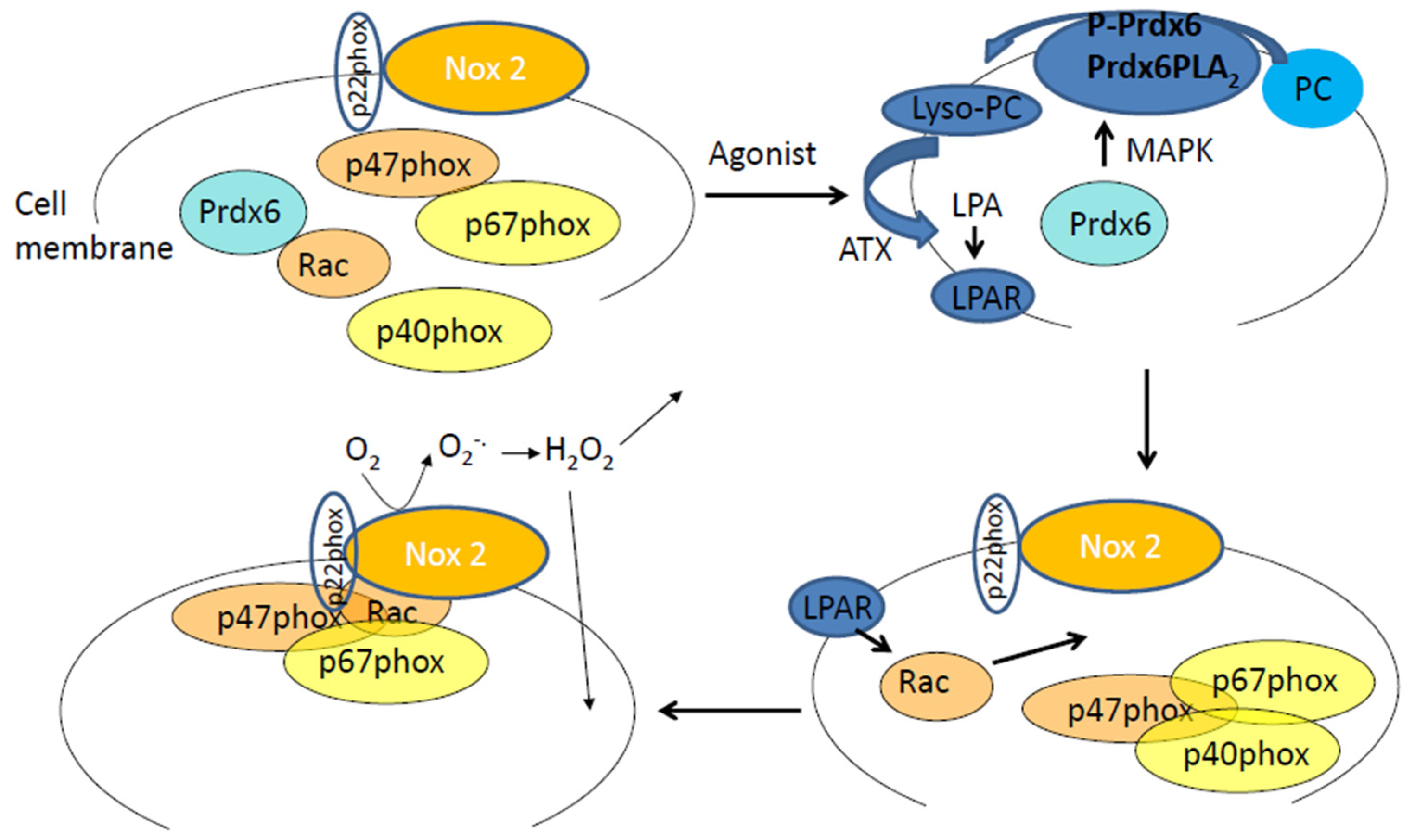
Figure 1.
The Prdx6–PLA2 activity and the production of reactive oxygen species (ROS) by endothelial cells in response to an agonist. In the cytosol are Prdx6 and cytosolic subunits of NADPH 2 oxidase (p40phox, p67phox, p47phox, and Rac). Upon phosphorylation (in response to an agonist, Angiotensin II, or LPS) Prdx6 translocates to the plasma membrane. P-Prdx6 has phosphospholipase A2 activity via which it hydrolyses membrane phosphatidylcholine (PC) to lysophosphatidylcholine (lyso-PC). Lyso-PC is catalyzed to lysophosphatidic acid (LPA) by the enzyme autotaxin (ATX). LPA binds to its receptor on the cell membrane, and the resulting signaling cascade leads to Rac phosphorylation that in turn enables assembly of the cytosolic components of NADPH oxidase 2 with the membrane bound components (gp91phox or Nox2 and p22phox). The assembled enzyme reduces molecular oxygen to superoxide which then dismutates to hydrogen peroxide. H2O2 can participate in extracellular and intracellular signaling cascades.
Figure 1.
The Prdx6–PLA2 activity and the production of reactive oxygen species (ROS) by endothelial cells in response to an agonist. In the cytosol are Prdx6 and cytosolic subunits of NADPH 2 oxidase (p40phox, p67phox, p47phox, and Rac). Upon phosphorylation (in response to an agonist, Angiotensin II, or LPS) Prdx6 translocates to the plasma membrane. P-Prdx6 has phosphospholipase A2 activity via which it hydrolyses membrane phosphatidylcholine (PC) to lysophosphatidylcholine (lyso-PC). Lyso-PC is catalyzed to lysophosphatidic acid (LPA) by the enzyme autotaxin (ATX). LPA binds to its receptor on the cell membrane, and the resulting signaling cascade leads to Rac phosphorylation that in turn enables assembly of the cytosolic components of NADPH oxidase 2 with the membrane bound components (gp91phox or Nox2 and p22phox). The assembled enzyme reduces molecular oxygen to superoxide which then dismutates to hydrogen peroxide. H2O2 can participate in extracellular and intracellular signaling cascades.

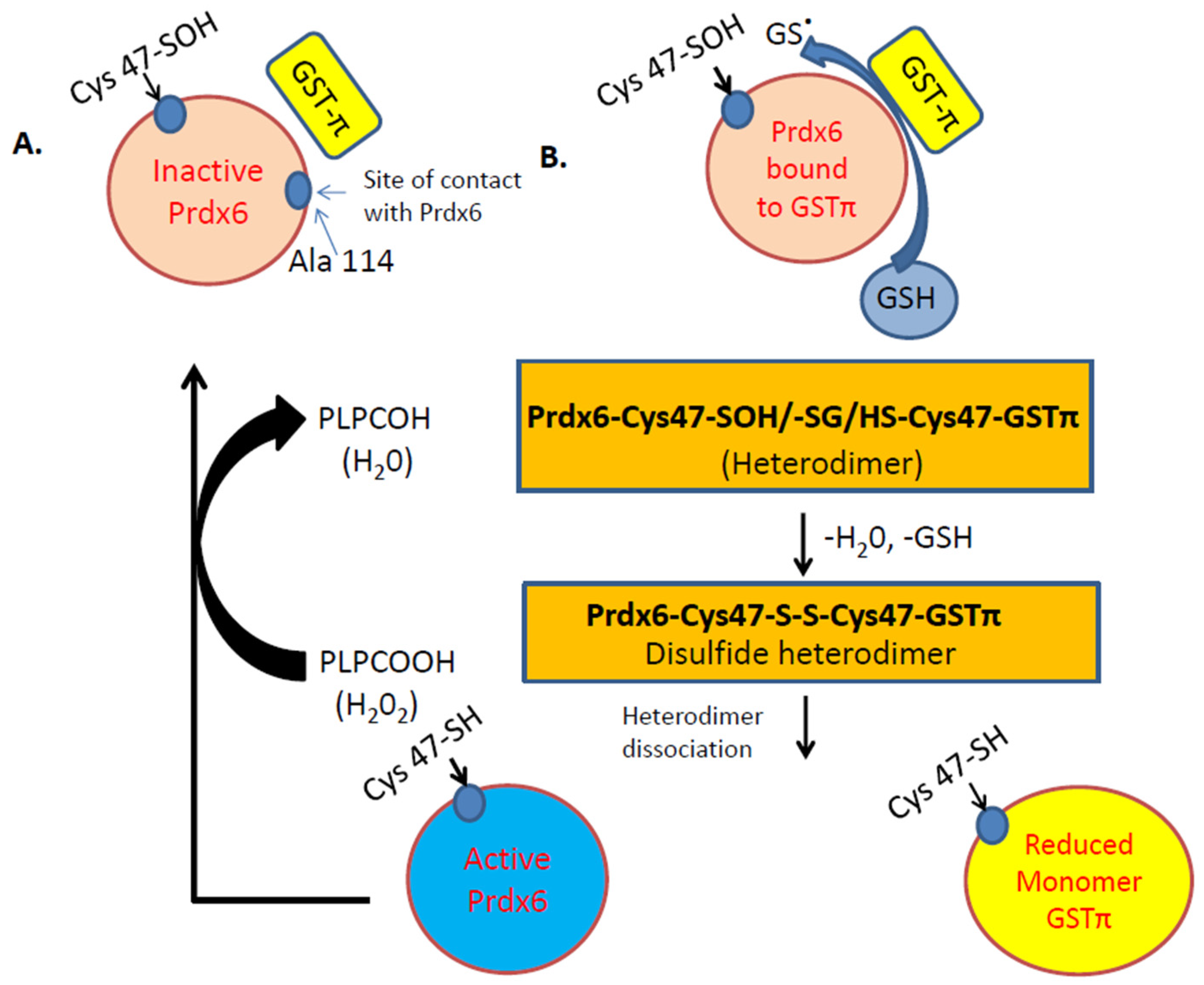
Figure 2.
Illustration of the Peroxidase activity of Prdx6. A. Inactive Prdx6: its active site (Cys 47) needs to be oxidized to Cys-sulfenic acid (Cys47-SOH). B. After oxidation of Cys47 to sulfenic acid (-SOH), the Prdx6 forms a heterodimer with thiolate anion (via GSTπ). S-glutathionylation of the heterodimer, followed by its alignment with the catalytic Cys47 of GSTπ, results in the formation of a disulfide-based heterodimer. Reduction of this disulfide bond by GSH causes heterodimer dissociation to active Prdx6 monomer. The active monomer reduces phospholipid hydroperoxide (PLPCOOH) to PLPCOH.
Figure 2.
Illustration of the Peroxidase activity of Prdx6. A. Inactive Prdx6: its active site (Cys 47) needs to be oxidized to Cys-sulfenic acid (Cys47-SOH). B. After oxidation of Cys47 to sulfenic acid (-SOH), the Prdx6 forms a heterodimer with thiolate anion (via GSTπ). S-glutathionylation of the heterodimer, followed by its alignment with the catalytic Cys47 of GSTπ, results in the formation of a disulfide-based heterodimer. Reduction of this disulfide bond by GSH causes heterodimer dissociation to active Prdx6 monomer. The active monomer reduces phospholipid hydroperoxide (PLPCOOH) to PLPCOH.

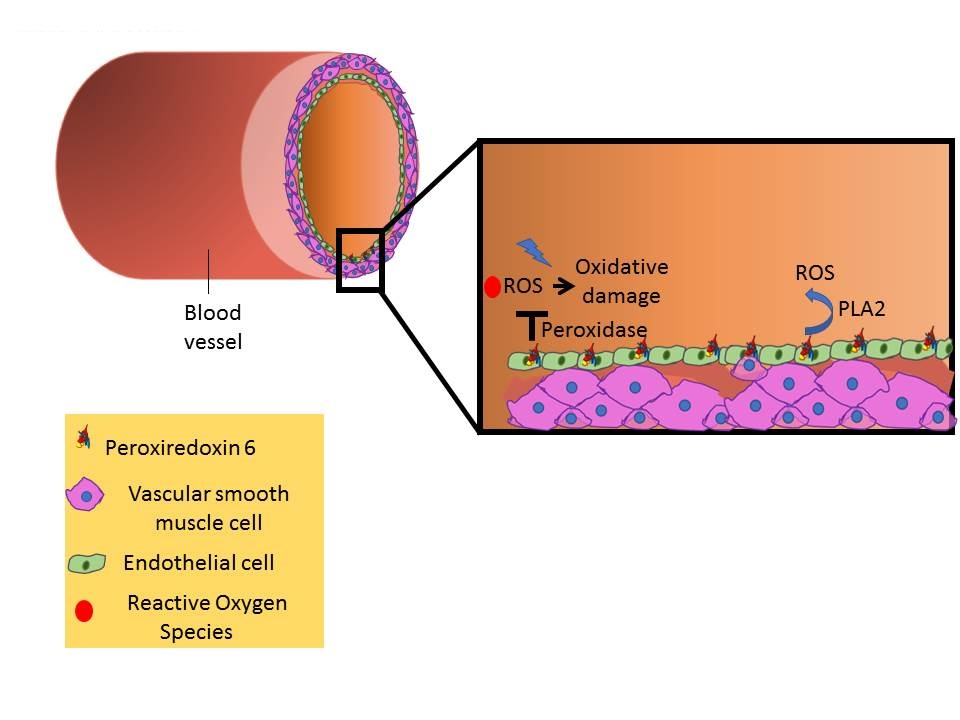
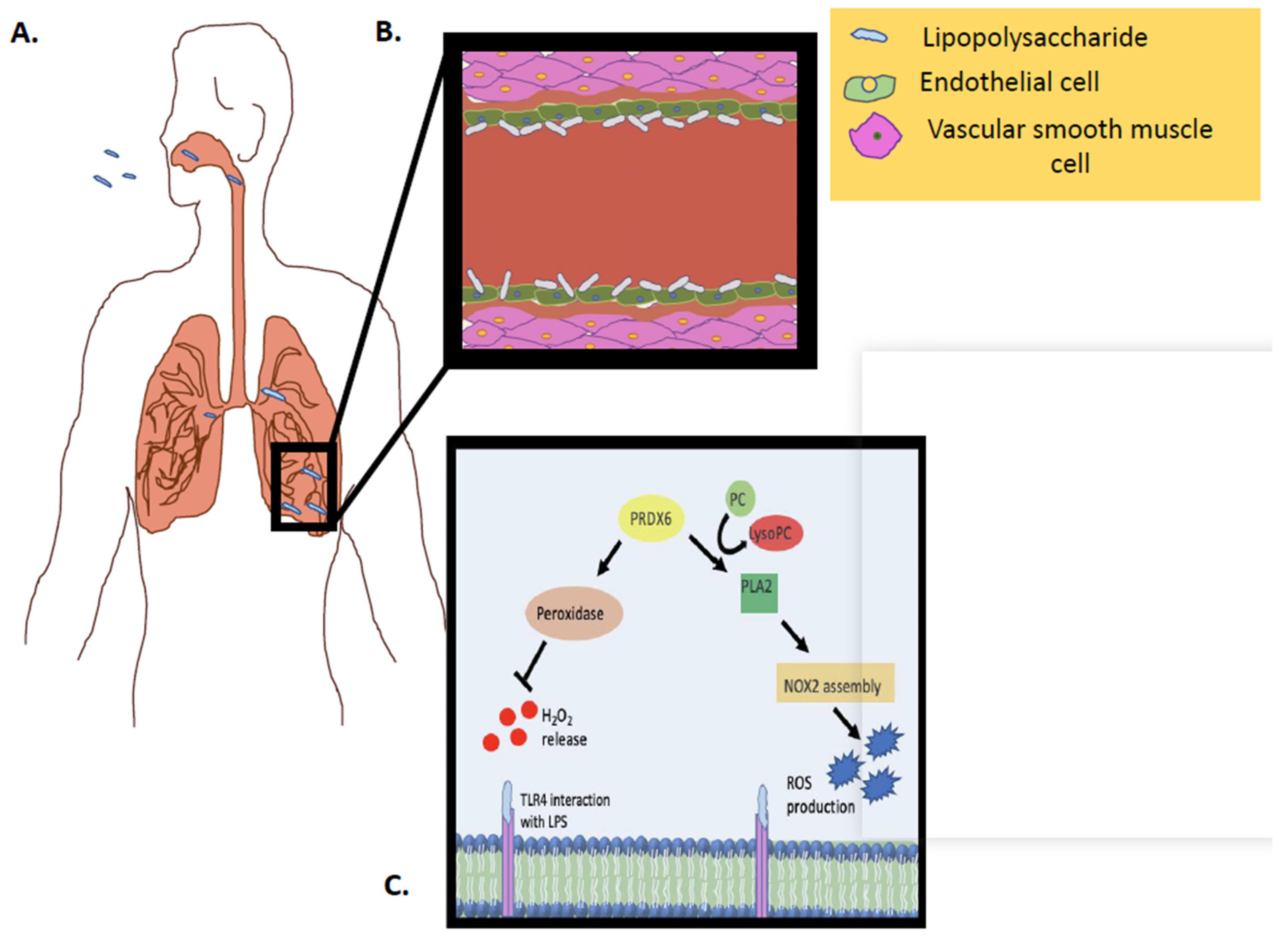
Figure 3.
Schematic representation of Prdx6 activity (PLA2 and peroxidase) in vivo, in response to an inflammatory stimulus. (A) LPS inhalation and interaction with pulmonary capillary. (B) LPS inhalation and interaction with endothelial cells in pulmonary capillary. (C) LPS interaction with the endothelial cell membrane and the Prdx6–PLA2 activity as well as the Prdx6–peroxidase activity.
Figure 3.
Schematic representation of Prdx6 activity (PLA2 and peroxidase) in vivo, in response to an inflammatory stimulus. (A) LPS inhalation and interaction with pulmonary capillary. (B) LPS inhalation and interaction with endothelial cells in pulmonary capillary. (C) LPS interaction with the endothelial cell membrane and the Prdx6–PLA2 activity as well as the Prdx6–peroxidase activity.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Patel, P.; Chatterjee, S. Peroxiredoxin6 in Endothelial Signaling. Antioxidants 2019, 8, 63. https://doi.org/10.3390/antiox8030063
AMA Style
Patel P, Chatterjee S. Peroxiredoxin6 in Endothelial Signaling. Antioxidants. 2019; 8(3):63. https://doi.org/10.3390/antiox8030063
Chicago/Turabian StylePatel, Priyal, and Shampa Chatterjee. 2019. "Peroxiredoxin6 in Endothelial Signaling" Antioxidants 8, no. 3: 63. https://doi.org/10.3390/antiox8030063
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.