Regulation of Oxidative Stress in Corneal Endothelial Cells by Prdx6

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Chemicals and Reagents
2.2. Induction of Oxidative Stress
2.3. Human Corneal Tissue and Cell Culture
2.4. Cell Fractionation and Western Blotting
Plasma Membrane Isolation
2.5. RNAi Knockdown of Prdx6
2.6. Lipid Peroxidation Assay
2.7. Cell Viability Assay: Flow Cytometry
2.7.1. Cell Viability Assay: xCELLigence
2.7.2. Cell Viability Assay: Mitochondrial Membrane Potential (ΔΨm)
3. Results
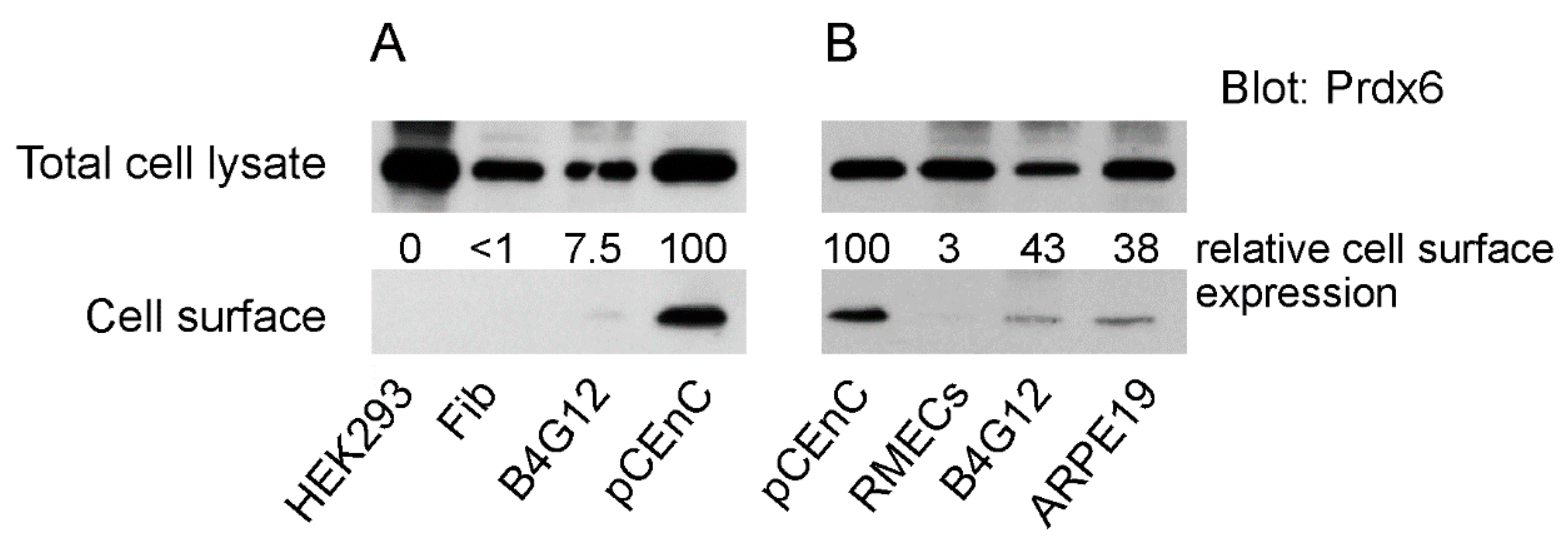
3.1. Membrane Expression of Prdx6 in Corneal Endothelial Cells
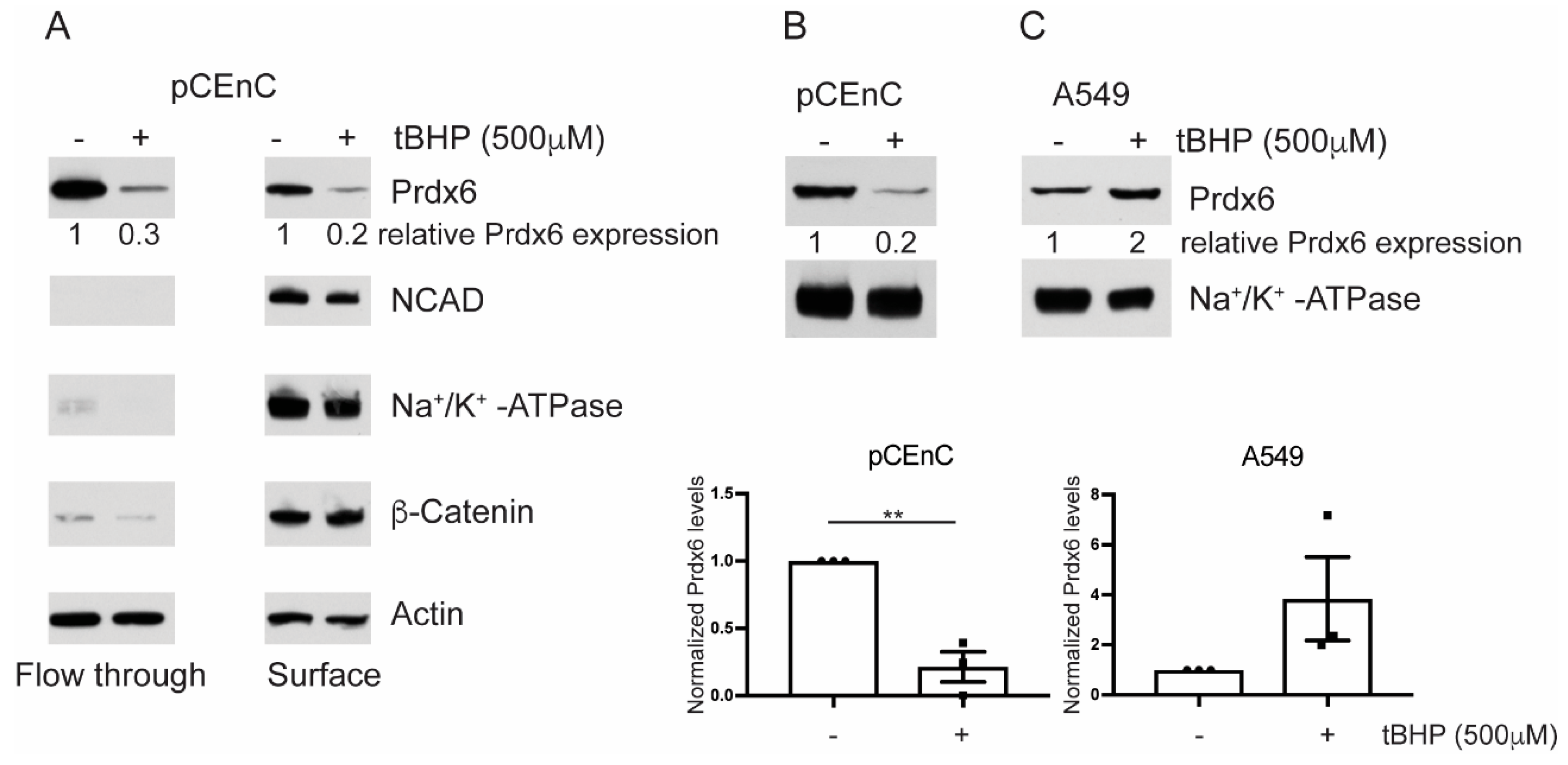
3.2. Prdx6 Levels are Sensitive to Oxidative Stress
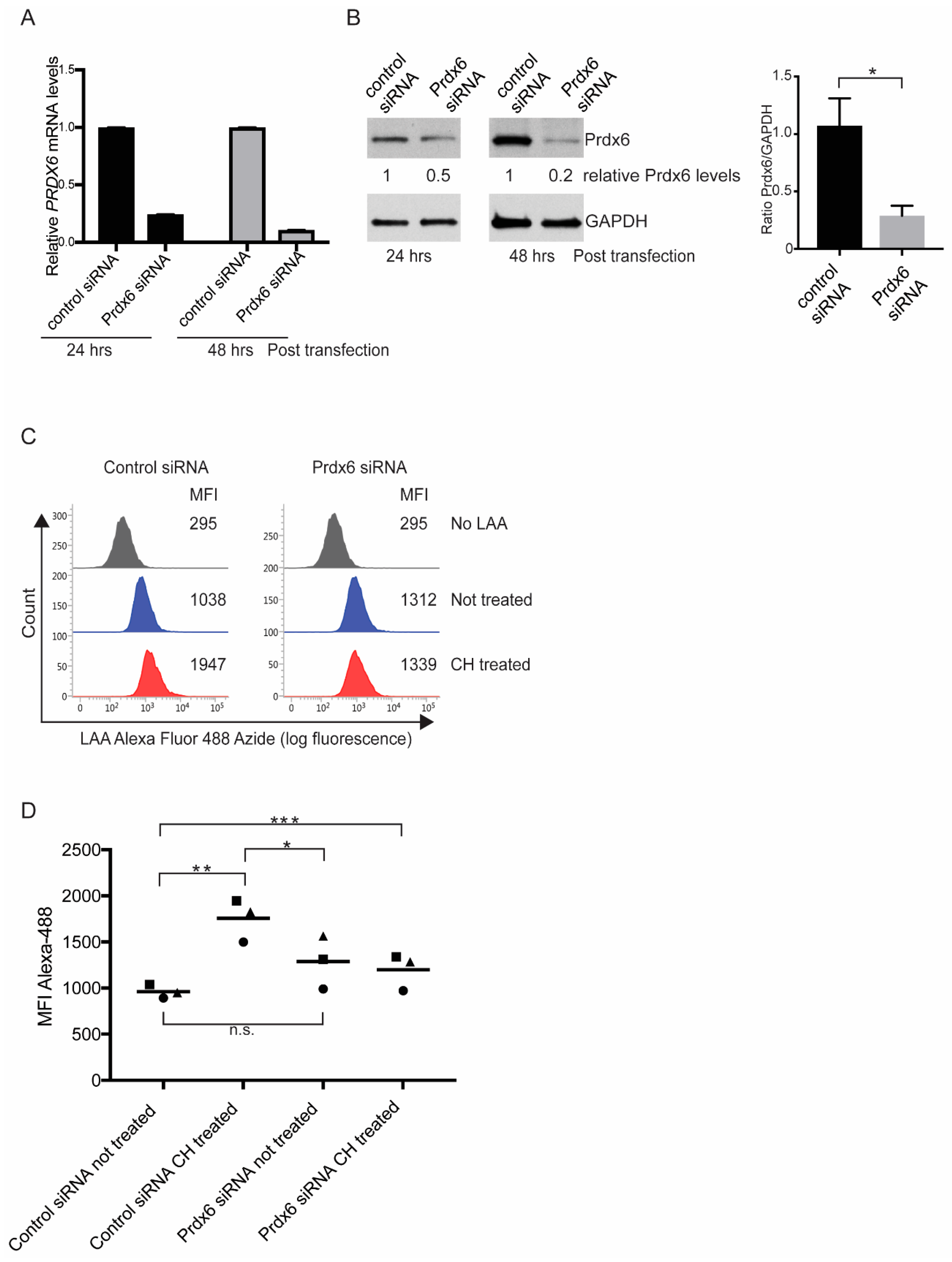
3.3. Prdx6 is Required for a Normal Lipid Peroxidation Response
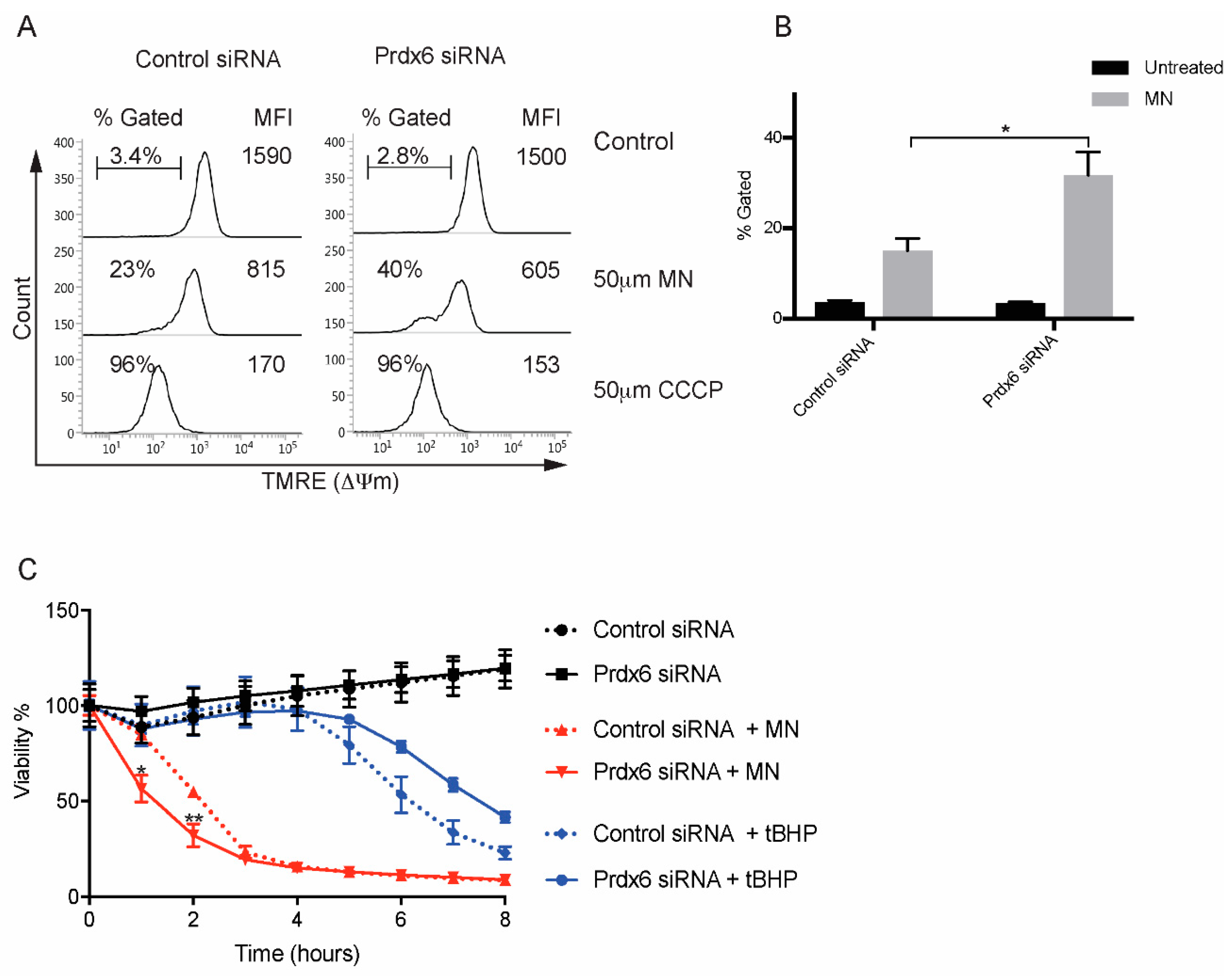
3.4. Loss of Prdx6 Does Not Affect Cell Viability in Response to Cumene Hydroperoxide
3.5. Regulation of Mitochondrial Membrane Potential by Prdx6
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of interest
References
- Peh, G.S.L.; Beuerman, R.W.; Colman, A.; Tan, D.T.; Mehta, J.S. Human corneal endothelial cell expansion for corneal endothelium transplantation: An overview. Transplantation 2011, 91, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Wahlig, S.; Lovatt, M.; Mehta, J.S. Functional role of peroxiredoxin 6 in the eye. Free Radic. Biol. Med. 2018, 126, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Bourne, W.M. Biology of the corneal endothelium in health and disease. Eye 2003, 17, 912–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laing, R.A.; Sanstrom, M.M.; Berrospi, A.R.; Leibowitz, H.M. Changes in the corneal endothelium as a function of age. Exp. Eye Res. 1976, 22, 587–594. [Google Scholar] [CrossRef]
- Galgauskas, S.; Norvydaitė, D.; Krasauskaitė, D.; Stech, S.; Ašoklis, R.S. Age-related changes in corneal thickness and endothelial characteristics. Clin. Interv. Aging 2013, 8, 1445–1450. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Vojnovic, D.; Kochevar, I.E.; Jurkunas, U.V. UV-A Irradiation Activates Nrf2-Regulated Antioxidant Defense and Induces p53/Caspase3-Dependent Apoptosis in Corneal Endothelial Cells. Investig. Ophthalmol. Vis. Sci. 2016, 57, 2319–2327. [Google Scholar] [CrossRef] [PubMed]
- Vedana, G.; Villarreal, G.; Jun, A.S. Fuchs endothelial corneal dystrophy: Current perspectives. Clin. Ophthalmol. 2016, 10, 321–330. [Google Scholar]
- Jurkunas, U.V.; Bitar, M.S.; Funaki, T.; Azizi, B. Evidence of oxidative stress in the pathogenesis of fuchs endothelial corneal dystrophy. Am. J. Pathol. 2010, 177, 2278–2289. [Google Scholar] [CrossRef]
- Jurkunas, U.V.; Rawe, I.; Bitar, M.S.; Zhu, C.; Harris, D.L.; Colby, K.; Joyce, N.C. Decreased expression of peroxiredoxins in Fuchs’ endothelial dystrophy. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2956–2963. [Google Scholar] [CrossRef]
- Fisher, A.B.; Dodia, C.; Sorokina, E.M.; Li, H.; Zhou, S.; Raabe, T.; Feinstein, S.I. A novel lysophosphatidylcholine acyl transferase activity is expressed by peroxiredoxin 6. J. Lipid Res. 2016, 57, 587–596. [Google Scholar] [CrossRef]
- Fisher, A.B. The phospholipase A2 activity of peroxiredoxin 6. J. Lipid Res. 2018, 59, 1132–1147. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.B. Peroxiredoxin 6: A bifunctional enzyme with glutathione peroxidase and phospholipase A₂ activities. Antioxid. Redox Signal. 2011, 15, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Benipal, B.; Zhou, S.; Dodia, C.; Chatterjee, S.; Tao, J.-Q.; Sorokina, E.M.; Raabe, T.; Feinstein, S.I.; Fisher, A.B. Critical role of peroxiredoxin 6 in the repair of peroxidized cell membranes following oxidative stress. Free Radic. Biol. Med. 2015, 87, 356–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, V.; Chin, A.; Peh, G.; Mehta, J.S.; Choo, A. Generation of novel monoclonal antibodies for the enrichment and characterization of human corneal endothelial cells (hCENC) necessary for the treatment of corneal endothelial blindness. MAbs 2014, 6, 1439–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambruso, D.R.; Ellison, M.A.; Thurman, G.W.; Leto, T.L. Peroxiredoxin 6 translocates to the plasma membrane during neutrophil activation and is required for optimal NADPH oxidase activity. Biochim. Biophys. Acta 2012, 1823, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Manevich, Y.; Shuvaeva, T.; Dodia, C.; Kazi, A.; Feinstein, S.I.; Fisher, A.B. Binding of peroxiredoxin 6 to substrate determines differential phospholipid hydroperoxide peroxidase and phospholipase A(2) activities. Arch. Biochem. Biophys. 2009, 485, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Vroegop, S.M.; Decker, D.E.; Buxser, S.E. Localization of damage induced by reactive oxygen species in cultured cells. Free Radic. Biol. Med. 1995, 18, 141–151. [Google Scholar] [CrossRef]
- Halilovic, A.; Schmedt, T.; Benischke, A.-S.; Hamill, C.; Chen, Y.; Santos, J.H.; Jurkunas, U.V. Menadione-Induced DNA Damage Leads to Mitochondrial Dysfunction and Fragmentation During Rosette Formation in Fuchs Endothelial Corneal Dystrophy. Antioxid. Redox Signal. 2016, 24, 1072–1083. [Google Scholar] [CrossRef] [Green Version]
- Peh, G.S.L.; Chng, Z.; Ang, H.-P.; Cheng, T.Y.D.; Adnan, K.; Seah, X.-Y.; George, B.L.; Toh, K.-P.; Tan, D.T.; Yam, G.H.F.; et al. Propagation of human corneal endothelial cells: A novel dual media approach. Cell Transplant. 2015, 24, 287–304. [Google Scholar] [CrossRef]
- Valtink, M.; Gruschwitz, R.; Funk, R.H.W.; Engelmann, K. Two clonal cell lines of immortalized human corneal endothelial cells show either differentiated or precursor cell characteristics. Cells Tissues Organs (Print) 2008, 187, 286–294. [Google Scholar] [CrossRef]
- Schindler, J.; Nothwang, H.G. Aqueous polymer two-phase systems: Effective tools for plasma membrane proteomics. Proteomics 2006, 6, 5409–5417. [Google Scholar] [CrossRef] [PubMed]
- Jurkunas, U.V. Fuchs Endothelial Corneal Dystrophy through the Prism of Oxidative Stress. Cornea 2018, 37 (Suppl. 1), S50–S54. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, H.A.; Yim, S.H.; Shin, D.H.; Kang, D.; Yu, D.-Y.; Rhee, S.G. Inactivation of peroxiredoxin I by phosphorylation allows localized H(2)O(2) accumulation for cell signaling. Cell 2010, 140, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Zhang, X.; Zheng, L.; Li, Z.; Zhao, X.; Lai, W.; Shen, H.; Lv, J.; Yang, G.; Wang, Q.; et al. Peroxiredoxin 6 Is a Crucial Factor in the Initial Step of Mitochondrial Clearance and Is Upstream of the PINK1-Parkin Pathway. Antioxid. Redox Signal. 2016, 24, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Eismann, T.; Huber, N.; Shin, T.; Kuboki, S.; Galloway, E.; Wyder, M.; Edwards, M.J.; Greis, K.D.; Shertzer, H.G.; Fisher, A.B.; et al. Peroxiredoxin-6 protects against mitochondrial dysfunction and liver injury during ischemia-reperfusion in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G266–G274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benischke, A.-S.; Vasanth, S.; Miyai, T.; Katikireddy, K.R.; White, T.; Chen, Y.; Halilovic, A.; Price, M.; Price, F.; Liton, P.B.; et al. Activation of mitophagy leads to decline in Mfn2 and loss of mitochondrial mass in Fuchs endothelial corneal dystrophy. Sci. Rep. 2017, 7, 6656. [Google Scholar] [CrossRef] [Green Version]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lovatt, M.; Adnan, K.; Peh, G.S.L.; Mehta, J.S. Regulation of Oxidative Stress in Corneal Endothelial Cells by Prdx6. Antioxidants 2018, 7, 180. https://doi.org/10.3390/antiox7120180
Lovatt M, Adnan K, Peh GSL, Mehta JS. Regulation of Oxidative Stress in Corneal Endothelial Cells by Prdx6. Antioxidants. 2018; 7(12):180. https://doi.org/10.3390/antiox7120180
Chicago/Turabian StyleLovatt, Matthew, Khadijah Adnan, Gary S. L. Peh, and Jodhbir S. Mehta. 2018. "Regulation of Oxidative Stress in Corneal Endothelial Cells by Prdx6" Antioxidants 7, no. 12: 180. https://doi.org/10.3390/antiox7120180