Stereoselectivity of Aldose Reductase in the Reduction of Glutathionyl-Hydroxynonanal Adduct

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Determination of AKR1B1 Activity
2.3. Expression and Purification of AKR1B1
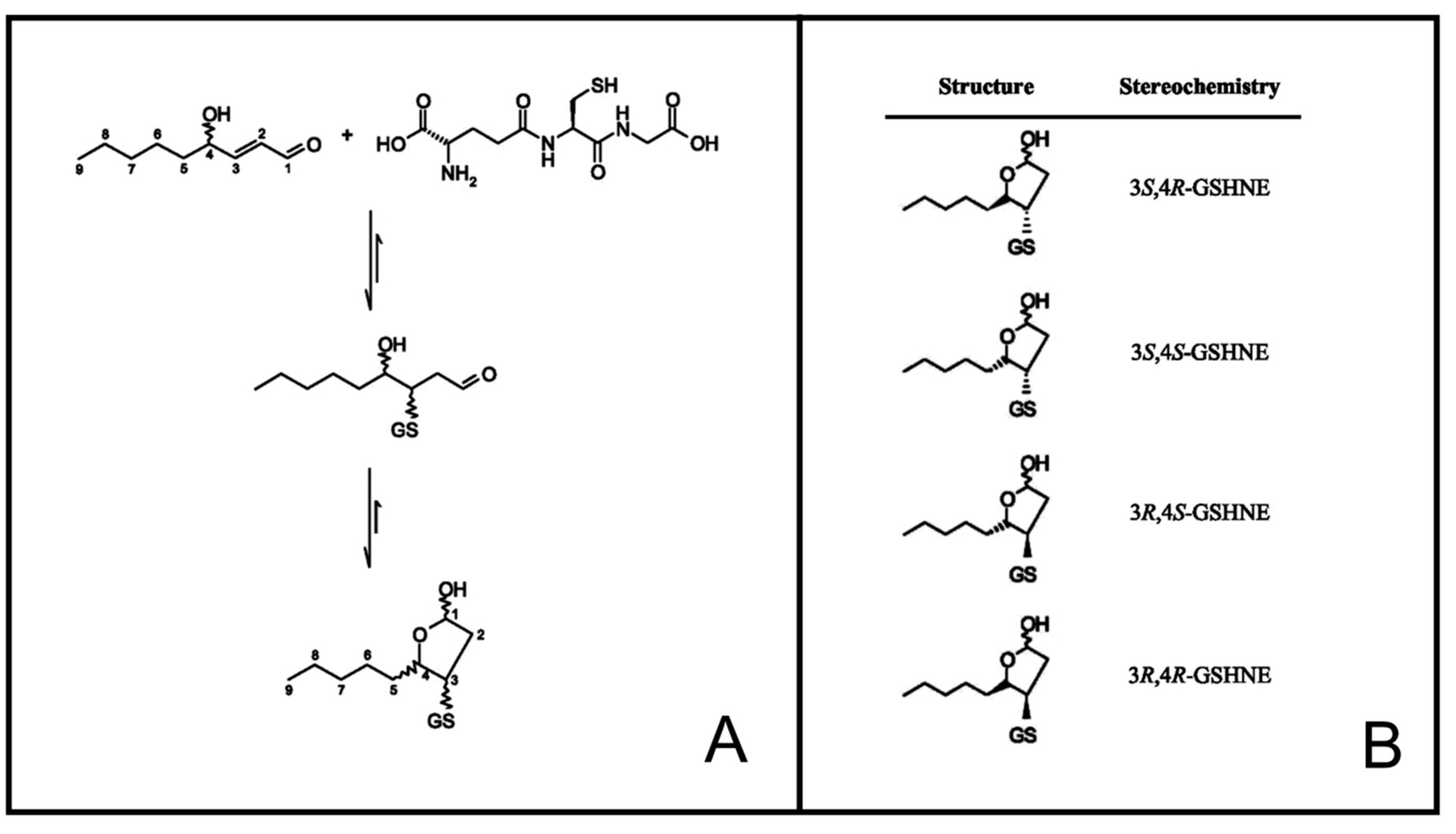
2.4. Synthesis of HNE Stereoisomers
2.4.1. Analytical Methods
2.4.2. Enzymatic Resolution of R/S-oct-1-en-3-ol into R-oct-1-en-3-ol- and S-oct-1-en-3-yl-Propionate
2.4.3. Preparation of S-oct-1-en-3-ol
2.4.4. Synthesis of (E)-1,1-Diethoxynon-2-en-4-ol
2.5. Preparation of GSHNE Adducts
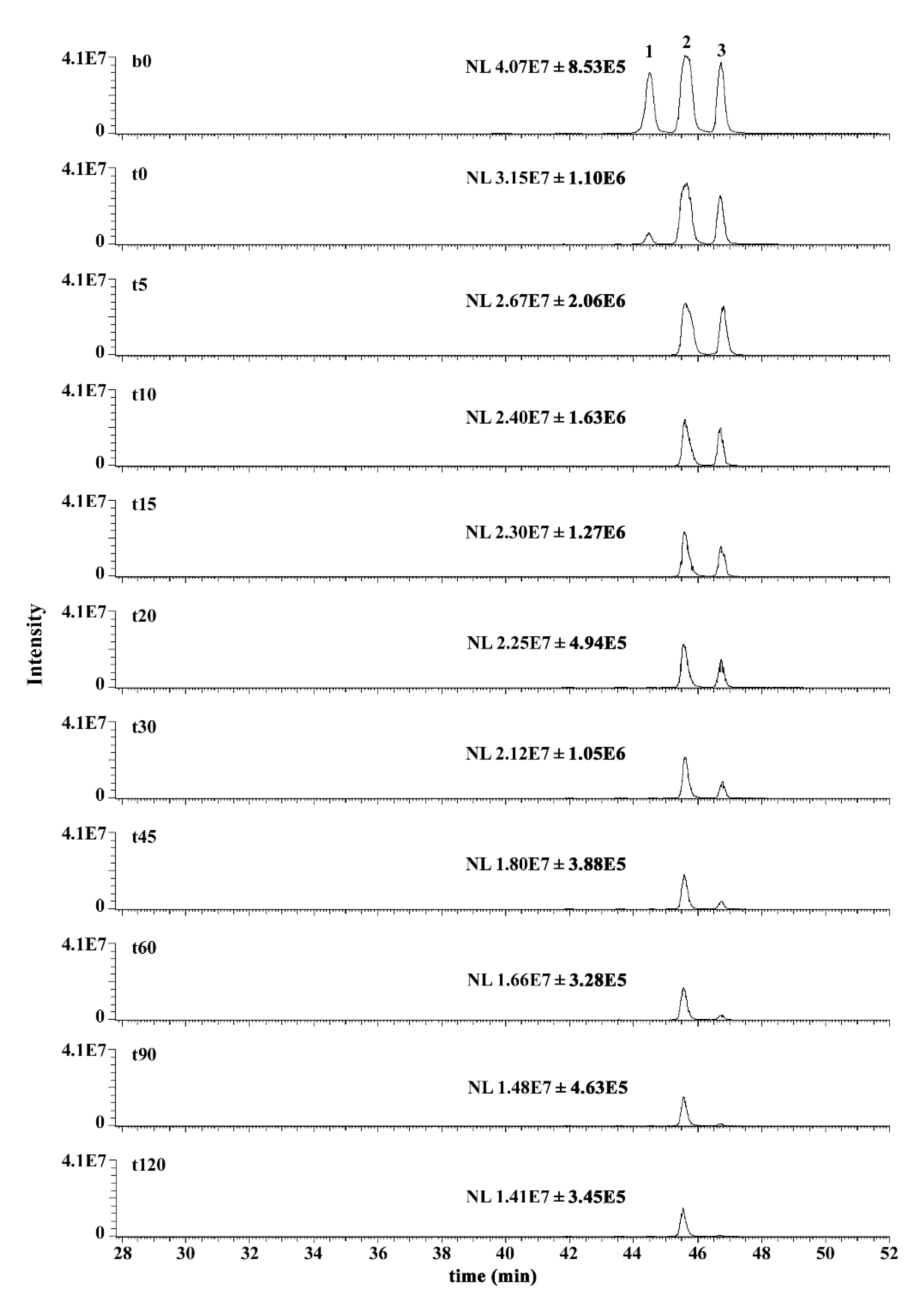
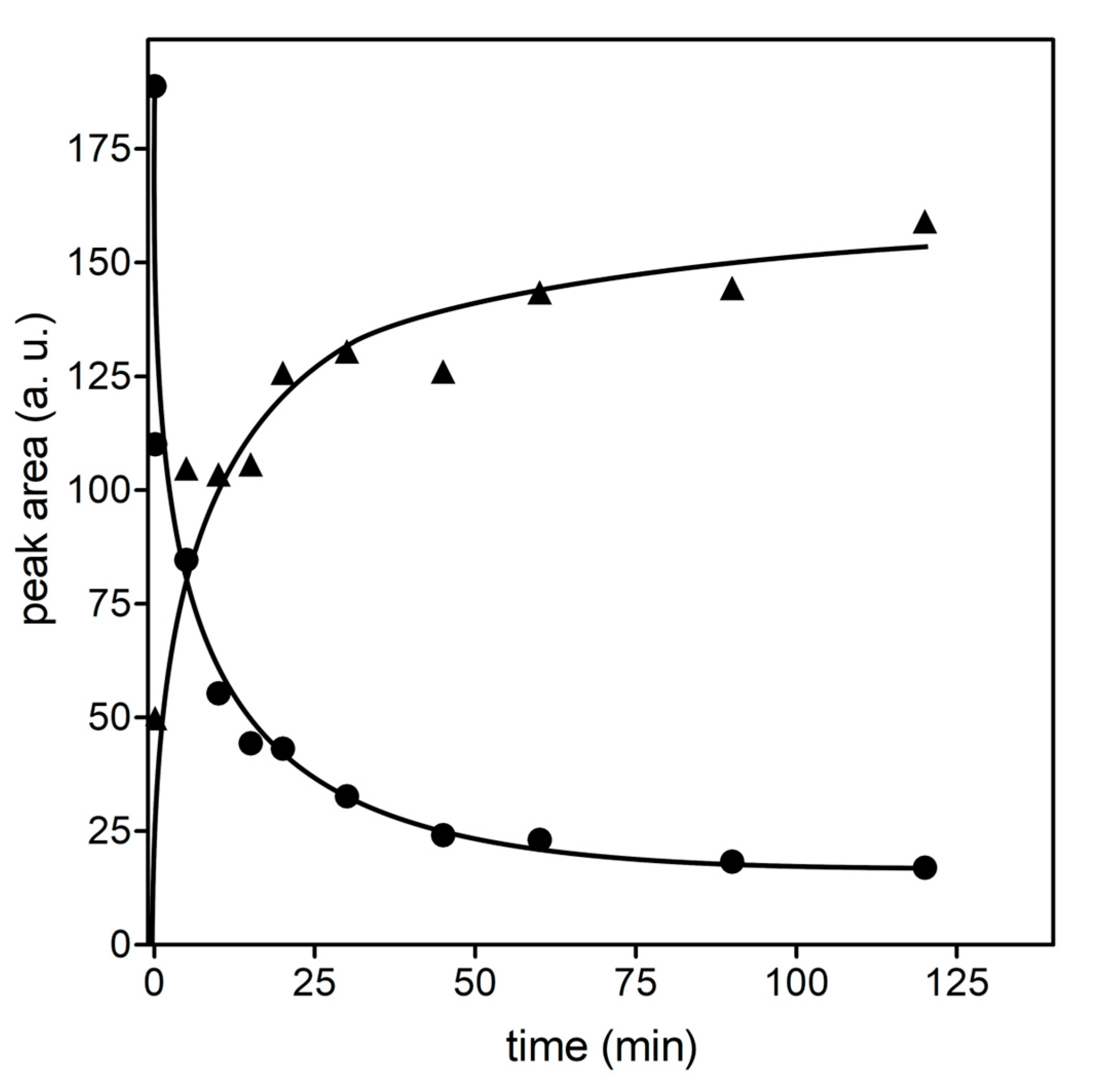
2.6. Mass Spectrometry Analysis of GSHNE Reaction Products Obtained after hAKR1B1 Catalysis
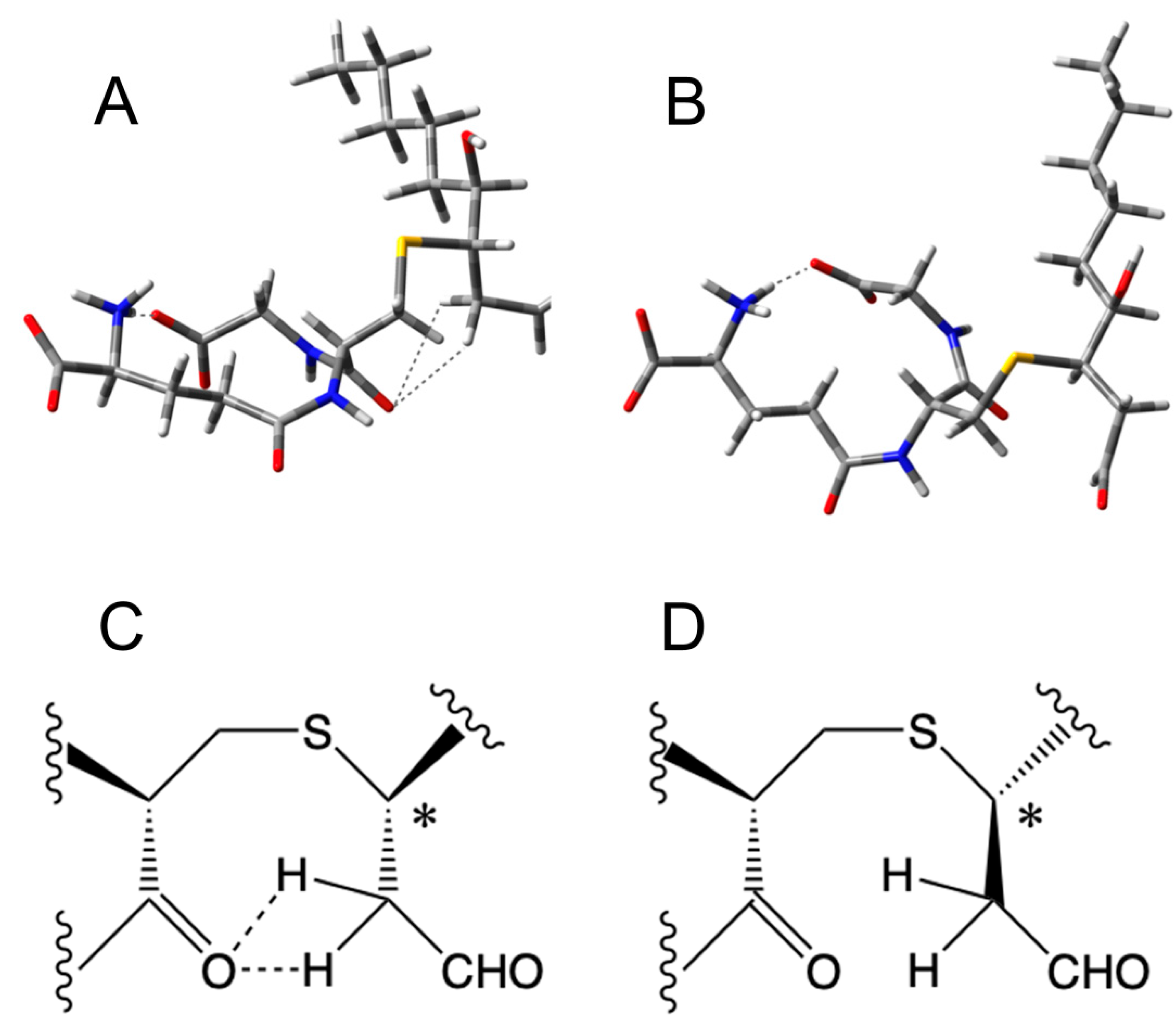
2.7. Molecular Modeling
2.8. Density Functional Theory (DFT) Calculations
2.9. Other Methods
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Niki, E.; Yoshida, Y.; Saito, Y.; Noguchi, N. Lipid peroxidation: Mechanisms, inhibition, and biological effects. Biochem. Biophys. Res. Commun. 2005, 338, 668–676. [Google Scholar] [CrossRef]
- Catala, A. Lipid peroxidation of membrane phospholipids generates hydroxy-alkenals and oxidized phospholipids active in physiological and/or pathological conditions. Chem. Phys. Lipids 2009, 157, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Schaur, R.J.; Siems, W.G.; Leonarduzzi, G. 4-Hydroxynonenal: A membrane lipid oxidation product of medicinal interest. Med. Res. Rev. 2008, 28, 569–631. [Google Scholar] [CrossRef] [PubMed]
- Zarkovic, K. 4-Hydroxynonenal and neurodegenerative diseases. Mol. Asp. Med. 2003, 24, 293–303. [Google Scholar] [CrossRef]
- Nègre-Salvayre, A.; Garoby-Salom, S.; Swiader, A.; Rouahi, M.; Pucelle, M.; Salvayre, R. Proatherogenic effects of 4-hydroxynonenal. Free Radic. Biol. Med. 2017, 111, 127–139. [Google Scholar] [CrossRef]
- Castro, J.P.; Jung, T.; Grune, T.; Siems, W. 4-Hydroxynonenal (HNE) modified proteins in metabolic diseases. Free Radic. Biol. Med. 2017, 111, 309–315. [Google Scholar] [CrossRef]
- Petersen, D.R.; Doorn, J.A. Reactions of 4-hydroxynonenal with proteins and cellular targets. Free Radic. Biol. Med. 2004, 37, 937–945. [Google Scholar] [CrossRef]
- Zarkovic, K.; Jakovcevic, A.; Zarkovic, N. Contribution of the HNE-immunohistochemistry to modern pathological concepts of major human diseases. Free Radic. Biol. Med. 2017, 111, 110–126. [Google Scholar] [CrossRef]
- Cesar, V.; Jozić, I.; Begović, L.; Vuković, T.; Mlinarić, S.; Lepeduš, H.; Borović, Š.S.; Žarković, N. Cell-type-specific modulation of hydrogen peroxide cytotoxicity and 4-hydroxynonenal binding to human cellular proteins in vitro by antioxidant Aloe vera extract. Antioxidants 2018, 7, 125. [Google Scholar] [CrossRef]
- Cherkas, A.; Zarkovic, K.; Cipak Gasparovic, A.; Jaganjac, M.; Milkovic, L.; Abrahamovych, O.; Yatskevych, O.; Waeg, G.; Yelisyeyeva, O.; Zarkovic, N. Amaranth oil reduces accumulation of 4-hydroxynonenal-histidine adducts in gastric mucosa and improves heart rate variability in duodenal peptic ulcer patients undergoing Helicobacter pylori eradication. Free Radic. Res. 2018, 52, 135–149. [Google Scholar] [CrossRef]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malondialdehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Pappa, A.; Estey, T.; Manzer, R.; Brown, D.; Vasiliou, V. Human aldehyde dehydrogenase 3A1 (ALDH3A1): Biochemical characterization and immunohistochemical localization in the cornea. Biochem. J. 2003, 376, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.C.; Amarnath, V.; Gibson, K.M.; Picklo, M.J. Oxidation of 4-hydroxy-2-nonenal by succinic semialdehyde dehydrogenase (ALDH5A). J. Neurochem. 2003, 86, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Kotraiah, V. Modulation of aldehyde dehydrogenase activity affects (±)-4-hydroxy-2E-nonenal (HNE) toxicity and HNE-protein adducts levels in PC12 cells. J. Mol. Neurosci. 2012, 47, 595–603. [Google Scholar] [CrossRef] [PubMed]
- VanderJagt, D.L.; Kolb, N.S.; VanderJagt, T.J.; Chino, J.; Martinez, F.J.; Hunsaker, L.A.; Royer, R.E. Substrate specificity of human aldose reductase: Identification of 4-hydroxynonenal as an endogenous substrate. Biochim. Biophys. Acta 1995, 1249, 117–126. [Google Scholar] [CrossRef]
- Srivastava, S.; Chandra, A.; Bhatnagar, A.; Srivastava, S.K.; Ansari, N.H. Lipid peroxidation product, 4-hydroxynonenal and its conjugate with GSH are excellent substrates of bovine lens aldose reductase. Biochem. Biophys. Res. Commun. 1995, 217, 741–746. [Google Scholar] [CrossRef]
- Shen, Y.; Zhong, L.; Johnson, S.; Cao, D. Human aldo-keto reductases 1B1 and 1B10: A comparative study on their enzyme activity toward electrophilic carbonyl compounds. Chem. Biol. Interact. 2011, 191, 192–198. [Google Scholar] [CrossRef]
- Hubatsch, I.; Ridderstrom, M.; Mannervik, B. Human glutathione transferase A4-4: An alpha class enzyme with high catalytic efficiency in the conjugation of 4-hydroxynonenal and other genotoxic products of lipid peroxidation. Biochem. J. 1998, 330, 175–179. [Google Scholar] [CrossRef]
- Hou, L.; Honaker, M.T.; Shireman, L.M.; Balogh, L.M.; Roberts, A.G.; Ng, K.C.; Nath, A.; Atkins, W.M. Functional promiscuity correlates with conformational heterogeneity in A-class glutathione S-transferases. J. Biol. Chem. 2007, 282, 23264–23274. [Google Scholar] [CrossRef]
- Ramana, K.V.; Bhatnagar, A.; Srivastava, S.; Yadav, U.C.; Awasthi, S.; Yogesh, C.; Awasthi, Y.C.; Srivastava, S.K. Mitogenic responses of vascular smooth muscle cells to lipid peroxidation-derived aldehyde 4-hydroxy-trans-2-nonenal (HNE): Role of aldose reductase-catalyzed reduction of the HNE-glutathione conjugates in regulating cell growth. J. Biol. Chem. 2006, 281, 17652–17660. [Google Scholar] [CrossRef]
- Frohnert, B.I.; Long, E.K.; Hahn, W.S.; Bernlohr, D.A. Glutathionylated lipid aldehydes are products of adipocyte oxidative stress and activators of macrophage inflammation. Diabetes 2014, 63, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.C.; Petrash, J.M. Aldo-keto reductases: Multifunctional proteins as therapeutic targets in diabetes and inflammatory disease. Adv. Exp. Med. Biol. 2018, 1032, 173–202. [Google Scholar] [PubMed]
- Moschini, R.; Peroni, E.; Rotondo, R.; Renzone, G.; Melck, D.; Cappiello, M.; Srebot, M.; Napolitano, E.; Motta, A.; Scaloni, A.; et al. NADP+-dependent dehydrogenase activity of carbonyl reductase on glutathionyl-hydroxynonanal as a new pathway for hydroxy nonenal detoxification. Free Radic. Biol. Med. 2015, 83, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Rotondo, R.; Moschini, R.; Renzone, G.; Tuccinardi, T.; Balestri, F.; Cappiello, M.; Scaloni, A.; Mura, U.; Del-Corso, A. Human carbonyl reductase 1 as efficient catalyst for the reduction of glutathionylated aldehydes derived from lipid peroxidation. Free Radic. Biol. Med. 2016, 99, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Ruef, J.; Liu, S.Q.; Bode, C.; Tocchi, M.; Srivastava, S.; Runge, M.S.; Bhatnagar, A. Involvement of aldose reductase in vascular smooth muscle cell growth and lesion formation after arterial injury. Arter. Thromb. Vasc. Biol. 2000, 20, 1745–1752. [Google Scholar] [CrossRef]
- Srivastava, S.; Conklin, D.J.; Liu, S.Q.; Prakash, N.; Boor, P.J.; Srivastava, S.K.; Bhatnagar, A. Identification of biochemical pathways for the metabolism of oxidized low-density lipoprotein derived aldehyde-4-hydroxy trans-2-nonenal in vascular smooth muscle cells. Atherosclerosis 2001, 158, 339–350. [Google Scholar] [CrossRef] [Green Version]
- Tammali, R.; Saxena, A.; Srivastava, S.K.; Ramana, K.V. Aldose reductase regulates vascular smooth muscle cell proliferation by modulating G1/S phase transition of cell cycle. Endocrinology 2010, 151, 2140–2150. [Google Scholar] [CrossRef]
- Li, M.; Qian, M.; Kyler, K.; Xu, J. Endothelial-vascular smooth muscle cells interactions in atherosclerosis. Front. Cardiovasc. Med. 2018, 5, 151. [Google Scholar] [CrossRef]
- Andrés, V. Control of vascular cell proliferation and migration by cyclin-dependent kinase signalling: New perspectives and therapeutic potential. Cardiovasc. Res. 2004, 63, 11–21. [Google Scholar] [CrossRef]
- Brichac, J.; Ho, K.K.; Honzatko, A.; Wang, R.; Lu, X.; Weiner, H.; Picklo, M.J. Enantioselective oxidation of trans-4-hydroxy-2-nonenal is aldehyde dehydrogenase isozyme and Mg2+ dependent. Chem. Res. Toxicol. 2007, 20, 887–895. [Google Scholar] [CrossRef]
- Hiratsuka, A.; Hirose, K.; Saito, H.; Watabe, T. 4-Hydroxy-2(E)-nonenal enantiomers: (S)-selective inactivation of glyceraldehyde-3-phosphate dehydrogenase and detoxification by rat glutathione S-transferase A4-4. Biochem. J. 2000, 349, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Balogh, L.M.; Roberts, A.G.; Shireman, L.M.; Greene, R.J.; Atkins, W.M. The stereochemical course of 4-hydroxy-2- nonenal metabolism by glutathione S-transferases. J. Biol. Chem. 2008, 283, 16702–16710. [Google Scholar] [CrossRef] [PubMed]
- Ji, B.; Ito, K.; Suzuki, H.; Sugiyama, Y.; Horie, T. Multidrug resistance-associated protein2 (MRP2) plays an important role in the biliary excretion of glutathione conjugates of 4-hydroxynonenal. Free Radic. Biol. Med. 2002, 33, 370–378. [Google Scholar] [CrossRef]
- Balestri, F.; Rotondo, R.; Moschini, R.; Pellegrino, M.; Cappiello, M.; Barracco, V.; Misuri, L.; Sorce, C.; Andreucci, A.; Del Corso, A.; et al. Zolfino landrace (Phaseolus vulgaris L.) from Pratomagno: General and specific features of a functional food. Food Nutr. Res. 2016, 60, 31792. [Google Scholar] [CrossRef] [PubMed]
- Balestri, F.; Cappiello, M.; Moschini, R.; Rotondo, R.; Buggiani, I.; Pelosi, P.; Mura, U.; Del Corso, A. L-Idose: An attractive substrate alternative to d-glucose for measuring aldose reductase activity. Biochem. Biophys. Res. Commun. 2015, 456, 891–895. [Google Scholar] [CrossRef]
- Komisarski, M.; Kaczmarska, Z.; Kuśmierek, J.T. Practical highly enantioselective synthesis of (R)- and (S)-(E)-4-hydroxynon-2-enal. Acta Biochim. Polonica 2009, 56, 189–193. [Google Scholar] [CrossRef]
- Badenhop, A.F.; Wilkens, W.F. The formation of 1-octen-3-o1 in soybeans during soaking. J. Oil Chem. Soc. 1969, 46, 179–182. [Google Scholar] [CrossRef]
- Sabitha, G.; Bhikshapathi, M.J.; Yadav, S. Radical cyclization route to the stereoselective synthesis of (+)-trans-cognac lactone and (+)-trans-aerangis lactone. Synth. Comm. 2007, 37, 559–567. [Google Scholar] [CrossRef]
- Icikawa, A.; Ono, H. Preparation of single-enantiomer semiochemicals using 2-methoxy-2-(1-naphthyl) propionic acid and 2-methoxy-2-(9-phenanthryl) propionic acid. Tetrah. Asymm. 2005, 16, 2559–2568. [Google Scholar] [CrossRef]
- Kanbayashi, N.; Onitsuka, K. Ruthenium-catalyzed regio- and enantioselective allylic substitution with water: Direct synthesis of chiral allylic alcohols. Angew. Chem. Int. Ed. 2011, 50, 5197–5199. [Google Scholar] [CrossRef]
- Arora, J.S.; Tomoyuki, O.; Blair, I.A. Synthesis of deuterium-labeled analogs of the lipid hydroperoxide-derived bifunctional electrophile 4-oxo-2(E)-nonenal. J. Label. Compd. Radiopharm. 2011, 54, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Soulere, L.; Queneau, Y.; Doutheau, A. An expeditious synthesis of 4-hydroxy-2(E)-nonenal (4-HNE), its dimethyl acetal and of related compounds. Chem. Phys. Lipids 2007, 150, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Del Corso, A.; Balestri, F.; Di Bugno, E.; Moschini, R.; Cappiello, M.; Sartini, S.; La Motta, C.; Da Settimo, F.; Mura, U. A new approach to control the enigmatic activity of aldose reductase. PLoS ONE 2013, 8, e74076. [Google Scholar] [CrossRef]
- Cappiello, M.; Peroni, E.; Lepore, A.; Moschini, R.; Del-Corso, A.; Balestri, F.; Mura, U. Rapid colorimetric determination of reduced and oxidized glutathione using an end point coupled enzymatic assay. Anal. Bioanal. Chem. 2013, 405, 1779–1785. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; White, M.A.; Ramana, K.V.; Petrash, J.M.; Watowich, S.J.; Bhatnagar, A.; Srivastava, S.K. Structure of a glutathione conjugate bound to the active site of aldose reductase. Proteins 2006, 64, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E.; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. The FF14SB force field. Amber 2014, 14, 29–31. [Google Scholar]
- Jorgensen, W.; Chandrasekhar, J.; Madura, J.; Klein, M. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comp. Chem. 1996, 17, 490–516. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Granchi, C.; Rizzolio, F.; Palazzolo, S.; Carmignani, S.; Macchia, M.; Saccomanni, G.; Manera, C.; Martinelli, A.; Minutolo, F.; Tuccinardi, T. Structural optimization of 4-chlorobenzoylpiperidine derivatives for the development of potent, reversible, and selective monoacylglycerol lipase (MAGL) inhibitors. J. Med. Chem. 2016, 59, 10299–10314. [Google Scholar] [CrossRef] [PubMed]
- Bononi, G.; Granchi, C.; Lapillo, M.; Giannotti, M.; Nieri, D.; Fortunato, S.; Boustani, M.; Caligiuri, I.; Poli, G.; Carlson, K.E.; et al. Discovery of long-chain salicylketoxime derivatives as monoacylglycerol lipase (MAGL) inhibitors. Eur. J. Med. Chem. 2018, 157, 817–836. [Google Scholar] [CrossRef]
- Maier, J.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Tuccinardi, T.; Manetti, F.; Schenone, S.; Martinelli, A.; Botta, M. Construction and validation of a ret tk catalytic domain by homology modeling. J. Chem. Inf. Model. 2007, 47, 644–655. [Google Scholar] [CrossRef] [PubMed]
- Cincinelli, R.; Cassinelli, G.; Dallavalle, S.; Lanzi, C.; Merlini, L.; Botta, M.; Tuccinardi, T.; Martinelli, A.; Penco, S.; Zunino, F. Synthesis, modeling, and RET protein kinase inhibitory activity of 3- and 4-substituted β-carbolin-1-ones. J. Med. Chem. 2008, 51, 7777–7787. [Google Scholar] [CrossRef] [PubMed]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.; Bayly, C. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Ufimtsev, I.S.; Martinez, T.J. Quantum chemistry on graphical processing units. 2. Direct Self-Consistent-Field (SCF) implementation. J. Chem. Theory Comput. 2009, 5, 3138. [Google Scholar] [CrossRef]
- Titov, A.V.; Ufimtsev, I.S.; Luehr, N.; Martinez, T.J. Generating efficient quantum chemistry codes for novel architectures. J. Chem. Theory Comput. 2013, 9, 213–221. [Google Scholar] [CrossRef]
- Kästner, J.; Carr, J.M.; Keal, T.W.; Thiel, W.; Wander, A.; Sherwood, P. DL-FIND: An open-source geometry optimizer for atomistic simulations. J. Phys. Chem. A 2009, 113, 11856–11865. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Inagaki, K.; Miwa, I.; Okuda, J. Affinity purification and glucose specificity of aldose reductase from bovine lens. Arch. Biochem. Biophys. 1982, 216, 337–344. [Google Scholar] [CrossRef]
- Grimshaw, C.E. Direct measurement of the rate of ring opening of D-glucose by enzyme catalyzed reduction. Carbohydr. Res. 1986, 148, 345–358. [Google Scholar] [CrossRef]
- Srivastava, S.K.; Ramana, K.V.; Bhatnagar, A. Role of aldose reductase and oxidative damage in diabetes and the consequent potential for therapeutic options. Endocr. Rev. 2005, 26, 380–392. [Google Scholar] [CrossRef]
- Ramana, K.V. Aldose reductase: New insights for an old enzyme. Biomol. Concepts 2011, 2, 103–114. [Google Scholar] [CrossRef]
- Ramana, K.V.; Friedrich, B.; Tammali, R.; West, M.B.; Bhatnagar, A.; Srivastava, S.K. Requirement of aldose reductase for the hyperglycemic activation of protein kinase C and formation of diacylglycerol in vascular smooth muscle cells. Diabetes 2005, 54, 818–829. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
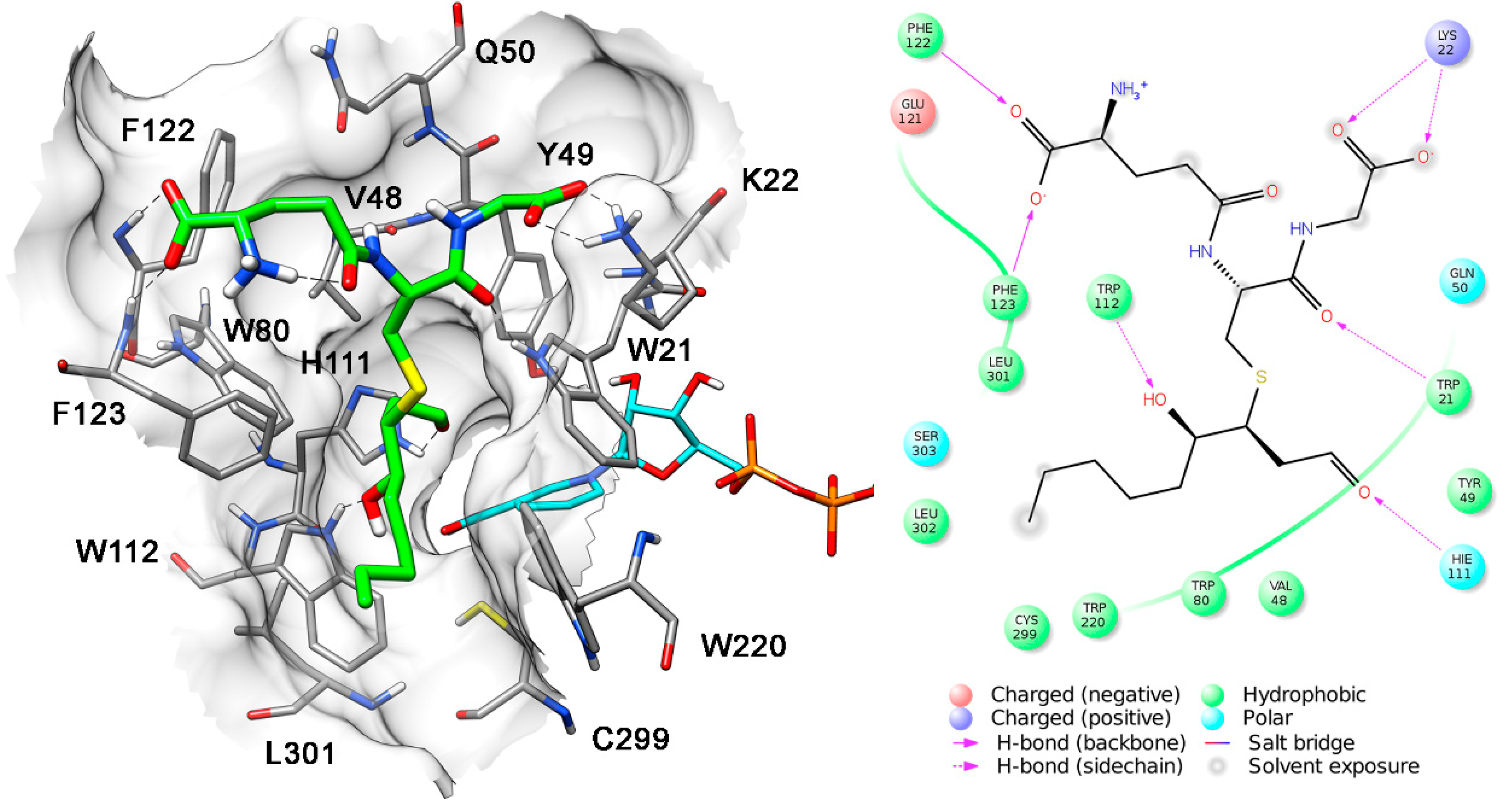{kind=link}
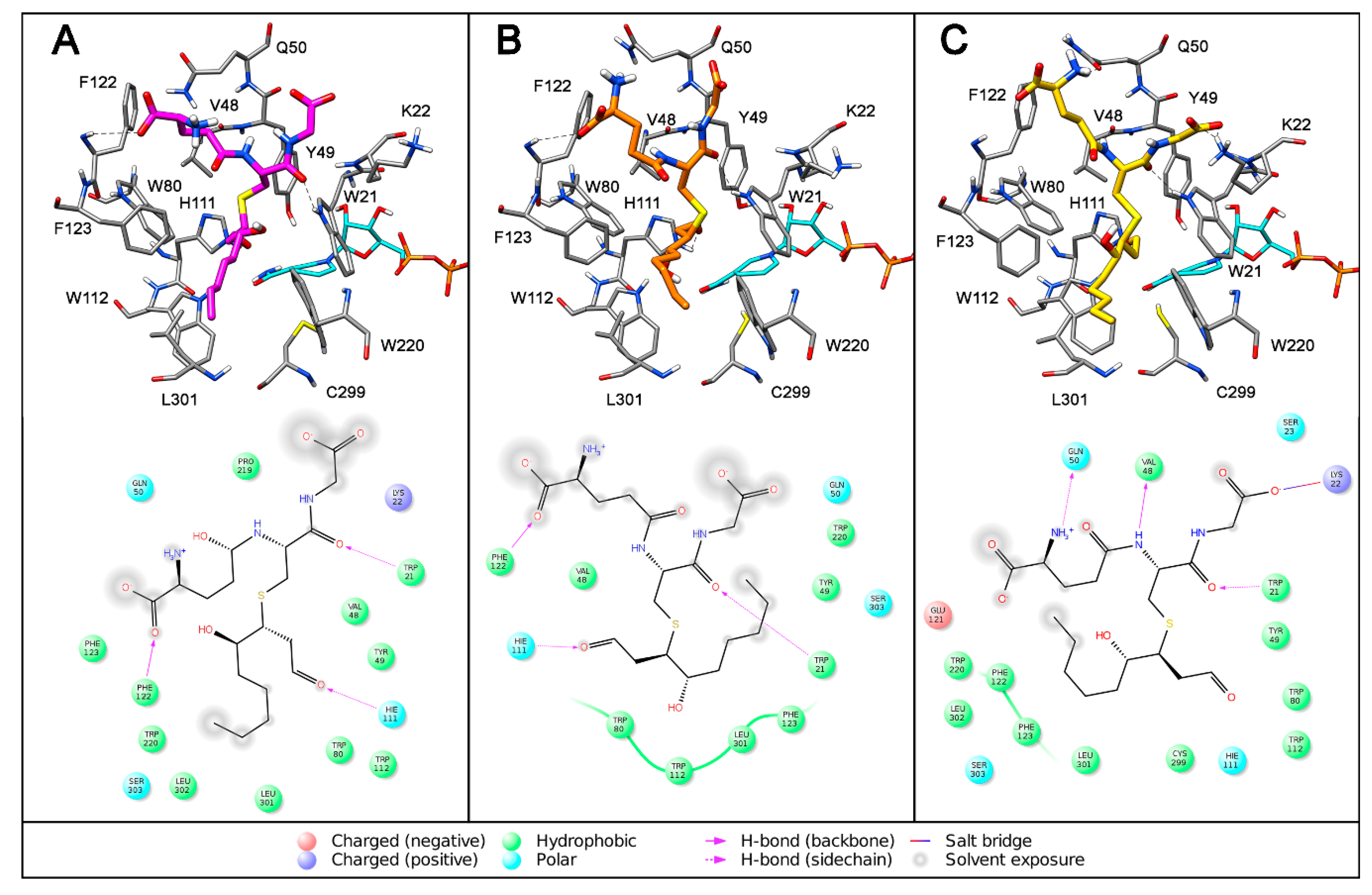{kind=link}
| Substrate | KM (µM) | kcat (min−1) | kcat/KM (µM−1 min−1) |
|---|---|---|---|
| 4R-HNE | 98.2 ± 6.7 | 87.5 ± 6.2 | 0.9 ± 0.1 |
| 4S-HNE | 47.6 ±5.7 | 91.3 ± 8.0 | 1.9 ± 0.3 |
| 3R,S,4R-GSHNE | 28.3 ± 4.6 | 120.9 ± 6.3 | 4.3 ± 0.7 |
| 3R,S,4S-GSHNE | 319.6 ± 55.7 | 70.0 ± 9.0 | 0.2 ± 0.1 |
| GSHNE Diasteroisomer | Relative Energies (KJ/mol) | Boltzmann Population (300 K) | R | ||||
|---|---|---|---|---|---|---|---|
| Open | Close (R) | Close (S) | Open | Close (R) | Close (S) | ||
| 3R,4R | 49.62 | 6.66 | 0.00 | 2.12 ∙10−9 | 0.06 | 0.94 | 139.77 |
| 3S,4S | 65.68 | 4.71 | 6.09 | 1.52 ∙10−11 | 0.63 | 0.37 | 1.00 |
| 3S,4R | 54.02 | 18.11 | 18.28 | 2.87 ∙10−7 | 0.52 | 0.48 | 18,932.25 |
| 3R,4S | 54.04 | 34.21 | 15.08 | 1.63 ∙10−7 | 4.65 ∙10−4 | 1.00 | 10,744.65 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balestri, F.; Barracco, V.; Renzone, G.; Tuccinardi, T.; Pomelli, C.S.; Cappiello, M.; Lessi, M.; Rotondo, R.; Bellina, F.; Scaloni, A.; et al. Stereoselectivity of Aldose Reductase in the Reduction of Glutathionyl-Hydroxynonanal Adduct. Antioxidants 2019, 8, 502. https://doi.org/10.3390/antiox8100502
Balestri F, Barracco V, Renzone G, Tuccinardi T, Pomelli CS, Cappiello M, Lessi M, Rotondo R, Bellina F, Scaloni A, et al. Stereoselectivity of Aldose Reductase in the Reduction of Glutathionyl-Hydroxynonanal Adduct. Antioxidants. 2019; 8(10):502. https://doi.org/10.3390/antiox8100502
Chicago/Turabian StyleBalestri, Francesco, Vito Barracco, Giovanni Renzone, Tiziano Tuccinardi, Christian Silvio Pomelli, Mario Cappiello, Marco Lessi, Rossella Rotondo, Fabio Bellina, Andrea Scaloni, and et al. 2019. "Stereoselectivity of Aldose Reductase in the Reduction of Glutathionyl-Hydroxynonanal Adduct" Antioxidants 8, no. 10: 502. https://doi.org/10.3390/antiox8100502