
Iron, Oxidative Stress, and Metabolic Dysfunction—Associated Steatotic Liver Disease


{kind=link}
Abstract
:1. Introduction
1.1. Iron Homeostasis
1.2. Iron, Oxidative Stress, and Ferroptosis
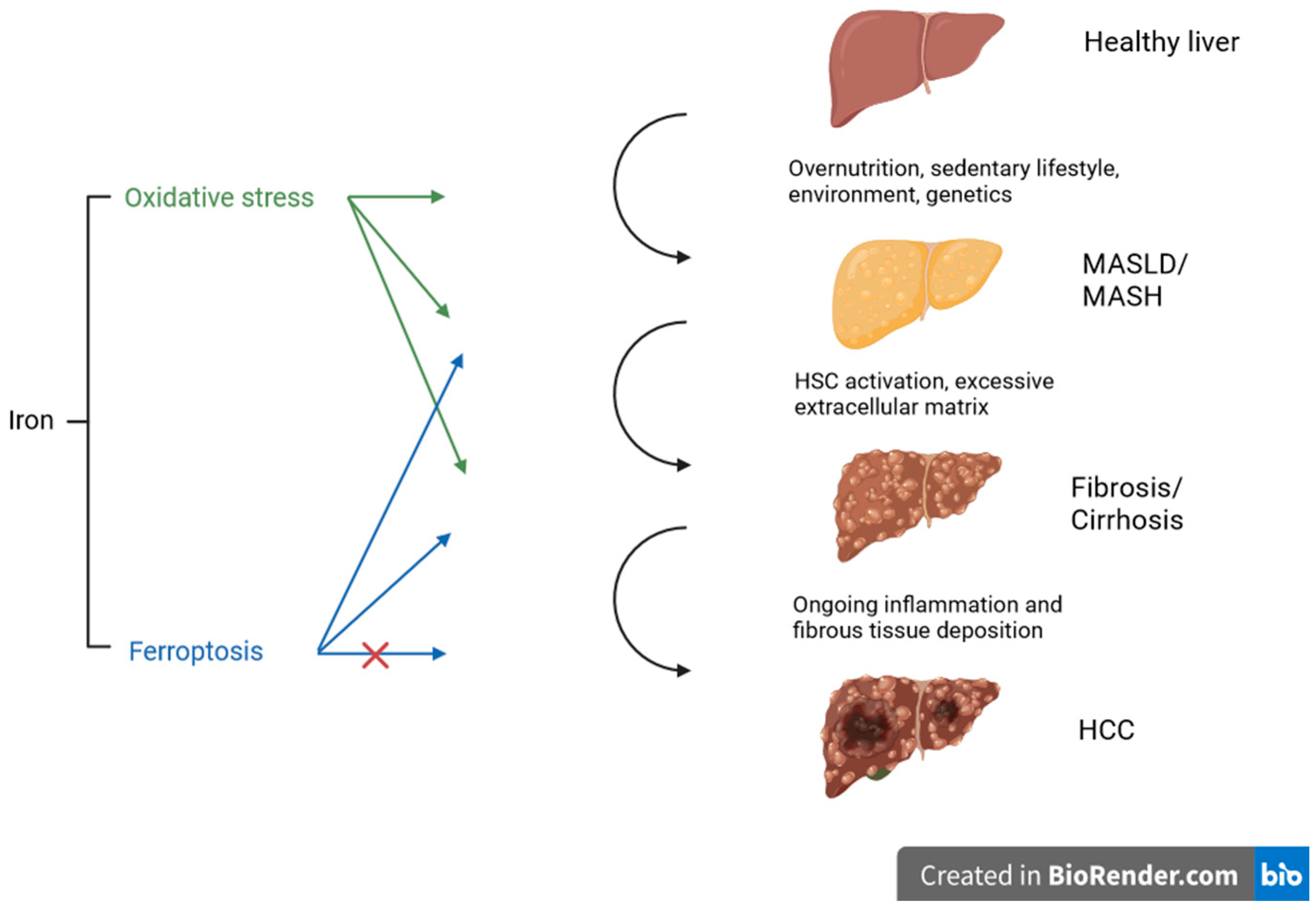
2. Oxidative Stress and Metabolic-Dysfunction-Associated Steatotic Liver Disease
3. Oxidative Stress and Liver Fibrosis
4. Oxidative Stress and Hepatocellular Carcinoma (HCC)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gozzelino, R.; Arosio, P. Iron Homeostasis in Health and Disease. Int. J. Mol. Sci. 2016, 17, 130. [Google Scholar] [CrossRef] [PubMed]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef]
- Papanikolaou, G.; Pantopoulos, K. Systemic iron homeostasis and erythropoiesis. IUBMB Life 2017, 69, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Katsarou, A.; Pantopoulos, K. Basics and principles of cellular and systemic iron homeostasis. Mol. Asp. Med. 2020, 75, 100866. [Google Scholar] [CrossRef] [PubMed]
- Kowdley, K.V.; Belt, P.; Wilson, L.A.; Yeh, M.M.; Neuschwander-Tetri, B.A.; Chalasani, N.; Sanyal, A.J.; Nelson, J.E.; The NASH Clinical Research Network. Serum ferritin is an independent predictor of histologic severity and advanced fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2012, 55, 77–85. [Google Scholar] [CrossRef]
- Wang, Q.; Zhu, M.; Li, H.; Chen, P.; Wang, M.; Gu, L.; Zhang, X.; Chen, L. Hyperferritinemia Correlates to Metabolic Dysregulation and Steatosis in Chinese Biopsy-Proven Nonalcoholic Fatty Liver Disease Patients. Diabetes Metab. Syndr. Obes. Targets Ther. 2022, 15, 1543–1552. [Google Scholar] [CrossRef]
- Dev, S.; Babitt, J.L. Overview of iron metabolism in health and disease: Iron metabolism in health and disease. Hemodial. Int. 2017, 21 (Suppl. S1), S6–S20. [Google Scholar] [CrossRef]
- Vogt, A.-C.S.; Arsiwala, T.; Mohsen, M.; Vogel, M.; Manolova, V.; Bachmann, M.F. On Iron Metabolism and Its Regulation. Int. J. Mol. Sci. 2021, 22, 4591. [Google Scholar] [CrossRef]
- Dutt, S.; Hamza, I.; Bartnikas, T.B. Molecular Mechanisms of Iron and Heme Metabolism. Annu. Rev. Nutr. 2022, 42, 311–335. [Google Scholar] [CrossRef]
- Kawabata, H. Transferrin and transferrin receptors update. Free. Radic. Biol. Med. 2019, 133, 46–54. [Google Scholar] [CrossRef]
- Plays, M.; Müller, S.; Rodriguez, R. Chemistry and biology of ferritin. Metallomics 2021, 13, mfab021. [Google Scholar] [CrossRef]
- Silva, B.; Faustino, P. An overview of molecular basis of iron metabolism regulation and the associated pathologies. Biochim. Biophys. Acta BBA—Mol. Basis Dis. 2015, 1852, 1347–1359. [Google Scholar] [CrossRef]
- Nemeth, E.; Ganz, T. Hepcidin-Ferroportin Interaction Controls Systemic Iron Homeostasis. Int. J. Mol. Sci. 2021, 22, 6493. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim. Biophys. Acta BBA—Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef]
- Nakamura, T.; Naguro, I.; Ichijo, H. Iron homeostasis and iron-regulated ROS in cell death, senescence and human diseases. Biochim. Biophys. Acta BBA—Gen. Subj. 2019, 1863, 1398–1409. [Google Scholar] [CrossRef]
- Angoro, B.; Motshakeri, M.; Hemmaway, C.; Svirskis, D.; Sharma, M. Non-transferrin bound iron. Clin. Chim. Acta 2022, 531, 157–167. [Google Scholar] [CrossRef]
- Jenkitkasemwong, S.; Wang, C.-Y.; Coffey, R.; Zhang, W.; Chan, A.; Biel, T.; Kim, J.-S.; Hojyo, S.; Fukada, T.; Knutson, M.D. SLC39A14 Is Required for the Development of Hepatocellular Iron Overload in Murine Models of Hereditary Hemochromatosis. Cell Metab. 2015, 22, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef] [PubMed]
- Sousa, L.; Oliveira, M.M.; Pessôa, M.T.C.; Barbosa, L.A. Iron overload: Effects on cellular biochemistry. Clin. Chim. Acta 2020, 504, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.; Huang, Z.; Lin, Z.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- He, J.; Li, Z.; Xia, P.; Shi, A.; FuChen, X.; Zhang, J.; Yu, P. Ferroptosis and ferritinophagy in diabetes complications. Mol. Metab. 2022, 60, 101470. [Google Scholar] [CrossRef]
- Qin, Y.; Qiao, Y.; Wang, D.; Tang, C.; Yan, G. Ferritinophagy and ferroptosis in cardiovascular disease: Mechanisms and potential applications. Biomed. Pharmacother. 2021, 141, 111872. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Dong, R.; Wu, Y.; Ni, B. Physiological Effects of Ferroptosis on Organ Fibrosis. Oxidative Med. Cell. Longev. 2022, 2022, 5295434. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Song, Y.; Wei, L.; Guo, J.; Xu, W.; Li, M. The emerging roles of ferroptosis in organ fibrosis and its potential therapeutic effect. Int. Immunopharmacol. 2023, 116, 109812. [Google Scholar] [CrossRef] [PubMed]
- Capelletti, M.M.; Manceau, H.; Puy, H.; Peoc’h, K. Ferroptosis in Liver Diseases: An Overview. Int. J. Mol. Sci. 2020, 21, 4908. [Google Scholar] [CrossRef]
- Powell, E.E.; Wong, V.W.-S.; Rinella, M. Non-alcoholic fatty liver disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wong, V.W.-S.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multi-society Delphi consensus statement on new fatty liver disease nomenclature. J. Hepatol. 2023, 78, 1966–1986. [Google Scholar] [CrossRef] [PubMed]
- Manne, V.; Handa, P.; Kowdley, K.V. Pathophysiology of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis. Clin. Liver Dis. 2018, 22, 23–37. [Google Scholar] [CrossRef]
- Datz, C.; Müller, E.; Aigner, E. Iron overload and non-alcoholic fatty liver disease. Minerva Endocrinol. 2017, 42, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F. NAFLD: Mechanisms, Treatments, and Biomarkers. Biomolecules 2022, 12, 824. [Google Scholar] [CrossRef]
- Bessone, F.; Razori, M.V.; Roma, M.G. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell. Mol. Life Sci. 2019, 76, 99–128. [Google Scholar] [CrossRef]
- Pierantonelli, I.; Svegliati-Baroni, G. Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression from NAFLD to NASH. Transplantation 2019, 103, e1–e13. [Google Scholar] [CrossRef]
- Martín-Fernández, M.; Arroyo, V.; Carnicero, C.; Sigüenza, R.; Busta, R.; Mora, N.; Antolín, B.; Tamayo, E.; Aspichueta, P.; Carnicero-Frutos, I.; et al. Role of Oxidative Stress and Lipid Peroxidation in the Pathophysiology of NAFLD. Antioxidants 2022, 11, 2217. [Google Scholar] [CrossRef]
- Nelson, J.E.; Klintworth, H.; Kowdley, K.V. Iron Metabolism in Nonalcoholic Fatty Liver Disease. Curr. Gastroenterol. Rep. 2012, 14, 8–16. [Google Scholar] [CrossRef]
- Aigner, E. Dysregulation of iron and copper homeostasis in nonalcoholic fatty liver. World J. Hepatol. 2014, 7, 177–188. [Google Scholar] [CrossRef]
- Okada, K.; Warabi, E.; Sugimoto, H.; Horie, M.; Tokushige, K.; Ueda, T.; Harada, N.; Taguchi, K.; Hashimoto, E.; Itoh, K.; et al. Nrf2 inhibits hepatic iron accumulation and counteracts oxidative stress-induced liver injury in nutritional steatohepatitis. J. Gastroenterol. 2012, 47, 924–935. [Google Scholar] [CrossRef]
- Handa, P.; Morgan-Stevenson, V.; Maliken, B.D.; Nelson, J.E.; Washington, S.; Westerman, M.; Yeh, M.M.; Kowdley, K.V. Iron overload results in hepatic oxidative stress, immune cell activation, and hepatocellular ballooning injury, leading to nonalcoholic steatohepatitis in genetically obese mice. Am. J. Physiol. Liver Physiol. 2016, 310, G117–G127. [Google Scholar] [CrossRef]
- Moris, W.; Verhaegh, P.; Jonkers, D.; van Deursen, C.; Koek, G. Hyperferritinemia in Nonalcoholic Fatty Liver Disease: Iron Accumulation or Inflammation? Semin. Liver Dis. 2019, 39, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Moirand, R.; Mortaji, A.M.; Loréal, O.; Paillard, F.; Brissot, P.; Deugnier, Y. A new syndrome of liver iron overload with normal transferrin saturation. Lancet 1997, 349, 95–97. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Corradini, E.; Adams, L.A.; Aigner, E.; Alqahtani, S.; Arrese, M.; Bardou-Jacquet, E.; Bugianesi, E.; Fernandez-Real, J.-M.; Girelli, D.; et al. Consensus Statement on the definition and classification of metabolic hyperferritinaemia. Nat. Rev. Endocrinol. 2023, 19, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Petta, S.; Manuguerra, R.; Luong, T.V.; Cabibi, D.; Corradini, E.; Craxì, A.; Pinzani, M.; Tsochatzis, E.; Pietrangelo, A. Evaluating the association of serum ferritin and hepatic iron with disease severity in non-alcoholic fatty liver disease. Liver Int. 2019, 39, 1325–1334. [Google Scholar] [CrossRef]
- Wang, H.; Sun, R.; Yang, S.; Ma, X.; Yu, C. Association between serum ferritin level and the various stages of non-alcoholic fatty liver disease: A systematic review. Front. Med. 2022, 9, 934989. [Google Scholar] [CrossRef] [PubMed]
- Gautheron, J.; Gores, G.J.; Rodrigues, C.M. Lytic cell death in metabolic liver disease. J. Hepatol. 2020, 73, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Tsurusaki, S.; Tsuchiya, Y.; Koumura, T.; Nakasone, M.; Sakamoto, T.; Matsuoka, M.; Imai, H.; Kok, C.Y.-Y.; Okochi, H.; Nakano, H.; et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 2019, 10, 449. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, T.; Huang, X.; Li, Y.; Sun, T.; Zang, S.; Guan, K.; Xiong, Y.; Liu, J.; Yuan, H. Targeting ferroptosis alleviates methionine-choline deficient (MCD)-diet induced NASH by suppressing liver lipotoxicity. Liver Int. 2020, 40, 1378–1394. [Google Scholar] [CrossRef]
- Qi, J.; Kim, J.-W.; Zhou, Z.; Lim, C.-W.; Kim, B. Ferroptosis Affects the Progression of Nonalcoholic Steatohepatitis via the Modulation of Lipid Peroxidation–Mediated Cell Death in Mice. Am. J. Pathol. 2020, 190, 68–81. [Google Scholar] [CrossRef]
- Tong, J.; Li, D.; Meng, H.; Sun, D.; Lan, X.; Ni, M.; Ma, J.; Zeng, F.; Sun, S.; Fu, J.; et al. Targeting a novel inducible GPX4 alternative isoform to alleviate ferroptosis and treat metabolic-associated fatty liver disease. Acta Pharm. Sin. B 2022, 12, 3650–3666. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Hu, J.; Chen, L.; Li, S.; Yuan, M.; Tian, X.; Cao, P.; Qiu, Z. Ferroptosis as an emerging therapeutic target in liver diseases. Front. Pharmacol. 2023, 14, 1196287. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.; Byrne, C.D.; Targher, G.; Wang, F.; Zheng, M. Ferroptosis and metabolic dysfunction-associated fatty liver disease: Is there a link? Liver Int. 2022, 42, 1496–1502. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Wang, M.-Y.; Wang, G.-D.; Lv, Q.-Y.; Huang, Y.-Q.; Zhang, P.; Wang, W.; Zhang, Y.; Bai, Y.-P.; Guo, L.-Q. Metformin improves nonalcoholic fatty liver disease in db/db mice by inhibiting ferroptosis. Eur. J. Pharmacol. 2024, 966, 176341. [Google Scholar] [CrossRef]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Tang, X. Use of a ferroptosis-related gene signature to construct diagnostic and prognostic models for assessing immune infiltration in metabolic dysfunction-associated fatty liver disease. Front. Cell Dev. Biol. 2023, 11, 1199846. [Google Scholar] [CrossRef] [PubMed]
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 875. [Google Scholar] [CrossRef]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, N. TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis—Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef]
- Liang, S.; Kisseleva, T.; Brenner, D.A. The Role of NADPH Oxidases (NOXs) in Liver Fibrosis and the Activation of Myofibroblasts. Front. Physiol. 2016, 7, 17. [Google Scholar] [CrossRef]
- Luedde, T.; Schwabe, R.F. NF-κB in the liver—Linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef]
- Gao, H.; Jin, Z.; Bandyopadhyay, G.; Wang, G.; Zhang, D.; Rocha, K.C.E.; Liu, X.; Zhao, H.; Kisseleva, T.; Brenner, D.A.; et al. Aberrant iron distribution via hepatocyte-stellate cell axis drives liver lipogenesis and fibrosis. Cell Metab. 2022, 34, 1201–1213.e5. [Google Scholar] [CrossRef]
- Mehta, K.J.; Farnaud, S.J.; Sharp, P.A. Iron and liver fibrosis: Mechanistic and clinical aspects. World J. Gastroenterol. 2019, 25, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Luo, Y.; Xia, Q.; He, K. Ferroptosis and Liver Fibrosis. Int. J. Med. Sci. 2021, 18, 3361–3366. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Guo, M.; Li, Y.; Shen, M.; Kong, D.; Shao, J.; Ding, H.; Tan, S.; Chen, A.; Zhang, F.; et al. RNA-binding protein ZFP36/TTP protects against ferroptosis by regulating autophagy signaling pathway in hepatic stellate cells. Autophagy 2020, 16, 1482–1505. [Google Scholar] [CrossRef]
- Zhang, Z.; Yao, Z.; Wang, L.; Ding, H.; Shao, J.; Chen, A.; Zhang, F.; Zheng, S. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy 2018, 14, 2083–2103. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Jiang, L.; Wang, H.; Shen, Z.; Cheng, Q.; Zhang, P.; Wang, J.; Wu, Q.; Fang, X.; Duan, L.; et al. Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood 2020, 136, 726–739. [Google Scholar] [CrossRef]
- Wu, A.; Feng, B.; Yu, J.; Yan, L.; Che, L.; Zhuo, Y.; Luo, Y.; Yu, B.; Wu, D.; Chen, D. Fibroblast growth factor 21 attenuates iron overload-induced liver injury and fibrosis by inhibiting ferroptosis. Redox Biol. 2021, 46, 102131. [Google Scholar] [CrossRef]
- Yuan, S.; Wei, C.; Liu, G.; Zhang, L.; Li, J.; Li, L.; Cai, S.; Fang, L. Sorafenib attenuates liver fibrosis by triggering hepatic stellate cell ferroptosis via HIF-1α/SLC7A11 pathway. Cell Prolif. 2022, 55, e13158. [Google Scholar] [CrossRef]
- Kong, Z.; Liu, R.; Cheng, Y. Artesunate alleviates liver fibrosis by regulating ferroptosis signaling pathway. Biomed. Pharmacother. 2019, 109, 2043–2053. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Z.; Li, M.; Wang, F.; Jia, Y.; Zhang, F.; Shao, J.; Chen, A.; Zheng, S. P53-dependent induction of ferroptosis is required for artemether to alleviate carbon tetrachloride-induced liver fibrosis and hepatic stellate cell activation. IUBMB Life 2019, 71, 45–56. [Google Scholar] [CrossRef]
- Yi, J.; Wu, S.; Tan, S.; Qin, Y.; Wang, X.; Jiang, J.; Liu, H.; Wu, B. Berberine alleviates liver fibrosis through inducing ferrous redox to activate ROS-mediated hepatic stellate cells ferroptosis. Cell Death Discov. 2021, 7, 374. [Google Scholar] [CrossRef]
- Zheng, Y.; Zheng, Y.; Zhao, T.; Zhao, T.; Wang, J.; Wang, J.; Jiang, R.; Jiang, R.; Huang, J.; Huang, J.; et al. Curcumol alleviates liver fibrosis through inducing autophagy and ferroptosis in hepatic stellate cells. FASEB J. 2022, 36, e22665. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Meyer, T.; Sapisochin, G.; Salem, R.; Saborowski, A. Hepatocellular carcinoma. Lancet 2022, 400, 1345–1362. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.J.H.; Ng, C.H.; Lin, S.Y.; Pan, X.H.; Tay, P.; Lim, W.H.; Teng, M.; Syn, N.; Lim, G.; Yong, J.N.; et al. Clinical characteristics, surveillance, treatment allocation, and outcomes of non-alcoholic fatty liver disease-related hepatocellular carcinoma: A systematic review and meta-analysis. Lancet Oncol. 2022, 23, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef] [PubMed]
- Alarcón-Sánchez, B.R.; Pérez-Carreón, J.I.; Villa-Treviño, S.; Arellanes-Robledo, J. Molecular alterations that precede the establishment of the hallmarks of cancer: An approach on the prevention of hepatocarcinogenesis. Biochem. Pharmacol. 2021, 194, 114818. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zheng, Q.; Chen, Z. The Nrf2 Pathway in Liver Diseases. Front. Cell Dev. Biol. 2022, 10, 826204. [Google Scholar] [CrossRef] [PubMed]
- Brahma, M.K.; Gilglioni, E.H.; Zhou, L.; Trépo, E.; Chen, P.; Gurzov, E.N. Oxidative stress in obesity-associated hepatocellular carcinoma: Sources, signaling and therapeutic challenges. Oncogene 2021, 40, 5155–5167. [Google Scholar] [CrossRef]
- Ngo, H.K.C.; Kim, D.-H.; Cha, Y.-N.; Na, H.-K.; Surh, Y.-J. Nrf2 Mutagenic Activation Drives Hepatocarcinogenesis. Cancer Res 2017, 77, 4797–4808. [Google Scholar] [CrossRef]
- Kitamura, H.; Motohashi, H. NRF2 addiction in cancer cells. Cancer Sci. 2018, 109, 900–911. [Google Scholar] [CrossRef]
- Peng, C.; Li, X.; Ao, F.; Li, T.; Guo, J.; Liu, J.; Zhang, X.; Gu, J.; Mao, J.; Zhou, B. Mitochondrial ROS driven by NOX4 upregulation promotes hepatocellular carcinoma cell survival after incomplete radiofrequency ablation by inducing of mitophagy via Nrf2/PINK1. J. Transl. Med. 2023, 21, 218. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Li, J.; Qu, K.; Wan, Y.; Liu, S.; Zheng, W.; Zhang, Z.; Liu, C. CRIF1 overexpression facilitates tumor growth and metastasis through inducing ROS/NFκB pathway in hepatocellular carcinoma. Cell Death Dis. 2020, 11, 332. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Geng, Z.; Li, H.; Zhang, Y.; Li, J.; Chang, H. Over-expression of TFB2M facilitates cell growth and metastasis via activating ROS-Akt-NF-κB signalling in hepatocellular carcinoma. Liver Int. 2020, 40, 1756–1769. [Google Scholar] [CrossRef] [PubMed]
- Asare, G.A.; Mossanda, K.S.; Kew, M.C.; Paterson, A.C.; Kahler-Venter, C.P.; Siziba, K. Hepatocellular carcinoma caused by iron overload: A possible mechanism of direct hepatocarcinogenicity. Toxicology 2006, 219, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Paganoni, R.; Lechel, A.; Spasic, M.V. Iron at the Interface of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 4097. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.C.; Jeffrey, G.; Ryan, J. Haemochromatosis. Lancet 2023, 401, 1811–1821. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, Y.; Patel, P.; Chu, J.; Yu, X.; Hernaez, R.; El-Serag, H.; Kanwal, F. Risk of Hepatocellular Carcinoma in Patients with Various HFE Genotypes. Dig. Dis. Sci. 2023, 68, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Lei, G.; Zhuang, L.; Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 2022, 22, 381–396. [Google Scholar] [CrossRef]
- Bekric, D.; Ocker, M.; Mayr, C.; Stintzing, S.; Ritter, M.; Kiesslich, T.; Neureiter, D. Ferroptosis in Hepatocellular Carcinoma: Mechanisms, Drug Targets and Approaches to Clinical Translation. Cancers 2022, 14, 1826. [Google Scholar] [CrossRef]
- Pan, F.; Lin, X.; Hao, L.; Wang, T.; Song, H.; Wang, R. The Critical Role of Ferroptosis in Hepatocellular Carcinoma. Front. Cell Dev. Biol. 2022, 10, 882571. [Google Scholar] [CrossRef]
- Huang, Z.; Xia, H.; Cui, Y.; Yam, J.W.P.; Xu, Y. Ferroptosis: From Basic Research to Clinical Therapeutics in Hepatocellular Carcinoma. J. Clin. Transl. Hepatol. 2023, 11, 207–218. [Google Scholar] [CrossRef]
- Louandre, C.; Ezzoukhry, Z.; Godin, C.; Barbare, J.; Mazière, J.; Chauffert, B.; Galmiche, A. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int. J. Cancer 2013, 133, 1732–1742. [Google Scholar] [CrossRef]
- Li, Q.; Chen, K.; Zhang, T.; Jiang, D.; Chen, L.; Jiang, J.; Zhang, C.; Li, S. Understanding sorafenib-induced ferroptosis and resistance mechanisms: Implications for cancer therapy. Eur. J. Pharmacol. 2023, 955, 175913. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhao, Y.; Li, L.; Jiang, J.; Li, W.; Mang, Y.; Gao, Y.; Dong, Y.; Zhu, J.; Yang, C.; et al. Metformin promotes ferroptosis and sensitivity to sorafenib in hepatocellular carcinoma cells via ATF4/STAT3. Mol. Biol. Rep. 2023, 50, 6399–6413. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.; Chen, Q.; Liu, Y.; Wang, L.; Lu, W. Combination of Metformin and Sorafenib Induces Ferroptosis of Hepatocellular Carcinoma Through p62-Keap1-Nrf2 Pathway. J. Cancer 2022, 13, 3234–3243. [Google Scholar] [CrossRef]
- Li, Y.; Yang, W.; Zheng, Y.; Dai, W.; Ji, J.; Wu, L.; Cheng, Z.; Zhang, J.; Li, J.; Xu, X.; et al. Targeting fatty acid synthase modulates sensitivity of hepatocellular carcinoma to sorafenib via ferroptosis. J. Exp. Clin. Cancer Res. 2023, 42, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, L.; Yang, Z.; Zhao, D.; Deng, Z.; Xu, J.; Wu, Y.; Hao, Y.; Dong, Z.; Feng, L.; et al. Self-fueling ferroptosis-inducing microreactors based on pH-responsive Lipiodol Pickering emulsions enable transarterial ferro-embolization therapy. Natl. Sci. Rev. 2024, 11, nwad257. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gensluckner, S.; Wernly, B.; Datz, C.; Aigner, E. Iron, Oxidative Stress, and Metabolic Dysfunction—Associated Steatotic Liver Disease. Antioxidants 2024, 13, 208. https://doi.org/10.3390/antiox13020208
Gensluckner S, Wernly B, Datz C, Aigner E. Iron, Oxidative Stress, and Metabolic Dysfunction—Associated Steatotic Liver Disease. Antioxidants. 2024; 13(2):208. https://doi.org/10.3390/antiox13020208
Chicago/Turabian StyleGensluckner, Sophie, Bernhard Wernly, Christian Datz, and Elmar Aigner. 2024. "Iron, Oxidative Stress, and Metabolic Dysfunction—Associated Steatotic Liver Disease" Antioxidants 13, no. 2: 208. https://doi.org/10.3390/antiox13020208