
Nanoencapsulation of Anthocyanins from Red Cabbage (Brassica oleracea L. var. Capitata f. rubra) through Coacervation of Whey Protein Isolate and Apple High Methoxyl Pectin
 ,
,  , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparations and Characterisation of Anthocyanin Water Extract
2.2.1. ACNs Extraction
2.2.2. Differential pH Method
2.2.3. HPLC-DAD-MS Analysis
2.3. Nanoparticles Formation
2.3.1. Solutions Preparation
2.3.2. WPI/HMP Complex Coacervation
2.4. Dynamic Light Scattering (DLS) Analysis
2.5. Encapsulation Efficiency (EE)
2.6. Effect of pH
2.7. Antioxidant Capacity
2.7.1. ABTS
2.7.2. Ferric-Reducing Antioxidant Power (FRAP)
2.8. Fourier Transform Infrared Spectroscopy (FTIR)
2.9. Atomic Force Microscopy (AFM)
2.10. Transmission Electron Microscopy (TEM)
2.11. X-ray Powder Diffraction (XRPD)
2.12. Statistical Analysis
3. Results and Discussion
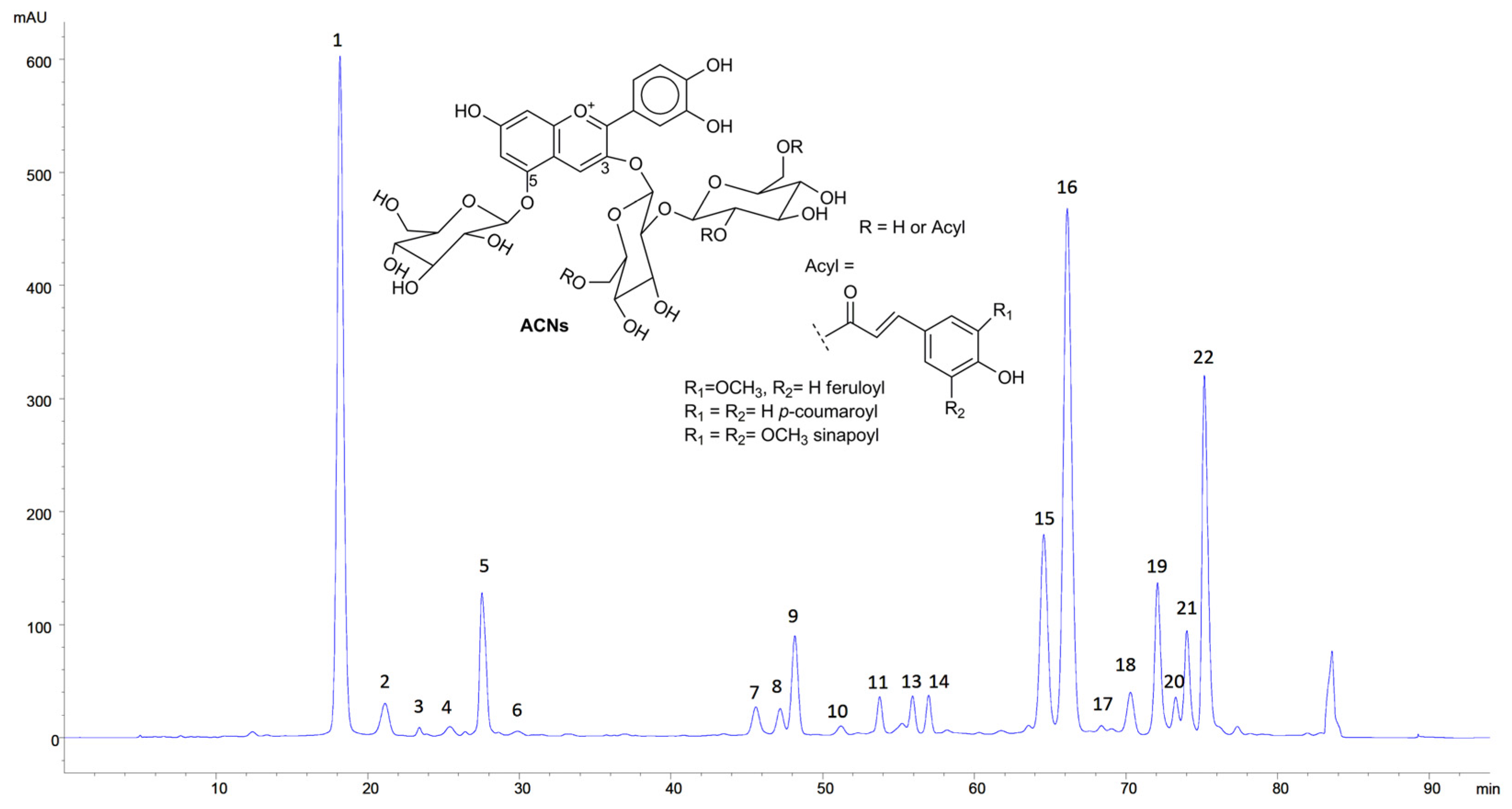
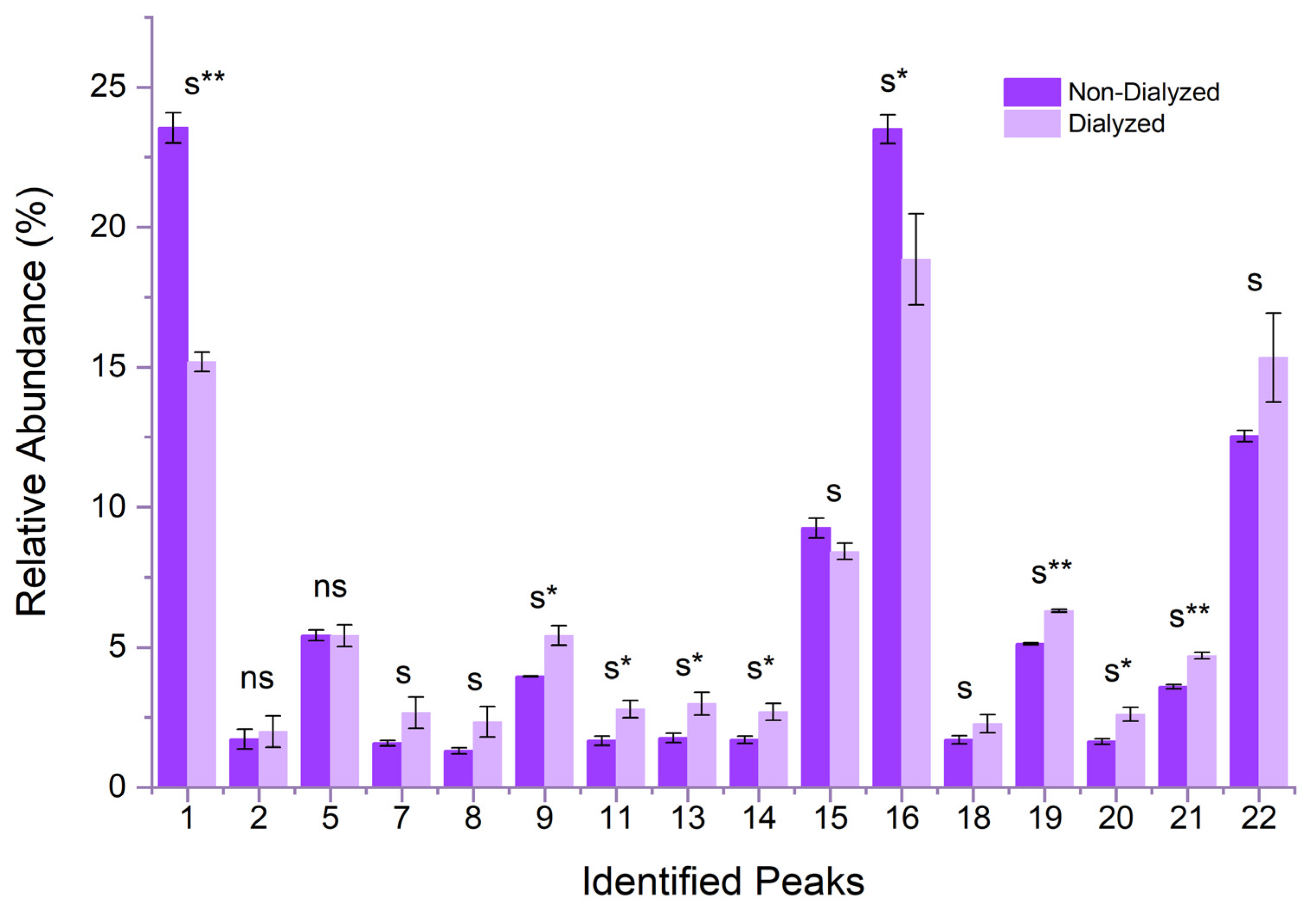
3.1. Characterisation of RCE


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peak No. | Peak Identity | M+ (m/z) |
|---|---|---|
| 1 | Cyanidin-3-diglucoside-5-glucoside | 773 |
| 2 | Cyanidin-3-glucoside-5-glucoside | 611 |
| 5 | Cyanidin-3-(sinapoyl)-diglucoside-5-glucoside | 979 |
| 5 | Cyanidin-3-(sinapoyl)-triglucoside-5-glucoside | 1141 |
| 7 | Cyanidin-3-(p-coumaroyl)-triglucoside-5-glucoside | 1081 |
| 8 | Cyanidin-3-(feruloyl)-triglucoside-5-glucoside | 1111 |
| 9 | Cyanidin-3-(sinapoyl)-triglucoside-5-glucoside | 1141 |
| 11 | Cyanidin-3-(feruloyl)(feruloyl)-triglucoside-5-glucoside | 1287 |
| 13 | Cyanidin-3-(feruloyl)(sinapoyl)-triglucoside-5-glucoside | 1317 |
| 14 | Cyanidin-3-(sinapoyl)(sinapoyl)-triglucoside-5-glucoside | 1347 |
| 15 | Cyanidin-3-(p-coumaroyl)-diglucoside-5-glucoside | 919 |
| 16 | Cyanidin-3-(sinapoyl)-diglucoside-5-glucoside | 979 |
| 17 | Cyanidin-3-(feruloyl)-diglucoside-5-glucoside | 949 |
| 18 | Cyanidin-3-(sinapoyl)-glucoside-5-glucoside | 817 |
| 19 | Cyanidin-3-(feruloyl)(feruloyl)-diglucoside-5-glucoside | 1125 |
| 20 | Cyanidin 3-(5-OHferuloyl)(sinapoyl)-triglucoside-5-glucoside | 1171 |
| 21 | Cyanidin-3-(feruloyl)(sinapoyl)-diglucoside-5-glucoside | 1155 |
| 22 | Cyanidin-3-(sinapoyl)(sinapoyl)-diglucoside-5-glucoside | 1185 |
3.2. Coacervation
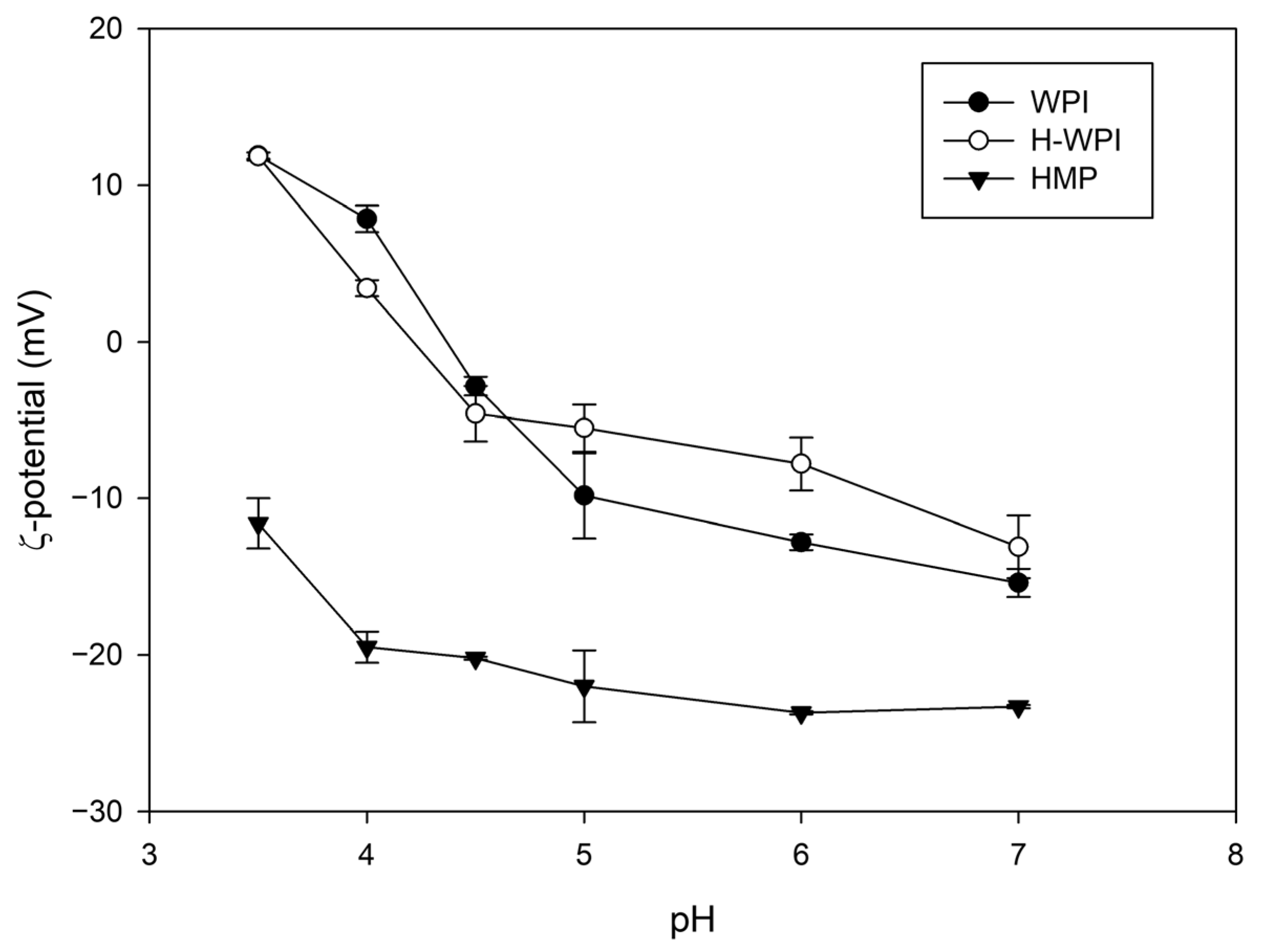
3.2.1. Electrical Characterisation of the Biopolymers
3.2.2. Coacervation
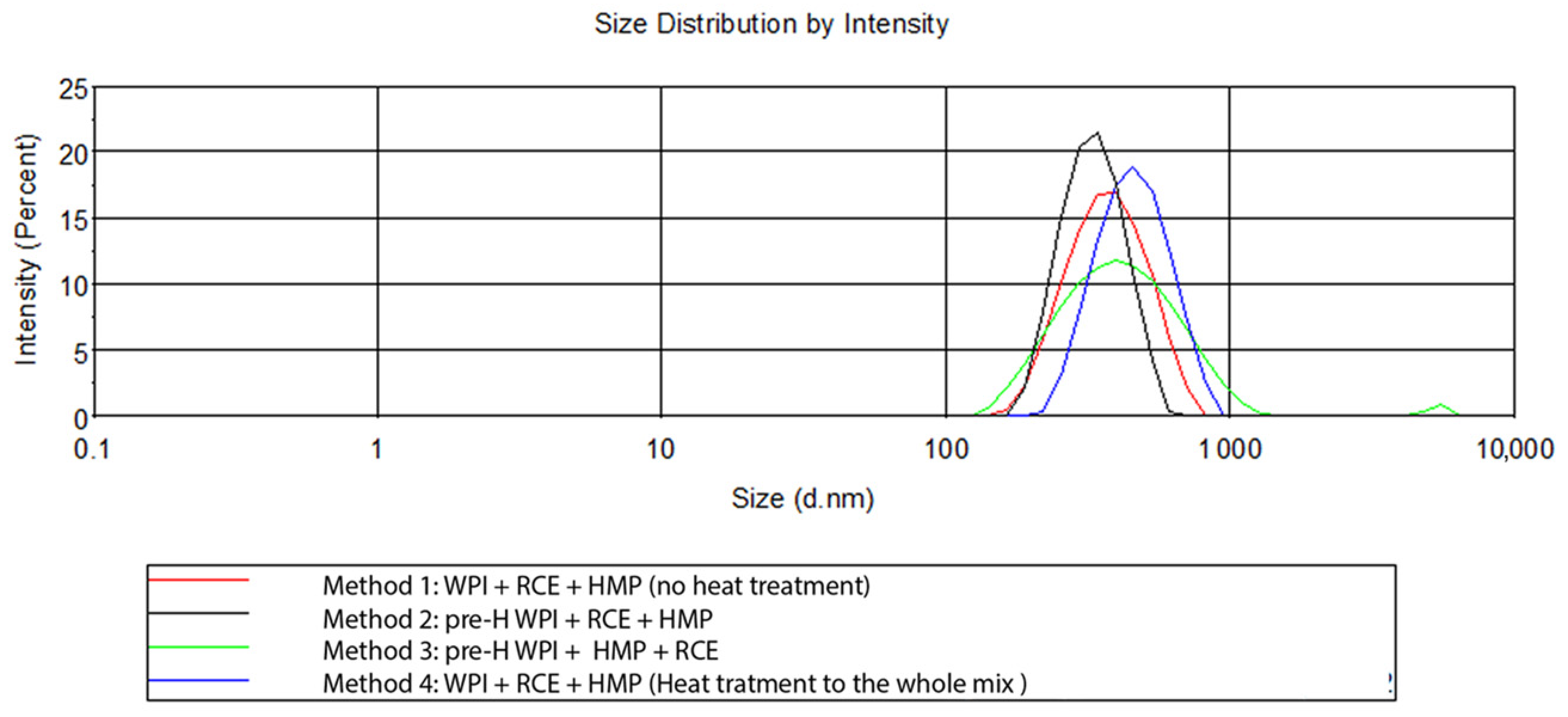
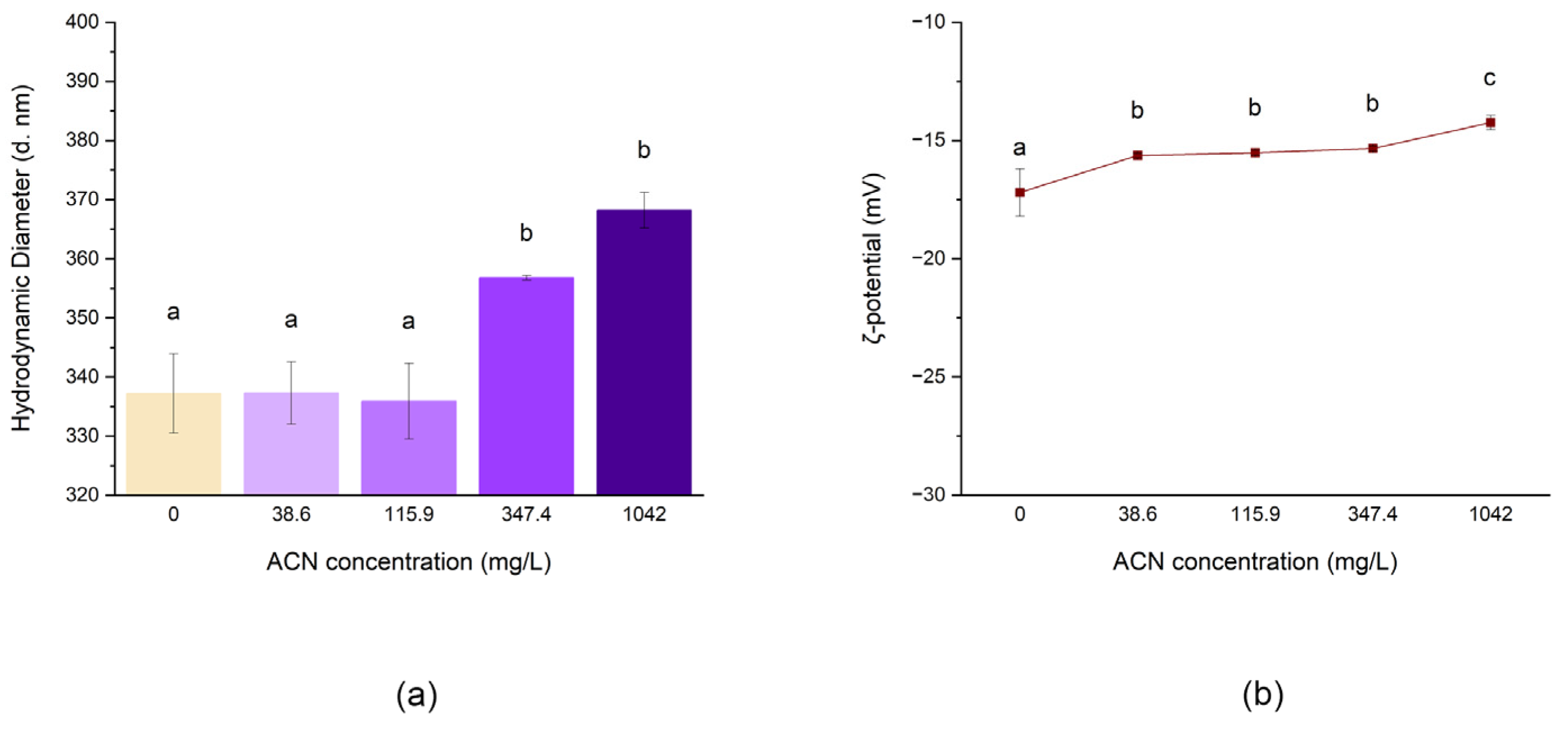
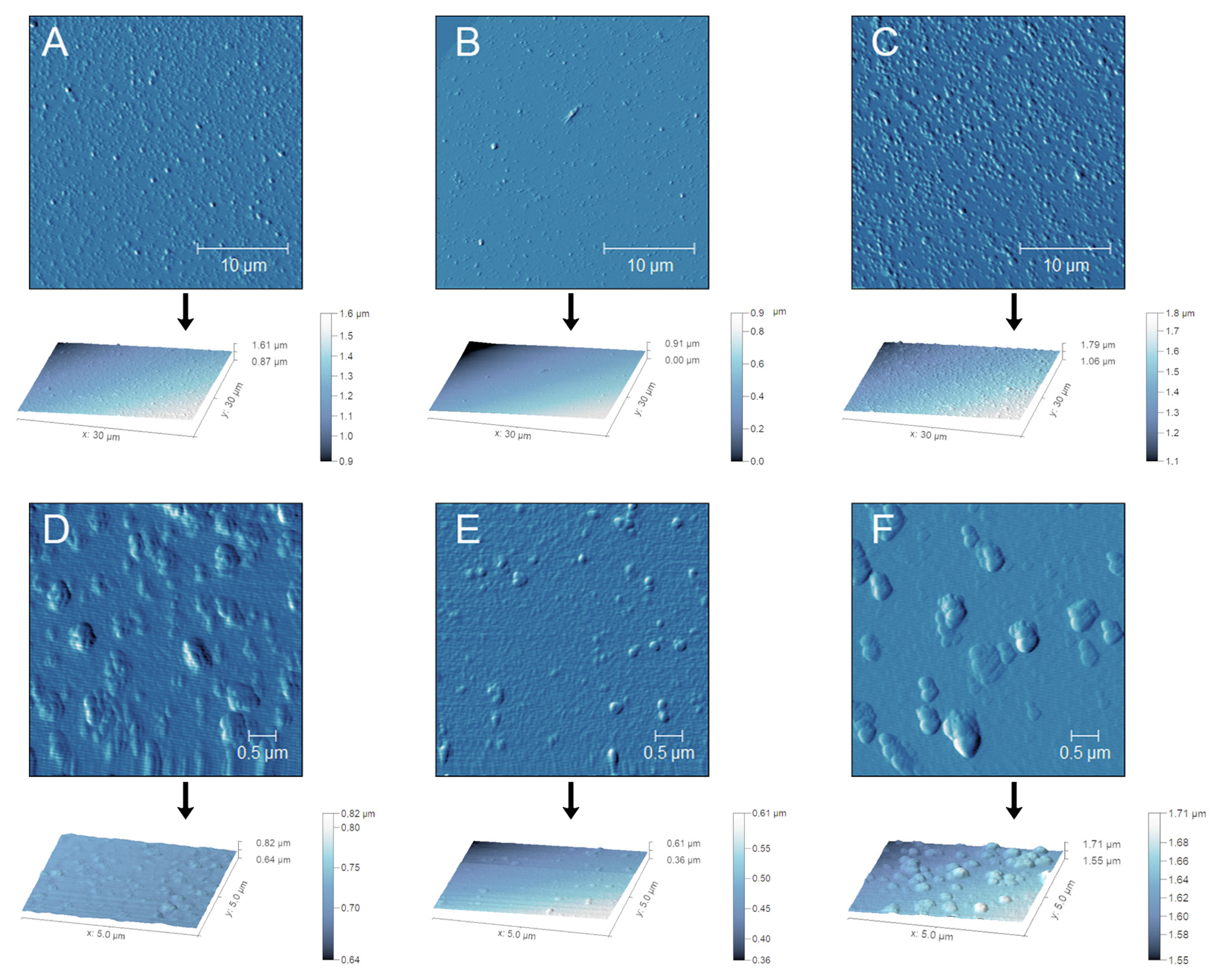
3.2.3. Effect of RCE Concentration on the Morphology of NPs
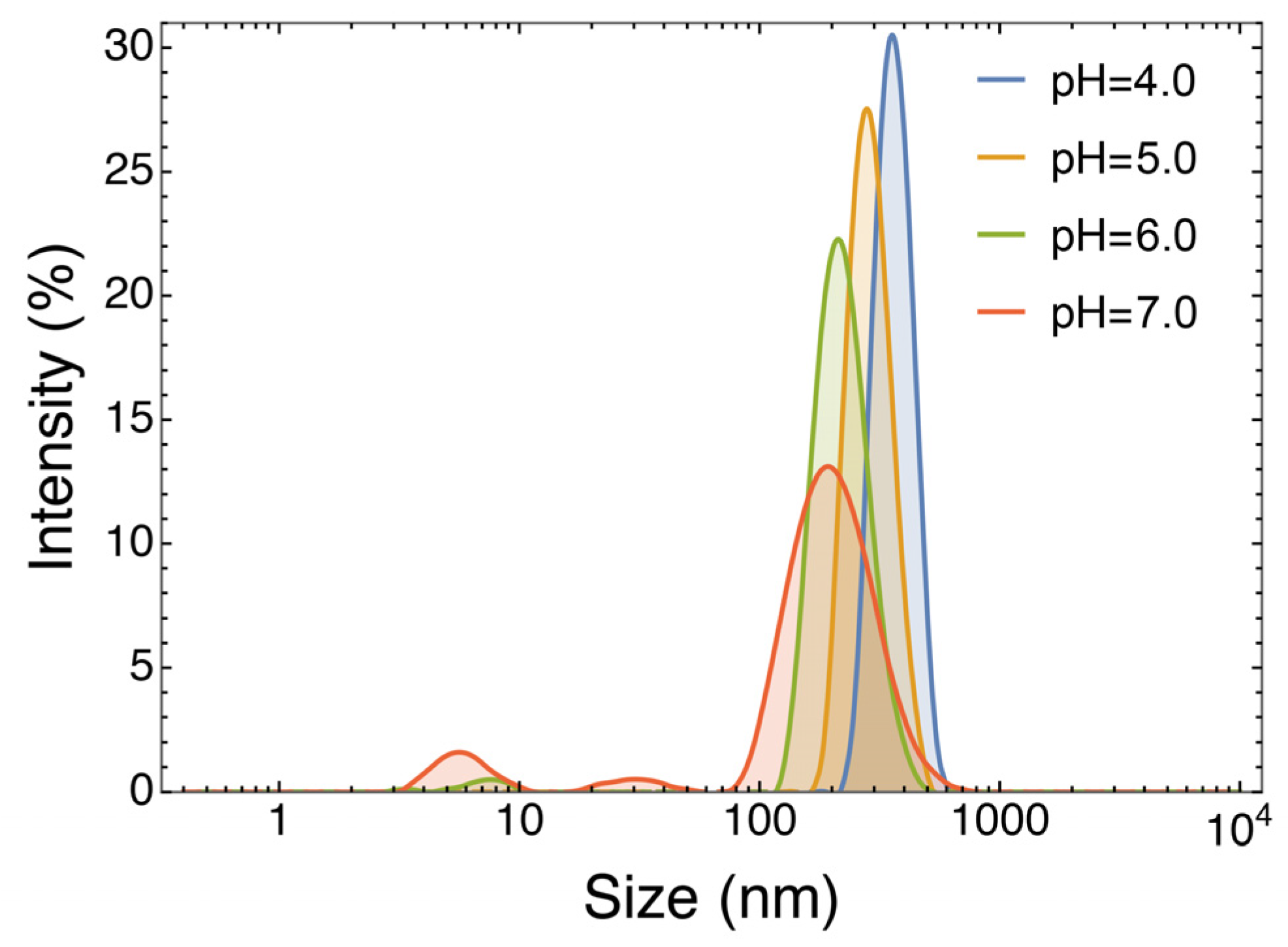
3.2.4. Effect of pH
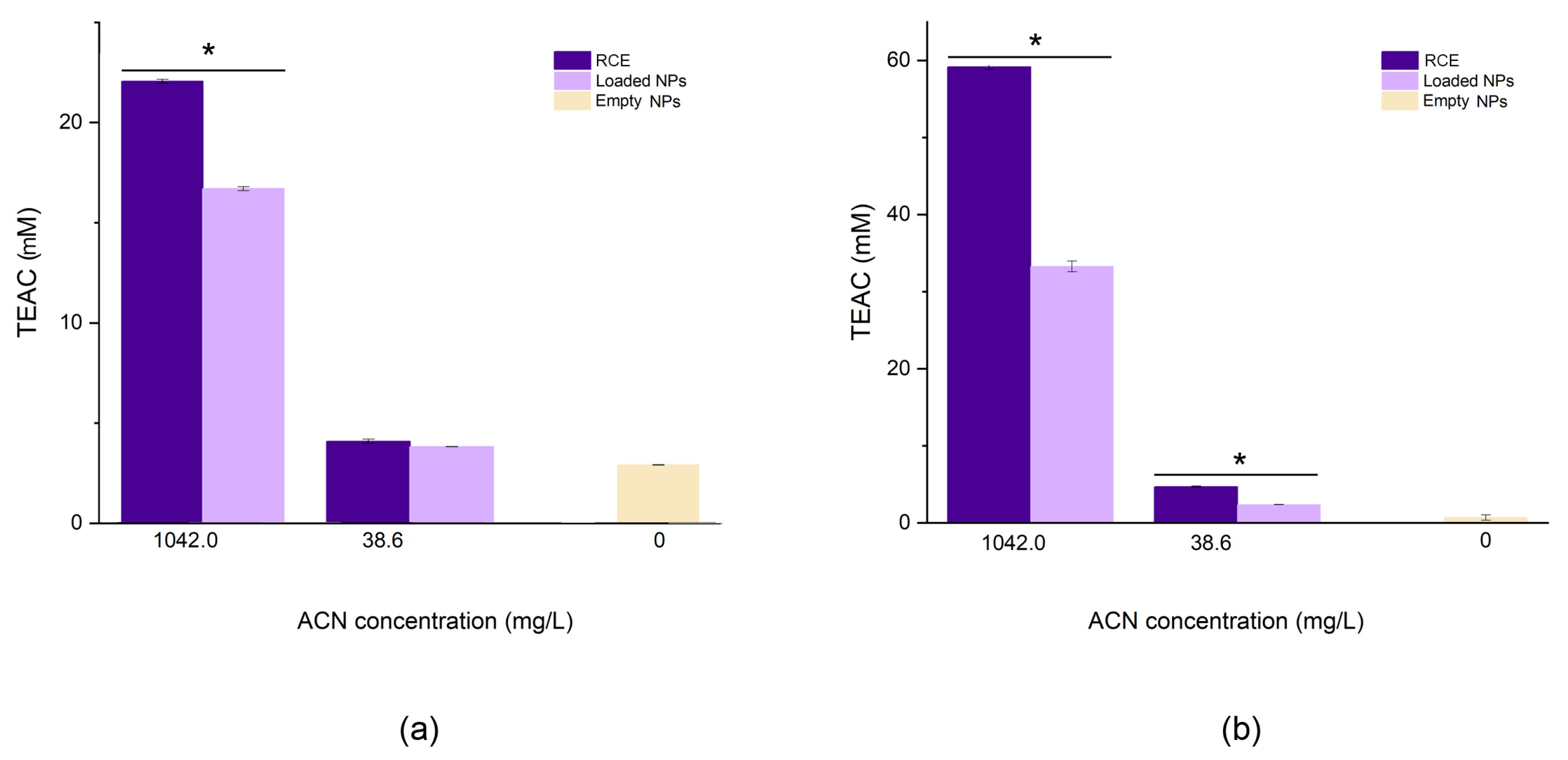
3.2.5. Antioxidant Capacity
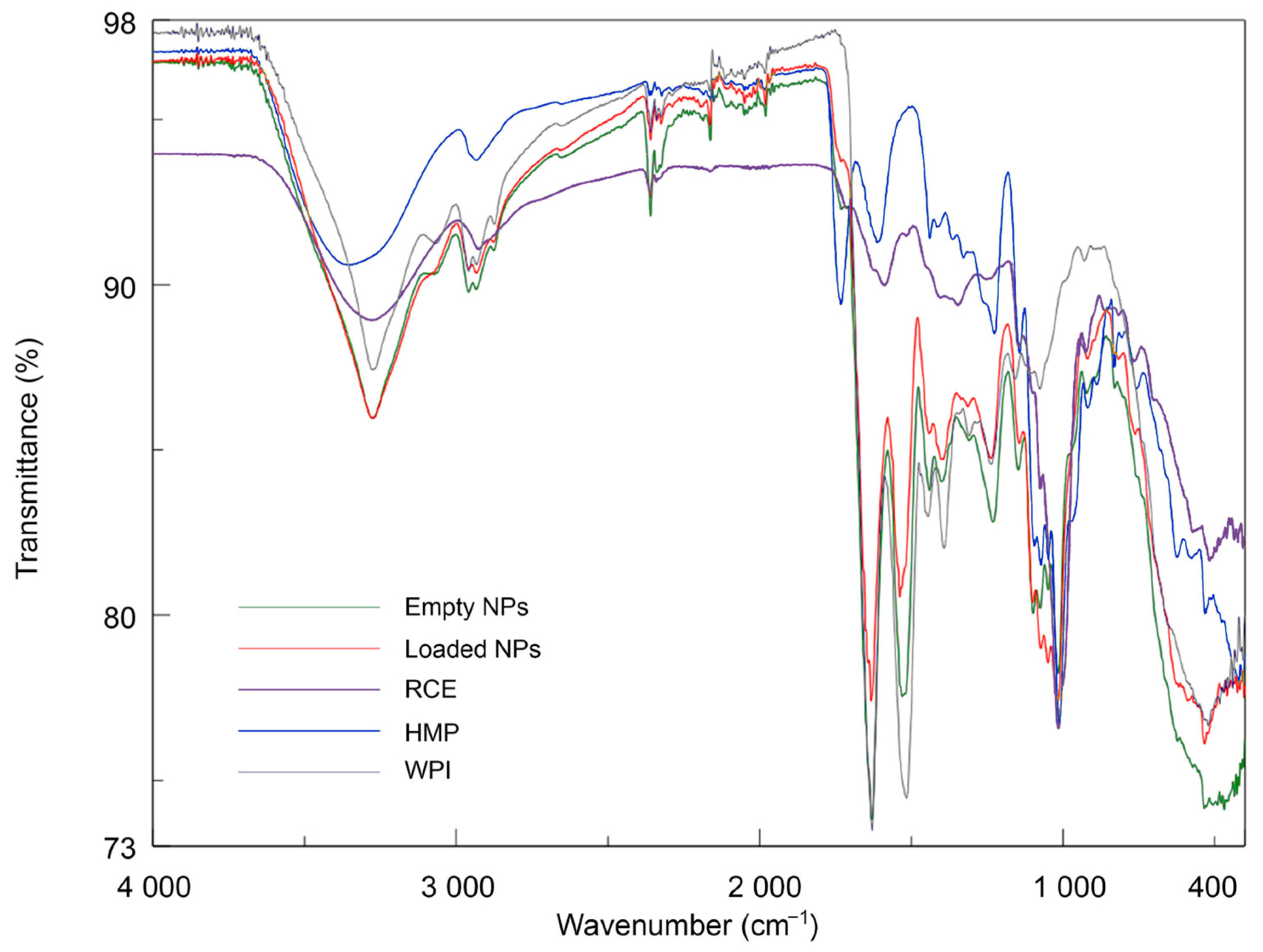
3.3. Fourier-Transform IR (FT-IR) Analysis of Coacervates
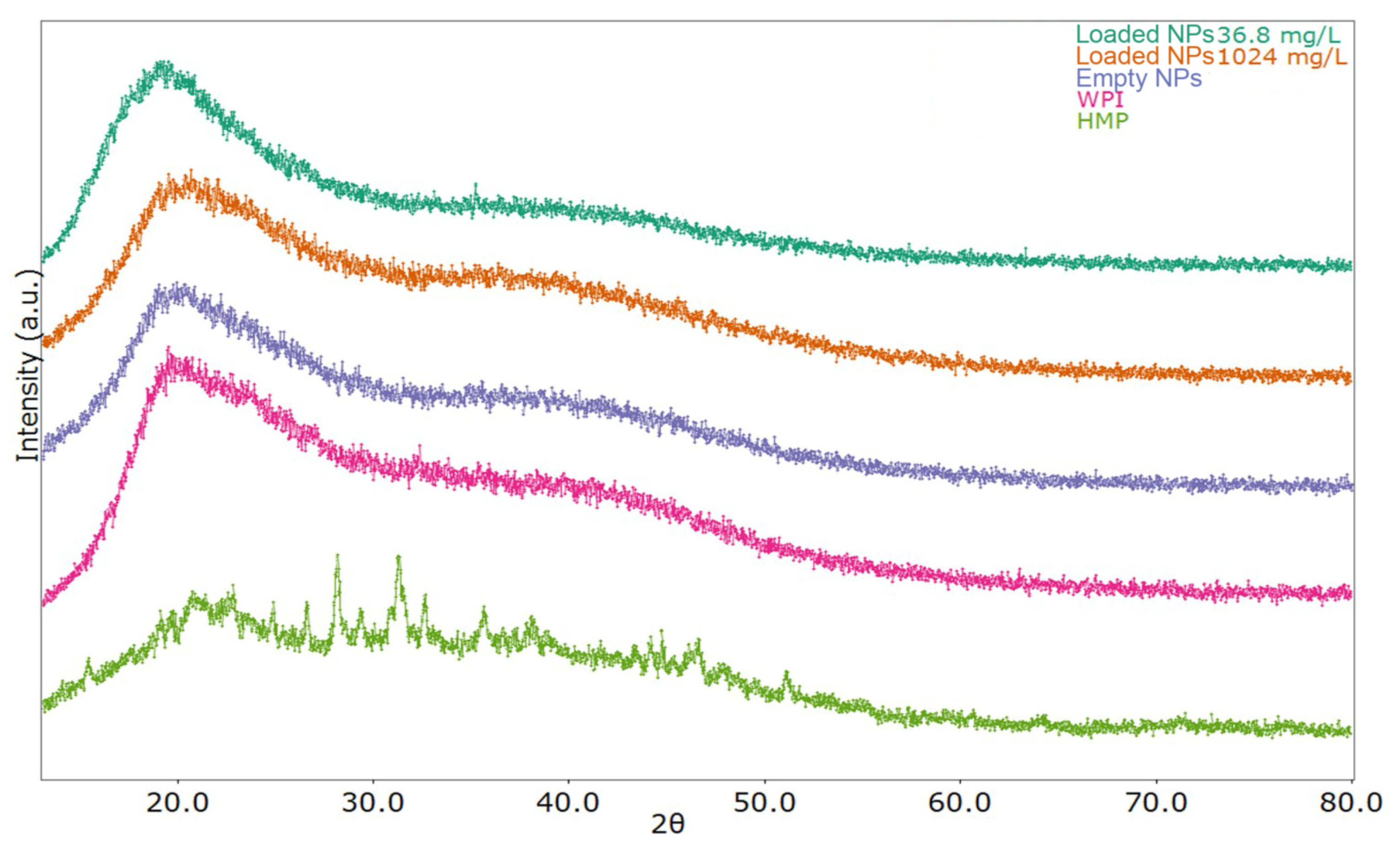
3.4. XRPD Analysis
3.5. Encapsulation Efficiency
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mattioli, R.; Francioso, A.; Mosca, L.; Silva, P. Anthocyanins: A comprehensive review of their chemical properties and health effects on cardiovascular and neurodegenerative diseases. Molecules 2020, 25, 3809. [Google Scholar] [CrossRef] [PubMed]
- Alappat, B.; Alappat, J. Anthocyanin Pigments: Beyond Aesthetics. Molecules 2020, 25, 5500. [Google Scholar] [CrossRef] [PubMed]
- Jackman, R.L.; Yada, R.Y.; Tung, M.A.; Speers, R.A. Anthocyanins as food colorants—A review. J. Food Biochem. 1987, 11, 201–247. [Google Scholar] [CrossRef]
- Salehi, B.; Sharifi-Rad, J.; Cappellini, F.; Reiner, A.; Zorzan, D.; Imran, M.; Sener, B.; Kilic, M.; El-Shazly, M.; Fahmy, N.M.; et al. The therapeutic potential of anthocyanins: Current approaches based on their molecular mechanism of action. Front. Pharmacol. 2020, 11, 1300. [Google Scholar] [CrossRef]
- Ockermann, P.; Headley, L.; Lizio, R.; Hansmann, J. A Review of the properties of anthocyanins and their influence on factors affecting cardiometabolic and cognitive health. Nutrients 2021, 13, 2831. [Google Scholar] [CrossRef]
- Enaru, B.; Dretcanu, G.; Pop, T.D.; Stanila, A.; Diaconeasa, Z. Anthocyanins: Factors affecting their stability and degradation. Antioxidants 2021, 10, 1967. [Google Scholar] [CrossRef] [PubMed]
- Song, J.H.; Yu, Y.; Chen, M.H.; Ren, Z.Y.; Chen, L.; Fu, C.L.; Ma, Z.F.; Li, Z.M. Advancement of protein- and polysaccharide-based biopolymers for anthocyanin encapsulation. Front. Nutr. 2022, 9, 938829. [Google Scholar] [CrossRef]
- Rosales, T.K.O.; Fabi, J.P. Nanoencapsulated anthocyanin as a functional ingredient: Technological application and future perspectives. Colloid Surf. B 2022, 218, 112707. [Google Scholar] [CrossRef]
- Cao, H.; Saroglu, O.; Karadag, A.; Diaconeasa, Z.; Zoccatelli, G.; Conte, C.A.; Gonzalez-Aguilar, G.A.; Ou, J.Y.; Bai, W.B.; Zamarioli, C.M.; et al. Available technologies on improving the stability of polyphenols in food processing. Food Front. 2021, 2, 109–139. [Google Scholar] [CrossRef]
- Comunian, T.A.; Silva, M.P.; Souza, C.J.F. The use of food by-products as a novel for functional foods: Their use as ingredients and for the encapsulation process. Trends Food Sci. Tech. 2021, 108, 269–280. [Google Scholar] [CrossRef]
- Arroyo-Maya, I.J.; McClements, D.J. Biopolymer nanoparticles as potential delivery systems for anthocyanins: Fabrication and properties. Food Res. Int. 2015, 69, 1–8. [Google Scholar] [CrossRef]
- Thongkaew, C.; Gibis, M.; Hinrichs, J.; Weiss, J. Polyphenol interactions with whey protein isolate and whey protein isolate-pectin coacervates. Food Hydrocolloid 2014, 41, 103–112. [Google Scholar] [CrossRef]
- Oliveira, A.; Ruiz-Henestrosa, V.M.P.; von Staszewski, M.; Pilosof, A.M.R.; Pintado, M. Behaviour of cyanidin-3-glucoside, beta-lactoglobulin and polysaccharides nanoparticles in bulk and oil-in-water interfaces. Carbohyd Polym. 2015, 132, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Ghareaghajlou, N.; Hallaj-Nezhadi, S.; Ghasempour, Z. Red cabbage anthocyanins: Stability, extraction, biological activities and applications in food systems. Food Chem. 2021, 365, 130482. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, F.; Primiterra, M.; Angeli, N.; Zoccatelli, G. Microencapsulation by spray-drying of polyphenols extracted from red chicory and red cabbage: Effects on stability and color properties. Food Chem. 2020, 307, 125535. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Durst, R.W.; Wrolstad, R.E. Determination of total monomeric anthocyanin pigment content of fruit juices, beverages, natural colorants, and wines by the pH differential method: Collaborative study. J. Aoac Int. 2005, 88, 1269–1278. [Google Scholar] [CrossRef] [PubMed]
- Bellumori, M.; De Marchi, L.; Mainente, F.; Zanoni, F.; Cecchi, L.; Innocenti, M.; Mulinacci, N.; Zoccatelli, G. A by-product from virgin olive oil production (pate) encapsulated by fluid bed coating: Evaluation of the phenolic profile after shelf-life test and in vitro gastrointestinal digestion. Int. J. Food Sci. Tech. 2021, 56, 3773–3783. [Google Scholar] [CrossRef]
- de la Torre, A.A.S.B.; Henderson, T.; Nigam, P.S.; Owusu-Apenten, R.K. A universally calibrated microplate ferric reducing antioxidant power (FRAP) assay for foods and applications to Manuka honey. Food Chem. 2015, 174, 119–123. [Google Scholar] [CrossRef]
- Wiczkowski, W.; Szawara-Nowak, D.; Topolska, J. Red cabbage anthocyanins: Profile, isolation, identification, and antioxidant activity. Food Res. Int. 2013, 51, 303–309. [Google Scholar] [CrossRef]
- Fang, S.; Lin, F.B.; Qu, D.F.; Liang, X.R.; Wang, L.P. Characterization of Purified Red Cabbage Anthocyanins: Improvement in HPLC Separation and Protective Effect against H2O2-Induced Oxidative Stress in HepG2 Cells. Molecules 2019, 24, 124. [Google Scholar] [CrossRef]
- Steingass, C.B.; Burkhardt, J.; Baumer, V.; Kumar, K.; Mibus-Schoppe, H.; Zinkernagel, J.; Esquivel, P.; Jimenez, V.M.; Schweiggert, R. Characterisation of acylated anthocyanins from red cabbage, purple sweet potato, and Tradescantia pallida leaves as natural food colourants by HPLC-DAD-ESI(+)-QTOF-MS/MS and ESI(+)-MS(n) analysis. Food Chem. 2023, 416, 135601. [Google Scholar] [CrossRef] [PubMed]
- Izzo, L.; Rodriguez-Carrasco, Y.; Pacifico, S.; Castaldo, L.; Narvaez, A.; Ritieni, A. Colon Bioaccessibility under in vitro gastrointestinal digestion of a red cabbage extract chemically profiled through UHPLC-Q-Orbitrap HRMS. Antioxidants 2020, 9, 955. [Google Scholar] [CrossRef] [PubMed]
- Strauch, R.C.; Mengist, M.F.; Pan, K.; Yousef, G.G.; Iorizzo, M.; Brown, A.F.; Lila, M.A. Variation in anthocyanin profiles of 27 genotypes of red cabbage over two growing seasons. Food Chem. 2019, 301, 125289. [Google Scholar] [CrossRef]
- Pelegrine, D.H.G.; Gasparetto, C.A. Whey proteins solubility as function of temperature and pH. LWT Food Sci. Technol. 2005, 38, 77–80. [Google Scholar] [CrossRef]
- Jones, O.; Decker, E.A.; McClements, D.J. Thermal analysis of beta-lactoglobulin complexes with pectins or carrageenan for production of stable biopolymer particles. Food Hydrocolloid 2010, 24, 239–248. [Google Scholar] [CrossRef]
- Gentes, M.C.; St-Gelais, D.; Turgeon, S.L. Stabilization of whey protein isolate-pectin complexes by heat. J. Agric. Food Chem. 2010, 58, 7051–7058. [Google Scholar] [CrossRef]
- Grgic, J.; Selo, G.; Planinic, M.; Tisma, M.; Bucic-Kojic, A. Role of the encapsulation in bioavailability of phenolic compounds. Antioxidants 2020, 9, 923. [Google Scholar] [CrossRef]
- Krzeminski, A.; Prell, K.A.; Weiss, J.; Hinrichs, J. Environmental response of pectin-stabilized whey protein aggregates. Food Hydrocolloid 2014, 35, 332–340. [Google Scholar] [CrossRef]
- Girard, M.; Sanchez, C.; Laneuville, S.I.; Turgeon, S.L.; Gauthier, S.E. Associative phase separation of beta-lactoglobulin/pectin solutions: A kinetic study by small angle static light scattering. Colloid Surf. B 2004, 35, 15–22. [Google Scholar] [CrossRef]
- Di Meo, F.; Garcia, J.C.S.; Dangles, O.; Trouillas, P. Highlights on Anthocyanin Pigmentation and Copigmentation: A Matter of Flavonoid pi-Stacking Complexation To Be Described by DFT-D. J. Chem. Theory Comput. 2012, 8, 2034–2043. [Google Scholar] [CrossRef]
- Devi, N.; Sarmah, M.; Khatun, B.; Maji, T.K. Encapsulation of active ingredients in polysaccharide-protein complex coacervates. Adv. Colloid Interface Sci. 2017, 239, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, A.; Ungell, A.L.; Knutson, L.; Lennernas, H. Characterization of fluids from the stomach and proximal jejunum in men and women. Pharm. Res. 1997, 14, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Tena, N.; Martin, J.; Asuero, A.G. State of the art of anthocyanins: Antioxidant activity, sources, bioavailability, and therapeutic effect in human health. Antioxidants 2020, 9, 451. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, F.; Vakarelova, M.; Zoccatelli, G. Development and characterization of astaxanthin-containing whey protein-based nanoparticles. Mar. Drugs 2019, 17, 627. [Google Scholar] [CrossRef]
- Ozgen, M.; Reese, R.N.; Tulio, A.Z.; Scheerens, J.C.; Miller, A.R. Modified 2,2-azino-bis-3-ethylbenzothiazoline-6-sulfonic acid (ABTS) method to measure antioxidant capacity of selected small fruits and comparison to ferric reducing antioxidant power (FRAP) and 2,2′-diphenyl-1-picrylhydrazyl (DPPH) methods. J. Agr. Food Chem. 2006, 54, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Barth, A. Infrared spectroscopy of proteins. Biochim. Biophys. Acta 2007, 1767, 1073–1101. [Google Scholar]
- Koziol, A.; Sroda-Pomianek, K.; Gorniak, A.; Wikiera, A.; Cyprych, K.; Malik, M. Structural determination of pectins by spectroscopy methods. Coatings 2022, 12, 546. [Google Scholar] [CrossRef]
- Fellah, A.; Anjukandi, P.; Waterland, M.R.; Williams, M.A.K. Determining the degree of methylesterification of pectin by ATR/FT-IR: Methodology optimisation and comparison with theoretical calculations. Carbohyd Polym. 2009, 78, 847–853. [Google Scholar] [CrossRef]
- Pismenskaya, N.; Sarapulova, V.; Klevtsova, A.; Mikhaylin, S.; Bazinet, L. Adsorption of anthocyanins by cation and anion exchange resins with aromatic and aliphatic polymer matrices. Int. J. Mol. Sci. 2020, 21, 7874. [Google Scholar] [CrossRef]
- Guerrero, P.; Kerry, J.P.; de la Caba, K. FTIR characterization of protein-polysaccharide interactions in extruded blends. Carbohyd Polym. 2014, 111, 598–605. [Google Scholar] [CrossRef]
- Carpenter, J.F.; Crowe, J.H. An infrared spectroscopic study of the interactions of carbohydrates with dried proteins. Biochemistry 1989, 28, 3916–3922. [Google Scholar] [CrossRef] [PubMed]
- Shakoury, N.; Aliyari, M.A.; Salami, M.; Emam-Djomeh, Z.; Vardhanabhuti, B.; Moosavi-Movahedi, A.A. Encapsulation of propolis extract in whey protein nanoparticles. LWT Food Sci. Technol. 2022, 158, 113138. [Google Scholar] [CrossRef]
- Kumar, A.; Chauhan, G.S. Extraction and characterization of pectin from apple pomace and its evaluation as lipase (steapsin) inhibitor. Carbohyd Polym. 2010, 82, 454–459. [Google Scholar] [CrossRef]
- Mainente, F.; Piovan, A.; Zanoni, F.; Chignola, R.; Cerantola, S.; Faggin, S.; Giron, M.C.; Filippini, R.; Seraglia, R.; Zoccatelli, G. Spray-drying microencapsulation of an extract from Tilia tomentosa Moench flowers: Physicochemical characterization and in vitro intestinal activity. Plant. Food Hum. Nutr. 2022, 77, 467–473. [Google Scholar] [CrossRef] [PubMed]










| Method | Blending Order | Heat Treatment | Z-Average (nm) | PDI | Diameter (nm) | ζ-Potential (mV) |
|---|---|---|---|---|---|---|
| 1 | WPI + RCE + HMP (1:0.2:1) | None | 396.80 ± 21.78 | 0.225 ± 0.03 | 385.85 ± 7.26 a | −16.83 ± 0.33 ab |
| 2 | WPI + RCE + HMP (1:0.2:1) | WPI, 65 °C, before blending | 323.71 ± 12.97 | 0.170 ± 0.09 | 337.28 ± 5.33 b | −17.76 ± 0.40 a |
| 3 | WPI + HMP + RCE (1:1:0.2) | WPI, 65 °C before blending | 428.61 ± 35.35 | 0.308 ± 0.04 | 384.31 ± 11.69 a | −17.99 ± 0.15 a |
| 4 | WPI + HMP + RCE (1:1:0.2) | Whole mix, 65 °C, after blending | 507.62 ± 13.8 | 0.283 ± 0.03 | 445.20 ± 2.23 c | −15.86 ± 0.38 b |
| ACN Concentration (mg/L) | Z-Average (nm) | PDI | Diameter (nm) | ζ-Potential (mV) |
|---|---|---|---|---|
| 0 | 320.4 ± 5.2 | 0.09 ± 0.1 | 337.2 ± 6.7 a | −17.20 ± 1.0 a |
| 38.6 | 323.7 ± 13.0 | 0.170 ± 0.1 | 337.3 ± 5.3 a | −15.64 ± 0.0 b |
| 115.9 | 322.7 ± 9.7 | 0.160 ± 0.1 | 335.9 ± 6.4 a | −15.52 ± 0.1 b |
| 347.4 | 343.0 ± 2.1 | 0.050 ± 0.0 | 356.8 ± 0.4 b | −15.34 ± 0.1 b |
| 1042.0 | 365.0 ± 8.5 | 0.250 ± 0.1 | 368.2 ± 3.0 b | −14.24 ± 0.3 c |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fierri, I.; De Marchi, L.; Chignola, R.; Rossin, G.; Bellumori, M.; Perbellini, A.; Mancini, I.; Romeo, A.; Ischia, G.; Saorin, A.; et al. Nanoencapsulation of Anthocyanins from Red Cabbage (Brassica oleracea L. var. Capitata f. rubra) through Coacervation of Whey Protein Isolate and Apple High Methoxyl Pectin. Antioxidants 2023, 12, 1757. https://doi.org/10.3390/antiox12091757
Fierri I, De Marchi L, Chignola R, Rossin G, Bellumori M, Perbellini A, Mancini I, Romeo A, Ischia G, Saorin A, et al. Nanoencapsulation of Anthocyanins from Red Cabbage (Brassica oleracea L. var. Capitata f. rubra) through Coacervation of Whey Protein Isolate and Apple High Methoxyl Pectin. Antioxidants. 2023; 12(9):1757. https://doi.org/10.3390/antiox12091757
Chicago/Turabian StyleFierri, Ilaria, Laura De Marchi, Roberto Chignola, Giacomo Rossin, Maria Bellumori, Anna Perbellini, Ines Mancini, Alessandro Romeo, Gloria Ischia, Asia Saorin, and et al. 2023. "Nanoencapsulation of Anthocyanins from Red Cabbage (Brassica oleracea L. var. Capitata f. rubra) through Coacervation of Whey Protein Isolate and Apple High Methoxyl Pectin" Antioxidants 12, no. 9: 1757. https://doi.org/10.3390/antiox12091757