The Association of Ascorbic Acid, Deferoxamine and N-Acetylcysteine Improves Cardiac Fibroblast Viability and Cellular Function Associated with Tissue Repair Damaged by Simulated Ischemia/Reperfusion

 , , and
, , and
Abstract
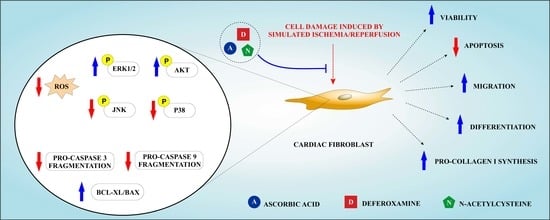
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Material and Methods
2.1. Reagents
2.2. Animals
2.3. Isolation and Culture of Cardiac Fibroblasts
2.4. Isolation and Culture of Cardiomyocytes
2.5. Simulated Ischemia/Reperfusion (sI/R) and Antioxidant Treatment
2.6. Cell Viability with Trypan Blue
2.7. Cell Viability with the Resazurin Reduction Assay
2.8. Determination of Intracellular ROS
2.9. Necrosis Assessment by Flow Cytometer
2.10. Sub-G1 Population Determination by Flow Cytometry
2.11. Western Blot Analysis
2.12. Evaluation of Cell Migration by Wound Healing Assay
2.13. Statistical Analysis
3. Results
3.1. Individual Effects of Ascorbic Acid, Deferoxamine, and N-Acetylcysteine on Viability of Cardiac Fibroblasts after Simulated Ischemia/Reperfusion
3.2. Ascorbic Acid, Deferoxamine, and N-Acetylcysteine Association Increased Cell Viability and Reduced Intracellular ROS Production in Cardiac Fibroblasts Subjected to Simulated Ischemia/Reperfusion
3.3. Association of Ascorbic Acid, Deferoxamine, and N-Acetylcysteine Reduced Apoptosis of CF Exposed to Simulated Ischemia/Reperfusion
3.4. Association of Ascorbic Acid, Deferoxamine and N-Acetylcysteine Activated the Pro-Survival Kinases ERK1/2 and Akt and Reduced the Phosphorylation of the Pro-Apoptotic Proteins p38.MAPK and JNK Induced by Simulated Ischemia/Reperfusion in CFs
3.5. Association of Ascorbic Acid, Deferoxamine, and N-Acetylcysteine Prevented the Loss of Function Associated with Tissue Repair Induced by Simulated Ischemia/Reperfusion in CF
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Chemical compounds studied in this article
Abbreviations
References
- GBD 2015 Mortality and Causes of Death Collaborators. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1459–1544. [Google Scholar] [CrossRef] [Green Version]
- Thygesen, K.; Alpert, J.S.; White, H.D.; Jaffe, A.S.; Katus, H.A.; Apple, F.S.; Lindahl, B.; Morrow, D.A.; Chaitman, B.R.; Clemmensen, P.M.; et al. Third universal definition of myocardial infarction. J. Am. Coll. Cardiol. 2012, 60, 1581–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, H.D.; Chew, D.P. Acute myocardial infarction. Lancet 2008, 372, 570–584. [Google Scholar] [CrossRef] [Green Version]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef]
- Vilahur, G.; Juan-Babot, O.; Peña, E.; Oñate, B.; Casaní, L.; Badimon, L. Molecular and cellular mechanisms involved in cardiac remodeling after acute myocardial infarction. J. Mol. Cell. Cardiol. 2011, 50, 522–533. [Google Scholar] [CrossRef]
- Braunersreuther, V.; Jaquet, V. Reactive oxygen species in myocardial reperfusion injury: From physiopathology to therapeutic approaches. Curr. Pharm. Biotechnol. 2012, 13, 97–114. [Google Scholar] [CrossRef]
- Guo, W.; Liu, X.; Li, J.; Shen, Y.; Zhou, Z.; Wang, M.; Xie, Y.; Feng, X.; Wang, L.; Wu, X. Prdx1 alleviates cardiomyocyte apoptosis through ROS-activated MAPK pathway during myocardial ischemia/reperfusion injury. Int. J. Biol. Macromol. 2018, 112, 608–615. [Google Scholar] [CrossRef]
- González-Montero, J.; Brito, R.; Gajardo, A.I.; Rodrigo, R. Myocardial reperfusion injury and oxidative stress: Therapeutic opportunities. World J. Cardiol. 2018, 10, 74–86. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. The role of cardiac fibroblasts in post-myocardial heart tissue repair. Exp. Mol. Pathol. 2016, 101, 231–240. [Google Scholar] [CrossRef]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting cardiac cellular composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef] [Green Version]
- Talman, V.; Ruskoaho, H. Cardiac fibrosis in myocardial infarction—From repair and remodeling to regeneration. Cell Tissue Res. 2016, 365, 563–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivar, R.; Humeres, C.; Varela, M.; Ayala, P.; Guzmán, N.; Olmedo, I.; Catalán, M.; Boza, P.; Muñoz, C.; Araya, G.D. Cardiac fibroblast death by ischemia/reperfusion is partially inhibited by IGF-1 through both PI3K/Akt and MEK–ERK pathways. Exp. Mol. Pathol. 2012, 93, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Vivar, R.; Humeres, C.; Ayala, P.; Olmedo, I.; Catalán, M.; García, L.; Lavandero, S.; Díaz-Araya, G. TGF-β1 prevents simulated ischemia/reperfusion-induced cardiac fibroblast apoptosis by activation of both canonical and non-canonical signaling pathways. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 754–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Richards, A.M.; Wang, P. Characterization and standardization of cultured cardiac fibroblasts for ex vivo models of heart fibrosis and heart ischemia. Tissue Eng. Part C Methods 2017, 23, 422–433. [Google Scholar] [CrossRef]
- Lefort, C.; Benoist, L.; Chadet, S.; Piollet, M.; Heraud, A.; Babuty, D.; Baron, C.; Ivanes, F.; Angoulvant, D. Stimulation of P2Y11 receptor modulates cardiac fibroblasts secretome toward immunomodulatory and protective roles after Hypoxia/Reoxygenation injury. J. Mol. Cell. Cardiol. 2018, 121, 212–222. [Google Scholar] [CrossRef]
- Ekelof, S.; Jensen, S.E.; Rosenberg, J.; Gogenur, I. Reduced oxidative stress in STEMI patients treated by primary percutaneous coronary intervention and with antioxidant therapy: A systematic review. Cardiovasc. Drugs Ther. 2014, 28, 173–181. [Google Scholar] [CrossRef]
- Williams, R.E.; Zweier, J.L.; Flaherty, J.T. Treatment with deferoxamine during ischemia improves functional and metabolic recovery and reduces reperfusion-induced oxygen radical generation in rabbit hearts. Circulation 1991, 83, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Hao, J.; Li, W.W.; Du, H.; Zhao, Z.F.; Liu, F.; Lu, J.C.; Yang, X.C.; Cui, W. Role of vitamin C in cardioprotection of ischemia/reperfusion injury by activation of mitochondrial KATP channel. Chem. Pharm. Bull. 2016, 64, 548–557. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.W.; Buller, C.L.; Charpie, J.R. Impact of N-acetylcysteine on neonatal cardiomyocyte ischemia-reperfusion injury. Pediatr. Res. 2011, 70, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Abe, M.; Takiguchi, Y.; Ichimaru, S.; Tsuchiya, K.; Wada, K. Comparison of the protective effect of N-acetylcysteine by different treatments on rat myocardial ischemia-reperfusion injury. J. Pharmacol. Sci. 2008, 106, 571–577. [Google Scholar] [CrossRef] [Green Version]
- Nishinaka, Y.; Sugiyama, S.; Yokota, M.; Saito, H.; Ozawa, T. The effects of a high dose of ascorbate on ischemia-reperfusion-induced mitochondrial dysfunction in canine hearts. Heart Vessel. 1992, 7, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.R.; Kloner, R.A.; Przyklenk, K. Early treatment with deferoxamine limits myocardial ischemic/reperfusion injury. Free Radic. Biol. Med. 1989, 7, 45–52. [Google Scholar] [CrossRef]
- National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academies Press: Washington, DC, USA, 2011. Available online: https://www.ncbi.nlm.nih.gov/books/NBK54050/ (accessed on 01 March 2016). [CrossRef]
- Bankowski, Z.; Howard-Jones, N. (Eds.) International Guiding Principles for Biomedical Research Involving Animals; CIOMS: Geneva, Switzerland, 1985. [Google Scholar]
- Mendoza-Torres, E.; Riquelme, J.A.; Vielma, A.; Sagredo, A.R.; Gabrielli, L.; Bravo-Sagua, R.; Jalil, J.E.; Rothermel, B.A.; Sanchez, G.; Ocaranza, M.P.; et al. Protection of the myocardium against ischemia/reperfusion injury by angiotensin-(1–9) through an AT2R and Akt-dependent mechanism. Pharmacol. Res. 2018, 135, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Asp. Med. 2019, 65, 70–99. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Ferdinandy, P.; Andreadou, I.; Bøtker, H.E.; Heusch, G.; Ibáñez, B.; Ovize, M.; Schulz, R.; Yellon, D.M.; Hausenloy, D.J.; et al. Multitarget strategies to reduce myocardial ischemia/reperfusion injury: JACC review topic of the week. J. Am. Coll. Cardiol. 2019, 73, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, M.; Zhang, X.; Li, J.; Wang, Y.; Fan, Y.; Shi, R. Limb ischemic preconditioning protects endothelium from oxidative stress by enhancing nrf2 translocation and upregulating expression of antioxidases. PLoS ONE 2015, 10, e0128455. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Yin, T.; Wang, Y. In vitro and in vivo assessment of high-dose vitamin C against murine tumors. Exp. Ther. Med. 2016, 12, 3058–3062. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Yao, C.L.; Gao, E.; Mo, Q.Z.; Yan, W.L.; McLaughlin, R.; Lopez, B.L.; Christopher, T.A.; Ma, X.L. Enhancement of glutathione cardioprotection by ascorbic acid in myocardial reperfusion injury. J. Pharmacol. Exp. Ther. 2002, 301, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Guaiquil, V.H.; Golde, D.W.; Beckles, D.L.; Mascareno, E.J.; Siddiqui, M. Vitamin C inhibits hypoxia-induced damage and apoptotic signaling pathways in cardiomyocytes and ischemic hearts. Free Radic. Biol. Med. 2004, 37, 1419–1429. [Google Scholar] [CrossRef]
- Karahaliou, A.; Katsouras, C.; Koulouras, V.; Nikas, D.; Niokou, D.; Papadopoulos, G.; Nakos, G.; Sideris, D.; Michalis, L. Ventricular arrhythmias and antioxidative medication: Experimental study. Hell. J. Cardiol. 2008, 49, 320–328. [Google Scholar]
- Phaelante, A.; Rohde, L.E.; Lopes, A.; Olsen, V.; Tobar, S.A.L.; Cohen, C.; Martinelli, N.; Biolo, A.; Dal-Pizzol, F.; Clausell, N.; et al. N-acetylcysteine plus deferoxamine improves cardiac function in wistar rats after non-reperfused acute myocardial infarction. J. Cardiovasc. Transl. Res. 2015, 8, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Nikas, D.N.; Chatziathanasiou, G.; Kotsia, A.; Papamichael, N.; Thomas, C.; Papafaklis, M.; Naka, K.K.; Kazakos, N.; Milionis, H.J.; Vakalis, K.; et al. Effect of intravenous administration of antioxidants alone and in combination on myocardial reperfusion injury in an experimental pig model. Curr. Ther. Res. Clin. Exp. 2008, 69, 423–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, J.M.; Qu, Z.C.; Neel, D.R.; Li, X. Recycling of vitamin C from its oxidized forms by human endothelial cells. Biochim. Biophys. Acta 2003, 1640, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Levine, M.; Padayatty, S.J.; Espey, M.G. Vitamin C: A concentration-function approach yields pharmacology and therapeutic discoveries. Adv. Nutr. 2011, 2, 78–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossello, X.; Yellon, D.M. The RISK pathway and beyond. Basic Res. Cardiol. 2018, 113, 2. [Google Scholar] [CrossRef] [Green Version]
- Rossello, X.; Riquelme, J.A.; Davidson, S.M.; Yellon, D.M. Role of PI3K in myocardial ischaemic preconditioning: Mapping pro-survival cascades at the trigger phase and at reperfusion. J. Cell. Mol. Med. 2018, 22, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Mao, X.; Li, H.; Qiao, S.; Xu, A.; Wang, J.; Lei, S.; Liu, Z.; Ng, K.F.; Wong, G.T.; et al. N-Acetylcysteine and allopurinol up-regulated the Jak/STAT3 and PI3K/Akt pathways via adiponectin and attenuated myocardial postischemic injury in diabetes. Free Radic. Biol. Med. 2013, 63, 291–303. [Google Scholar] [CrossRef]
- Wang, D.; Chen, T.; Liu, F. Betulinic acid alleviates myocardial hypoxia/reoxygenation injury via inducing Nrf2/HO-1 and inhibiting p38 and JNK pathways. Eur. J. Pharmacol. 2018, 838, 53–59. [Google Scholar] [CrossRef]
- Tobiume, K.; Matsuzawa, A.; Takahashi, T.; Nishitoh, H.; Morita, K.I.; Takeda, K.; Minowa, O.; Miyazono, K.; Noda, T.; Ichijo, H. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001, 2, 222–228. [Google Scholar] [CrossRef]
- Dobšák, P.; Siegelova, J.; Wolf, J.; Rochette, L.; Eicher, J.; Vasku, J.; Kuchtickova, S.; Horky, M. Prevention of apoptosis by deferoxamine during 4 h of cold cardioplegia and reperfusion: In vitro study of isolated working rat heart model. Pathophysiology 2002, 9, 27–32. [Google Scholar] [CrossRef]
- Humeres, C.; Vivar, R.; Boza, P.; Muñoz, C.; Bolivar, S.; Anfossi, R.; Osorio, J.M.; Olivares-Silva, F.; García, L.; Díaz-Araya, G. Cardiac fibroblast cytokine profiles induced by proinflammatory or profibrotic stimuli promote monocyte recruitment and modulate macrophage M1/M2 balance in vitro. J. Mol. Cell. Cardiol. 2016, 101, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.F.; Stachon, T.; Seitz, B.; Langenbucher, A.; Szentmáry, N. Effect of human autologous serum and fetal bovine serum on human corneal epithelial cell viability, migration and proliferation in vitro. Int. J. Ophthalmol. 2017, 10, 908–913. [Google Scholar] [CrossRef] [PubMed]
- Olmedo, I.; Muñoz, C.; Guzmán, N.; Catalán, M.; Vivar, R.; Ayala, P.; Humeres, C.; Aránguiz, P.; García, L.; Velarde, V.; et al. EPAC expression and function in cardiac fibroblasts and myofibroblasts. Toxicol. Appl. Pharmacol. 2013, 272, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Colston, J.T.; Sam, D.; Freeman, G.L. Impact of brief oxidant stress on primary adult cardiac fibroblasts. Biochem. Biophys. Res. Commun. 2004, 316, 256–262. [Google Scholar] [CrossRef]
- Shinde, A.V.; Humeres, C.; Frangogiannis, N.G. The role of α-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 298–309. [Google Scholar] [CrossRef]
- Driesen, R.B.; Nagaraju, C.K.; Abi-Char, J.; Coenen, T.; Lijnen, P.J.; Fagard, R.H.; Sipido, K.R.; Petrov, V.V. Reversible and irreversible differentiation of cardiac fibroblasts. Cardiovasc. Res. 2013, 101, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Xing, D.; Bonanno, J.A. Hypoxia reduces TGFβ1-induced corneal keratocyte myofibroblast transformation. Mol. Vis. 2009, 15, 1827–1834. [Google Scholar]
- Yan, Z.; Shen, D.; Liao, J.; Zhang, Y.; Chen, Y.; Shi, G.; Gao, F. Hypoxia suppresses TGF-B1-induced cardiac myocyte myofibroblast transformation by inhibiting Smad2/3 and rhoa signaling pathways. Cell. Physiol. Biochem. 2018, 45, 250–257. [Google Scholar] [CrossRef] [Green Version]
- Shu, Q.; Lai, S.; Wang, X.M.; Zhang, Y.L.; Yang, X.L.; Bi, H.L.; Li, H.H. Administration of ubiquitin-activating enzyme UBA1 inhibitor PYR-41 attenuates angiotensin II-induced cardiac remodeling in mice. Biochem. Biophys. Res. Commun. 2018, 505, 317–324. [Google Scholar] [CrossRef]
- Wang, H.X.; Li, W.J.; Hou, C.L.; Lai, S.; Zhang, Y.L.; Tian, C.; Yang, H.; Du, J.; Li, H.H. CD1d-dependent natural killer T cells attenuate angiotensin II-induced cardiac remodelling via IL-10 signalling in mice. Cardiovasc. Res. 2018, 115, 83–93. [Google Scholar] [CrossRef]
- Tian, H.P.; Sun, Y.H.; He, L.; Yi, Y.F.; Gao, X.; Xu, D.L. Single-stranded DNA-binding protein 1 abrogates cardiac fibroblast proliferation and collagen expression induced by angiotensin II. Int. Heart J. 2018, 59, 1398–1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.; Wang, W.; Jin, X. Hirudin protects Ang II-induced myocardial fibroblasts fibrosis by inhibiting the extracellular signal-regulated Kinase1/2 (ERK1/2) pathway. Med. Sci. Monit. 2018, 24, 6264–6272. [Google Scholar] [CrossRef] [PubMed]
- Siwik, D.A.; Pagano, P.J.; Colucci, W.S. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am. J. Physiol. Cell Physiol. 2001, 280, C53–C60. [Google Scholar] [CrossRef] [PubMed]
- Agocha, A.; Lee, H.W.; Eghbali-Webb, M. Hypoxia regulates basal and induced DNA synthesis and collagen type I production in human cardiac fibroblasts: Effects of transforming growth factor-β1, thyroid hormone, angiotensin II and basic fibroblast growth factor. J. Mol. Cell. Cardiol. 1997, 29, 2233–2244. [Google Scholar] [CrossRef] [PubMed]
- Shyu, K.G.; Wang, B.W.; Chen, W.J.; Kuan, P.; Lin, C.M. Angiotensin II mediates urotensin II expression by hypoxia in cultured cardiac fibroblast. Eur. J. Clin. Investig. 2012, 42, 17–26. [Google Scholar] [CrossRef]
- Grobe, J.L.; der Sarkissian, S.; Stewart, J.M.; Meszaros, J.G.; Raizada, M.K.; Katovich, M.J. ACE2 overexpression inhibits hypoxia-induced collagen production by cardiac fibroblasts. Clin. Sci. 2007, 113, 357–364. [Google Scholar] [CrossRef] [Green Version]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parra-Flores, P.; Riquelme, J.A.; Valenzuela-Bustamante, P.; Leiva-Navarrete, S.; Vivar, R.; Cayupi-Vivanco, J.; Castro, E.; Espinoza-Pérez, C.; Ruz-Cortés, F.; Pedrozo, Z.; et al. The Association of Ascorbic Acid, Deferoxamine and N-Acetylcysteine Improves Cardiac Fibroblast Viability and Cellular Function Associated with Tissue Repair Damaged by Simulated Ischemia/Reperfusion. Antioxidants 2019, 8, 614. https://doi.org/10.3390/antiox8120614
Parra-Flores P, Riquelme JA, Valenzuela-Bustamante P, Leiva-Navarrete S, Vivar R, Cayupi-Vivanco J, Castro E, Espinoza-Pérez C, Ruz-Cortés F, Pedrozo Z, et al. The Association of Ascorbic Acid, Deferoxamine and N-Acetylcysteine Improves Cardiac Fibroblast Viability and Cellular Function Associated with Tissue Repair Damaged by Simulated Ischemia/Reperfusion. Antioxidants. 2019; 8(12):614. https://doi.org/10.3390/antiox8120614
Chicago/Turabian StyleParra-Flores, Pablo, Jaime A Riquelme, Paula Valenzuela-Bustamante, Sebastian Leiva-Navarrete, Raúl Vivar, Jossete Cayupi-Vivanco, Esteban Castro, Claudio Espinoza-Pérez, Felipe Ruz-Cortés, Zully Pedrozo, and et al. 2019. "The Association of Ascorbic Acid, Deferoxamine and N-Acetylcysteine Improves Cardiac Fibroblast Viability and Cellular Function Associated with Tissue Repair Damaged by Simulated Ischemia/Reperfusion" Antioxidants 8, no. 12: 614. https://doi.org/10.3390/antiox8120614