
Redox Regulation of Nrf2 in Cisplatin-Induced Kidney Injury

 , ,
, , 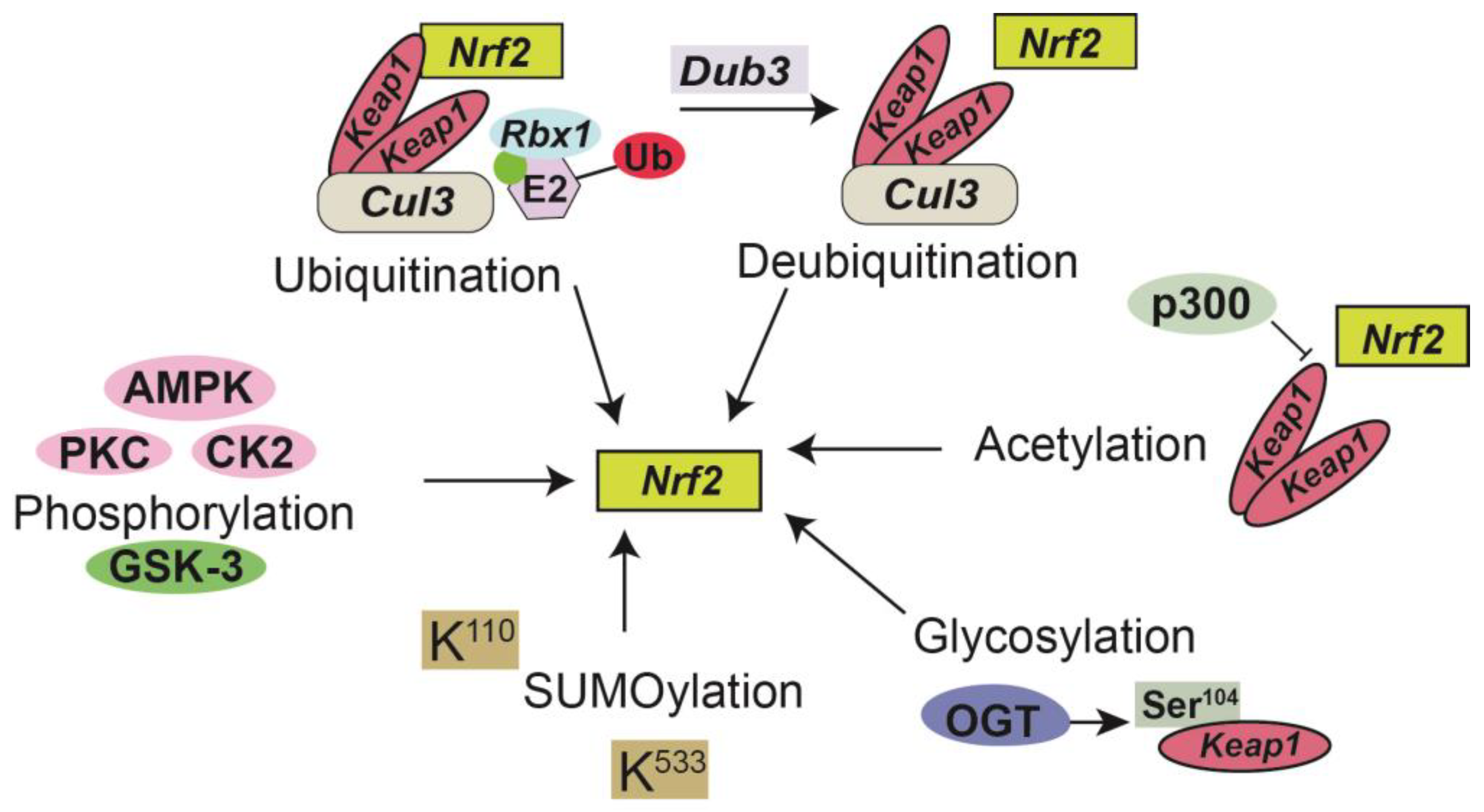{kind=link}
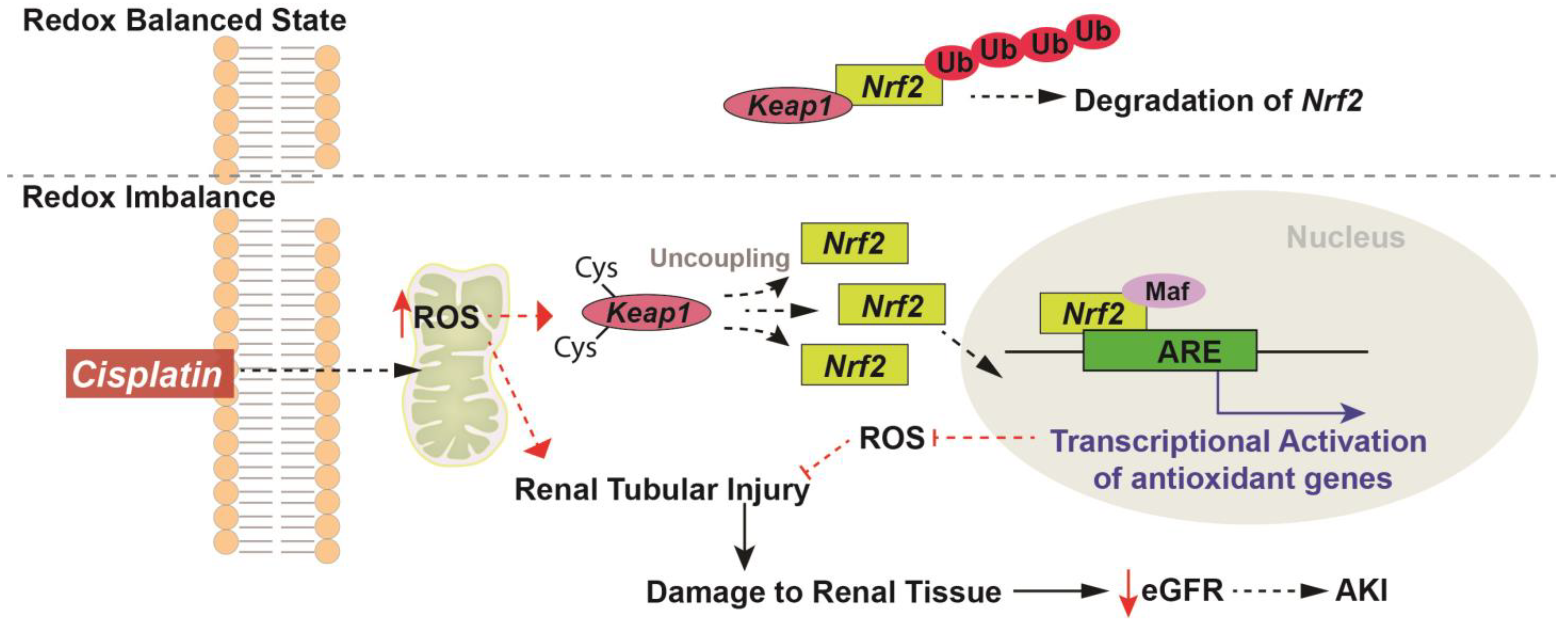{kind=link}
Abstract
:1. Introduction
2. Chemotherapy-Induced Kidney Injury
3. Role of Nrf2 in Antineoplastic Therapy
4. Signaling and Post-Translational Modifications of Nrf2
5. Redox Regulation of Nrf2 in Cisplatin-Induced Kidney Injury
6. Role of Nrf2 in Autophagy and Cisplatin-Induced Kidney Injury
7. Redox Based Interventions for Nrf2 Modulation in Cisplatin-Induced Kidney Injury
8. Limitations and off Target Effects of Nrf2 in Kidney Injury
9. Nrf2-Targeted Cancer Clinical Trials and Kidney Outcomes
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ahles, T.A. Brain vulnerability to chemotherapy toxicities. Psycho-Oncology 2012, 21, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Ahles, T.A.; Saykin, A.J.; McDonald, B.C.; Furstenberg, C.T.; Cole, B.F.; Hanscom, B.S.; Mulrooney, T.J.; Schwartz, G.N.; Kaufman, P.A. Cognitive function in breast cancer patients prior to adjuvant treatment. Breast Cancer Res. Treat. 2008, 110, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Bodenheimer, H.; Charland, C.; Leith, J. Alteration of rat Kupffer cell function following mitomycin-C administration. J. Leukoc. Biol. 1988, 43, 265–270. [Google Scholar] [CrossRef]
- Adachi, Y.; Arii, S.; Funaki, N.; Higashitsuji, H.; Fujita, S.; Furutani, M.; Mise, M.; Zhang, W.; Tobe, T. Tumoricidal activity of Kupffer cells augmented by anticancer drugs. Life Sci. 1992, 51, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Ramadori, G.; Cameron, S. Effects of systemic chemotherapy on the liver Ann Hepatol. Ann. Hepatol. 2010, 9, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, G.; Faqah, A.; Konowitz, N.; Lu, H.; Kuruvilla, A.; Adhikari, S. Acute Pancreatitis Related to a Chemotherapy Drug. World J. Oncol. 2017, 8, 18–19. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.-J.; Zheng, J.; Dang, F.-T.; Wan, Y.-M.; Yang, J. Acute pancreatitis induced by combination chemotherapy used for the treatment of acute myeloid leukemia. Medicine 2020, 99, e21848. [Google Scholar] [CrossRef]
- Oh, G.-S.; Kim, H.-J.; Shen, A.; Bin Lee, S.; Khadka, D.; Pandit, A.; So, H.-S. Cisplatin-induced Kidney Dysfunction and Perspectives on Improving Treatment Strategies. Electrolytes Blood Press. 2014, 12, 55–65. [Google Scholar] [CrossRef]
- Sharbaf, F.G.; Farhangi, H.; Assadi, F. Prevention of Chemotherapy-Induced Nephrotoxicity in Children with Cancer. Int. J. Prev. Med. 2017, 8, 76. [Google Scholar] [CrossRef]
- Cosmai, L.; Porta, C.; Foramitti, M.; Perrone, V.; Mollica, L.; Gallieni, M.; Capasso, G. Preventive strategies for acute kidney injury in cancer patients. Clin. Kidney J. 2021, 14, 70–83. [Google Scholar] [CrossRef]
- Chiruvella, V.; Annamaraju, P.; Guddati, A.K. Management of nephrotoxicity of chemotherapy and targeted agents: 2020. Am. J. Cancer Res. 2020, 10, 4151–4164. [Google Scholar] [PubMed]
- Rosner, M.H.; Jhaveri, K.D.; McMahon, B.A.; Perazella, M.A. Onconephrology: The intersections between the kidney and cancer. CA Cancer J. Clin. 2021, 71, 47–77. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M.C. Oxygen free radicals and iron in relation to biology and medicine: Some problems and concepts. Arch. Biochem. Biophys. 1986, 246, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Spitz, D.R.; Azzam, E.I.; Li, J.J.; Gius, D. Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: A unifying concept in stress response biology. Cancer Metastasis Rev. 2004, 23, 311–322. [Google Scholar] [CrossRef]
- Cadenas, E.; Boveris, A.; Ragan, C.I.; Stoppani, A.O. Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef-heart mitochondria. Arch. Biochem. Biophys. 1977, 180, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Mapuskar, K.A.; Wen, H.; Holanda, D.G.; Rastogi, P.; Steinbach, E.; Han, R.; Coleman, M.C.; Attanasio, M.; Riley, D.P.; Spitz, D.R.; et al. Persistent increase in mitochondrial superoxide mediates cisplatin-induced chronic kidney disease. Redox Biol. 2019, 20, 98–106. [Google Scholar] [CrossRef]
- Cimmino, T.P.; Ammendola, R.; Cattaneo, F.; Esposito, G. NOX Dependent ROS Generation and Cell Metabolism. Int. J. Mol. Sci. 2023, 24, 2086. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Hotter, G.; Viñas, J.L.; Sola, A. Cisplatin upregulates mitochondrial nitric oxide synthase and peroxynitrite formation to promote renal injury. Toxicol. Appl. Pharmacol. 2009, 234, 236–246. [Google Scholar] [CrossRef]
- Barakat, L.A.; Barakat, N.; Zakaria, M.M.; Khirallah, S.M. Protective role of zinc oxide nanoparticles in kidney injury induced by cisplatin in rats. Life Sci. 2020, 262, 118503. [Google Scholar] [CrossRef]
- Fouad, A.A.; Al-Sultan, A.I.; Refaie, S.M.; Yacoubi, M.T. Coenzyme Q10 treatment ameliorates acute cisplatin nephrotoxicity in mice. Toxicology 2010, 274, 49–56. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Adinolfi, S.; Patinen, T.; Deen, A.J.; Pitkänen, S.; Härkönen, J.; Kansanen, E.; Küblbeck, J.; Levonen, A.-L. The KEAP1-NRF2 pathway: Targets for therapy and role in cancer. Redox Biol. 2023, 63, 102726. [Google Scholar] [CrossRef] [PubMed]
- Lingappan, K. NF-κB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B.; Liby, K.T. NRF2 and cancer: The good, the bad and the importance of context. Nat. Rev. Cancer 2012, 12, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Oh, G.-S.; Kim, H.-J.; Shen, A.; Lee, S.-B.; Yang, S.-H.; Shim, H.; Cho, E.-Y.; Kwon, K.-B.; Kwak, T.H.; So, H.-S. New Therapeutic Concept of NAD Redox Balance for Cisplatin Nephrotoxicity. BioMed Res. Int. 2016, 2016, 4048390. [Google Scholar] [CrossRef]
- Dewaeles, E.; Carvalho, K.; Fellah, S.; Sim, J.; Boukrout, N.; Caillierez, R.; Ramakrishnan, H.; Van der Hauwaert, C.; Shankara, J.V.; Martin, N.; et al. Istradefylline protects from cisplatin-induced nephrotoxicity and peripheral neuropathy while preserving cisplatin antitumor effects. J. Clin. Investig. 2022, 132, e152924. [Google Scholar] [CrossRef]
- Holditch, S.J.; Brown, C.N.; Lombardi, A.M.; Nguyen, K.N.; Edelstein, C.L. Recent Advances in Models, Mechanisms, Biomarkers, and Interventions in Cisplatin-Induced Acute Kidney Injury. Int. J. Mol. Sci. 2019, 20, 3011. [Google Scholar] [CrossRef]
- Heney, D.; Wheeldon, J.; Rushworth, P.; Chapman, C.; Lewis, I.J.; Bailey, C.C. Progressive renal toxicity due to ifosfamide. Arch. Dis. Child. 1991, 66, 966–970. [Google Scholar] [CrossRef]
- Oberlin, O.; Fawaz, O.; Rey, A.; Niaudet, P.; Ridola, V.; Orbach, D.; Bergeron, C.; Defachelles, A.S.; Gentet, J.-C.; Schmitt, C.; et al. Long-term evaluation of Ifosfamide-related nephrotoxicity in children. J. Clin. Oncol. 2009, 27, 5350–5355. [Google Scholar] [CrossRef]
- Skinner, R.; Kaplan, R.; Nathan, P.C. Renal and pulmonary late effects of cancer therapy. Semin. Oncol. 2013, 40, 757–773. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V.K.; Lewis, I.J.; Aparicio, S.R.; Heney, D.; Hale, J.P.; Bailey, C.C.; Kinsey, S.E. Progressive glomerular toxicity of ifosfamide in children. Med. Pediatr. Oncol. 1996, 27, 149–155. [Google Scholar] [CrossRef]
- Latcha, S.; Gupta, M.; Lin, I.-H.; Jaimes, E.A. High Dose Methotrexate-Induced Acute Kidney Injury: Incidence, Risk Factors, and Recovery. Kidney Int. Rep. 2023, 8, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Sparks, J.A.; Vanni, K.M.M.; Sparks, M.A.; Xu, C.; Santacroce, L.M.; Glynn, R.J.; Ridker, P.M.; Solomon, D.H. Effect of Low-Dose Methotrexate on eGFR and Kidney Adverse Events: A Randomized Clinical Trial. J. Am. Soc. Nephrol. 2021, 32, 3197–3207. [Google Scholar] [CrossRef] [PubMed]
- Ayza, M.A.; Zewdie, K.A.; Yigzaw, E.F.; Ayele, S.G.; Tesfaye, B.A.; Tafere, G.G.; Abrha, M.G. Potential Protective Effects of Antioxidants against Cyclophosphamide-Induced Nephrotoxicity. Int. J. Nephrol. 2022, 2022, 5096825. [Google Scholar] [CrossRef]
- Zhao, N.; Xu, Q.; Wang, M.; Fei, X.; Pan, Y.; Chen, X.; Ma, S. Mechanism of kidney injury caused by bevacizumab in rats. Int. J. Clin. Exp. Pathol. 2014, 7, 8675–8683. [Google Scholar] [PubMed]
- Nakai, K.; Umehara, M.; Minamida, A.; Yamauchi-Sawada, H.; Sunahara, Y.; Matoba, Y.; Okuno-Ozeki, N.; Nakamura, I.; Nakata, T.; Yagi-Tomita, A.; et al. Streptozotocin induces renal proximal tubular injury through p53 signaling activation. Sci. Rep. 2023, 13, 8705. [Google Scholar] [CrossRef]
- Glezerman, I.; Kris, M.; Miller, V.; Seshan, S.; Flombaum, C. Gemcitabine nephrotoxicity and hemolytic uremic syndrome: Report of 29 cases from a single institution. Clin. Nephrol. 2009, 71, 130–139. [Google Scholar] [CrossRef]
- Chan, B.A.; Coward, J.I.G. Chemotherapy advances in small-cell lung cancer. J. Thorac. Dis. 2013, 5 (Suppl. 5), S565–S578. [Google Scholar] [CrossRef]
- Einhorn, L.H.; Donohue, J. Cis-diamminedichloroplatinum, vinblastine, and bleomycin combination chemotherapy in disseminated testicular cancer. Ann. Intern. Med. 1977, 87, 293–298. [Google Scholar] [CrossRef]
- Wang, S.; Xie, J.; Li, J.; Liu, F.; Wu, X.; Wang, Z. Cisplatin suppresses the growth and proliferation of breast and cervical cancer cell lines by inhibiting integrin β5-mediated glycolysis. Am. J. Cancer. Res. 2016, 6, 1108–1117. [Google Scholar] [PubMed]
- McSweeney, K.R.; Gadanec, L.K.; Qaradakhi, T.; Ali, B.A.; Zulli, A.; Apostolopoulos, V. Mechanisms of Cisplatin-Induced Acute Kidney Injury: Pathological Mechanisms, Pharmacological Interventions, and Genetic Mitigations. Cancers 2021, 13, 1572. [Google Scholar] [CrossRef] [PubMed]
- Price, P.M.; Yu, F.; Kaldis, P.; Aleem, E.; Nowak, G.D.A.; Safirstein, R.L.; Megyesi, J. Dependence of cisplatin-induced cell death in vitro and in vivo on cyclin-dependent kinase 2. J. Am. Soc. Nephrol. 2006, 17, 2434–2442. [Google Scholar] [CrossRef] [PubMed]
- Pabla, N.; Dong, Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Callejo, A.; Sedó-Cabezón, L.; Juan, I.D.; Llorens, J. Cisplatin-Induced Ototoxicity: Effects, Mechanisms and Protection Strategies. Toxics 2015, 3, 268–293. [Google Scholar] [CrossRef] [PubMed]
- Rybak, L.P.; Mukherjea, D.; Jajoo, S.; Ramkumar, V. Cisplatin ototoxicity and protection: Clinical and experimental studies. Tohoku J. Exp. Med. 2009, 219, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Sheth, S.; Mukherjea, D.; Rybak, L.P.; Ramkumar, V. Mechanisms of Cisplatin-Induced Ototoxicity and Otoprotection. Front. Cell Neurosci. 2017, 11, 338. [Google Scholar] [CrossRef]
- Dos Santos, N.A.G.; Rodrigues, M.A.C.; Martins, N.M.; dos Santos, A.C. Cisplatin-induced nephrotoxicity and targets of nephroprotection: An update. Arch. Toxicol. 2012, 86, 1233–1250. [Google Scholar] [CrossRef]
- Dos Santos, N.A.G.; Ferreira, R.S.; dos Santos, A.C. Overview of cisplatin-induced neurotoxicity and ototoxicity, and the protective agents. Food Chem. Toxicol. 2020, 136, 111079. [Google Scholar] [CrossRef]
- Cetinkaya-Fisgin, A.; Luan, X.; Reed, N.; Jeong, Y.E.; Oh, B.C.; Hoke, A. Cisplatin induced neurotoxicity is mediated by Sarm1 and calpain activation. Sci. Rep. 2020, 10, 21889. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.P.; Tadagavadi, R.K.; Ramesh, G.; Reeves, W.B. Mechanisms of Cisplatin nephrotoxicity. Toxins 2010, 2, 2490–2518. [Google Scholar] [CrossRef] [PubMed]
- Mapuskar, K.A.; Steinbach, E.J.; Zaher, A.; Riley, D.P.; Beardsley, R.A.; Keene, J.L.; Holmlund, J.T.; Anderson, C.M.; Zepeda-Orozco, D.; Buatti, J.M.; et al. Mitochondrial Superoxide Dismutase in Cisplatin-Induced Kidney Injury. Antioxidants 2021, 10, 1329. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Anderson, M.D.; Sutton, T.A. Pathophysiology of acute kidney injury. Compr. Physiol. 2012, 2, 1303–1353. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, N.; Nakai, K.; Nakata, T.; Nakamura, I.; Kirita, Y.; Matoba, S.; Humphreys, B.D.; Tamagaki, K.; Kusaba, T. Cumulative DNA damage by repeated low-dose cisplatin injection promotes the transition of acute to chronic kidney injury in mice. Sci. Rep. 2021, 11, 20920. [Google Scholar] [CrossRef]
- Oda, H.; Mizuno, T.; Ikejiri, M.; Nakamura, M.; Tsunoda, A.; Ishihara, M.; Saito, K.; Tamaru, S.; Yamashita, Y.; Nishimura, Y.; et al. Risk factors for cisplatin-induced acute kidney injury: A pilot study on the usefulness of genetic variants for predicting nephrotoxicity in clinical practice. Mol. Clin. Oncol. 2020, 13, 58. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Cronstein, B.N. Understanding the mechanisms of action of methotrexate: Implications for the treatment of rheumatoid arthritis. Bull. NYU Hosp. Jt. Dis. 2007, 65, 168–173. [Google Scholar]
- Houser, M.E.L.; Stewart, J.R.; Brewer, J.D. Psoriasis Patients Treated with Methotrexate Have an Increased Risk of Nonmelanoma Skin Cancer: A Systematic Review and Meta-Analysis. Cureus 2023, 15, e37174. [Google Scholar] [CrossRef]
- Radwan, S.M.; Alqulaly, M.; Elsaeed, M.Y.; Elshora, S.Z.; Atwa, A.H.; Wasfey, E.F. L-carnitine reverses methotrexate-induced nephrotoxicity in experimental rat model: Insight on SIRT1/PGC-1α/Nrf2/HO-1 axis. J. Appl. Toxicol. 2023. [Google Scholar] [CrossRef]
- Pannu, A.K. Methotrexate overdose in clinical practice. Curr. Drug Metab. 2019, 20, 714–719. [Google Scholar] [CrossRef]
- Aldossary, S.A.; Chohan, M.S.; Mohaini, M.A.; Tasleem Rasool, S. Capsaicin ameliorate the nephrotoxicity induced by methotrexate. Pak. J. Pharm. Sci. 2021, 34, 2191–2195. [Google Scholar] [PubMed]
- Haubitz, M.; Bohnenstengel, F.; Brunkhorst, R.; Schwab, M.; Hofmann, U.; Busse, D. Cyclophosphamide pharmacokinetics and dose requirements in patients with renal insufficiency. Kidney Int. 2002, 61, 1495–1501. [Google Scholar] [CrossRef]
- Emadi, A.; Jones, R.J.; Brodsky, R.A. Cyclophosphamide and cancer: Golden anniversary. Nat. Rev. Clin. Oncol. 2009, 6, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Zalupski, M.; Baker, L.H. Ifosamide. Gynecol. Oncol. 1988, 80, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Boogaard, W.M.C.v.D.; Komninos, D.S.J.; Vermeij, W.P. Chemotherapy Side-Effects: Not All DNA Damage Is Equal. Cancers 2022, 14, 627. [Google Scholar] [CrossRef] [PubMed]
- Willits, I.; Price, L.; Parry, A.; Tilby, M.J.; Ford, D.; Cholerton, S.; Pearson, A.D.J.; Boddy, A.V. Pharmacokinetics and metabolism of ifosfamide in relation to DNA damage assessed by the COMET assay in children with cancer. Br. J. Cancer 2005, 92, 1626–1635. [Google Scholar] [CrossRef]
- Martinez, D.; Rodelo, J.; García, S.P. Ifosfamide as a Cause of Fanconi Syndrome. Cureus 2022, 14, e22755. [Google Scholar] [CrossRef]
- Ensergueix, G.; Pallet, N.; Joly, D.; Levi, C.; Chauvet, S.; Trivin, C.; Augusto, J.-F.; Boudet, R.; Aboudagga, H.; Touchard, G.; et al. Ifosfamide nephrotoxicity in adult patients. Clin. Kidney J. 2020, 13, 660–665. [Google Scholar] [CrossRef]
- Dorairajan, L.N.; Manikandan, R.; Kumar, S. Hemorrhagic cystitis: A challenge to the urologist. Indian J. Urol. 2010, 26, 159–166. [Google Scholar] [CrossRef]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharm. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef]
- Afsar, T.; Razak, S.; Almajwal, A.; Al-Disi, D. Doxorubicin-induced alterations in kidney functioning, oxidative stress, DNA damage, and renal tissue morphology; Improvement by Acacia hydaspica tannin-rich ethyl acetate fraction. Saudi J. Biol. Sci. 2020, 27, 2251–2260. [Google Scholar] [CrossRef] [PubMed]
- Danmaigoro, A.; Selvarajah, G.T.; Noor, M.H.M.; Mahmud, R.; Abu Bakar, Z. Toxicity and Safety Evaluation of Doxorubicin-Loaded Cockleshell-Derived Calcium Carbonate Nanoparticle in Dogs. Adv. Pharmacol. Sci. 2018, 2018, 4848602. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Ricksten, S.-E.; Bragadottir, G.; Redfors, B.; Nordquist, L. Renal oxygenation and haemodynamics in acute kidney injury and chronic kidney disease. Clin. Exp. Pharmacol. Physiol. 2013, 40, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Palm, F.; Nordquist, L. Renal tubulointerstitial hypoxia: Cause and consequence of kidney dysfunction. Clin. Exp. Pharmacol. Physiol. 2011, 38, 474–480. [Google Scholar] [CrossRef]
- Seethapathy, H.; Herrmann, S.M.; Sise, M.E. Immune Checkpoint Inhibitors and Kidney Toxicity: Advances in Diagnosis and Management. Kidney Med. 2021, 3, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, D.; Capili, A.; Choi, M.E. Mitochondrial dysfunction in kidney injury, inflammation, and disease: Potential therapeutic approaches. Kidney Res. Clin. Pract. 2020, 39, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant Mechanisms in Renal Injury and Disease. Antioxid. Redox Signal. 2016, 25, 119–146. [Google Scholar] [CrossRef]
- He, F.; Antonucci, L.; Karin, M. NRF2 as a regulator of cell metabolism and inflammation in cancer. Carcinogenesis 2020, 41, 405–416. [Google Scholar] [CrossRef]
- Jayakumar, S.; Pal, D.; Sandur, S.K. Nrf2 facilitates repair of radiation induced DNA damage through homologous recombination repair pathway in a ROS independent manner in cancer cells. Mutat. Res. Mol. Mech. Mutagen. 2015, 779, 33–45. [Google Scholar] [CrossRef]
- Schneider, C.A.; Lin, T.Y.; Falzone, A.; Caldwell, S.; Johnson, J.L.; DeNicola, G.M. ATM phosphorylation of NRF2 promotes DNA damage repair. In Advancing the frontiers of cancer science and medicine, Orlando. Cancer Res. 2023, 83, 309. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Pedraza-Chaverri, J.; Scholze, A. Nrf2 Activation in Chronic Kidney Disease: Promises and Pitfalls. Antioxidants 2022, 11, 1112. [Google Scholar] [CrossRef] [PubMed]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Aparicio-Trejo, O.E.; Pedraza-Chaverri, J. Mitochondrial Redox Signaling and Oxidative Stress in Kidney Diseases. Biomolecules 2021, 11, 1144. [Google Scholar] [CrossRef] [PubMed]
- Fox, D.B.; Garcia, N.M.G.; McKinney, B.J.; Lupo, R.; Noteware, L.C.; Newcomb, R.; Liu, J.; Locasale, J.W.; Hirschey, M.D.; Alvarez, J.V. NRF2 activation promotes the recurrence of dormant tumour cells through regulation of redox and nucleotide metabolism. Nat. Metab. 2020, 2, 318–334. [Google Scholar] [CrossRef]
- Liu, T.; Lv, Y.-F.; Zhao, J.-L.; You, Q.-D.; Jiang, Z.-Y. Regulation of Nrf2 by phosphorylation: Consequences for biological function and therapeutic implications. Free. Radic. Biol. Med. 2021, 168, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Lo, S.-C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.-L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [PubMed]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef]
- Venugopal, R.; Jaiswal, A.K. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H: Quinone oxidoreductase 1 gene. Proc. Natl. Acad. Sci. USA 1996, 93, 14960–14965. [Google Scholar] [CrossRef]
- Alam, J.; Stewart, D.; Touchard, C.; Boinapally, S.; Choi, A.M.K.; Cook, J.L. Nrf2, a Cap'n'Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J. Biol. Chem. 1999, 274, 26071–26078. [Google Scholar] [CrossRef]
- Sihvola, V.; Levonen, A.-L. Keap1 as the redox sensor of the antioxidant response. Arch. Biochem. Biophys. 2017, 617, 94–100. [Google Scholar] [CrossRef]
- Walsh, C.T.; Garneau-Tsodikova, S.; Gatto, G.J., Jr. Protein posttranslational modifications: The chemistry of proteome diversifications. Angew. Chem. Int. Ed. 2005, 44, 7342–7372. [Google Scholar] [CrossRef] [PubMed]
- Gajjala, P.R.; Fliser, D.; Speer, T.; Jankowski, V.; Jankowski, J. Emerging role of posttranslational modifications in chronic kidney disease and cardiovascular disease. Nephrol. Dial. Transplant. 2015, 30, 1814–1824. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, N.F.; Lau, A.; Zhang, D.D.; Li, X.; Chatterjee, N.; Spirohn, K.; Boutros, M.; Bohmann, D.; de la Vega, M.R.; Dodson, M.; et al. Regulation of the Nrf2-Keap1 antioxidant response by the ubiquitin proteasome system: An insight into cullin-ring ubiquitin ligases. Antioxid. Redox Signal. 2010, 13, 1699–1712. [Google Scholar] [CrossRef] [PubMed]
- Ganner, A.; Pfeiffer, Z.-C.; Wingendorf, L.; Kreis, S.; Klein, M.; Walz, G.; Neumann-Haefelin, E. The acetyltransferase p300 regulates NRF2 stability and localization. Biochem. Biophys. Res. Commun. 2020, 524, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Walters, T.S.; McIntosh, D.J.; Ingram, S.M.; Tillery, L.; Motley, E.D.; Arinze, I.J.; Misra, S. SUMO-Modification of Human Nrf2 at K110 and K533 Regulates Its Nucleocytoplasmic Localization, Stability and Transcriptional Activity. Cell Physiol. Biochem. 2021, 55, 141–159. [Google Scholar] [CrossRef] [PubMed]
- DeBlasi, J.M.; DeNicola, G.M. Dissecting the Crosstalk between NRF2 Signaling and Metabolic Processes in Cancer. Cancers 2020, 12, 2023. [Google Scholar] [CrossRef] [PubMed]
- Eggler, A.L.; Small, E.; Hannink, M.; Mesecar, A.D. Cul3-mediated Nrf2 ubiquitination and antioxidant response element (ARE) activation are dependent on the partial molar volume at position 151 of Keap1. Biochem. J. 2009, 422, 171–180. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, Z.-Y.; Du, H.; Li, S.-Z.; Tu, R.; Jia, Y.-F.; Zheng, Z.; Song, X.-M.; Du, R.-L.; Zhang, X.-D. DUB3 deubiquitinates and stabilizes NRF2 in chemotherapy resistance of colorectal cancer. Cell Death Differ. 2019, 26, 2300–2313. [Google Scholar] [CrossRef]
- Chen, P.; Smith, T.J.; Wu, J.; Siesser, P.F.; Bisnett, B.J.; Khan, F.; Hogue, M.; Soderblom, E.; Tang, F.; Marks, J.R.; et al. Glycosylation of KEAP1 links nutrient sensing to redox stress signaling. EMBO J. 2017, 36, 2233–2250. [Google Scholar] [CrossRef]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol. Cell Biol. 2009, 29, 2658–2672. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Ahmadi, Z.; Farkhondeh, T.; Samarghandian, S. Back to Nucleus: Combating with Cadmium Toxicity Using Nrf2 Signaling Pathway as a Promising Therapeutic Target. Biol. Trace Element Res. 2020, 197, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Wu, H.; Wang, X.; He, J.; He, S.; Yin, Y. Resveratrol Attenuates Oxidative Stress-Induced Intestinal Barrier Injury through PI3K/Akt-Mediated Nrf2 Signaling Pathway. Oxidative Med. Cell Longev. 2019, 2019, 7591840. [Google Scholar] [CrossRef]
- Bolisetty, S.; Traylor, A.M.; Joseph, R.; Zarjou, A.; Agarwal, A. Proximal tubule-targeted heme oxygenase-1 in cisplatin-induced acute kidney injury. Am. J. Physiol. Physiol. 2016, 310, F385–F394. [Google Scholar] [CrossRef] [PubMed]
- Oh, G.-S.; Kim, H.-J.; Choi, J.-H.; Shen, A.; Choe, S.-K.; Karna, A.; Lee, S.H.; Jo, H.-J.; Yang, S.-H.; Kwak, T.H.; et al. Pharmacological activation of NQO1 increases NAD+ levels and attenuates cisplatin-mediated acute kidney injury in mice. Kidney Int. 2014, 85, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.-Q.; Chu, L.-K.; Cao, X.; Xiong, Q.-W.; Mao, Y.-M.; Chen, C.-H.; Bi, Y.-L.; Liu, J.; Yan, X.-M. Glutathione metabolism rewiring protects renal tubule cells against cisplatin-induced apoptosis and ferroptosis. Redox Rep. 2023, 28, 2152607. [Google Scholar] [CrossRef] [PubMed]
- Fujieda, M.; Naruse, K.; Hamauzu, T.; Miyazaki, E.; Hayashi, Y.; Enomoto, R.; Lee, E.; Ohta, K.; Wakiguchi, H.; Enzan, H. Effect of selenium on Cisplatin-induced nephrotoxicity in rats. Nephron Exp. Nephrol. 2006, 104, e112–e122. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhang, M.; Wang, W.; Zhou, S.; Yu, M.; Qiu, X.; Jiang, S.; Wang, X.; Tang, C.; Li, S.; et al. Dihydromyricetin attenuates cisplatin-induced acute kidney injury by reducing oxidative stress, inflammation and ferroptosis. Toxicol. Appl. Pharmacol. 2023, 473, 116595. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, L.; Guo, K.; Zheng, L.; Liu, B.; Yu, W.; Guo, C.; Liu, Z.; Chen, Y.; Tang, Z. Effects of different selenium levels on gene expression of a subset of selenoproteins and antioxidative capacity in mice. Biol. Trace Element Res. 2013, 154, 255–261. [Google Scholar] [CrossRef]
- Kang, J.-S.; Nam, L.B.; Yoo, O.-K.; Keum, Y.-S. Molecular mechanisms and systemic targeting of NRF2 dysregulation in cancer. Biochem. Pharmacol. 2020, 177, 114002. [Google Scholar] [CrossRef]
- Ren, D.; Villeneuve, N.F.; Jiang, T.; Wu, T.; Lau, A.; Toppin, H.A.; Zhang, D.D. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 1433–1438. [Google Scholar] [CrossRef]
- Hirayama, A.; Yoh, K.; Nagase, S.; Ueda, A.; Itoh, K.; Morito, N.; Hirayama, K.; Takahashi, S.; Yamamoto, M.; Koyama, A. EPR imaging of reducing activity in Nrf2 transcriptional factor-deficient mice. Free. Radic. Biol. Med. 2003, 34, 1236–1242. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Aleksunes, L.M.; Goedken, M.J.; Chen, C.; Reisman, S.A.; Manautou, J.E.; Klaassen, C.D. Coordinated induction of Nrf2 target genes protects against iron nitrilotriacetate (FeNTA)-induced nephrotoxicity. Toxicol. Appl. Pharmacol. 2008, 231, 364–373. [Google Scholar] [CrossRef]
- Aleksunes, L.M.; Goedken, M.J.; Rockwell, C.E.; Thomale, J.; Manautou, J.E.; Klaassen, C.D. Transcriptional regulation of renal cytoprotective genes by Nrf2 and its potential use as a therapeutic target to mitigate cisplatin-induced nephrotoxicity. J. Pharmacol. Exp. Ther. 2010, 335, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Grigoryev, D.N.; Crow, M.T.; Haas, M.; Yamamoto, M.; Reddy, S.P.; Rabb, H. Transcription factor Nrf2 is protective during ischemic and nephrotoxic acute kidney injury in mice. Kidney Int. 2009, 76, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.-T.; Chen, R.; Chen, C.; Su, K.; Li, W.; Tang, L.-H.; Liu, H.-M.; Xue, R.; Sun, Q.; Leng, Y.; et al. Transcription factors Nrf2 and NF-κB contribute to inflammation and apoptosis induced by intestinal ischemia-reperfusion in mice. Int. J. Mol. Med. 2017, 40, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Huang, C.; Liu, J.; Meng, C.; Gu, Q.; Du, X.; Yan, M.; Yu, Y.; Liu, F.; Xia, C. Nrf2 and its dependent autophagy activation cooperatively counteract ferroptosis to alleviate acute liver injury. Pharmacol. Res. 2023, 187, 106563. [Google Scholar] [CrossRef] [PubMed]
- Ong, A.J.S.; Bladen, C.E.; Tigani, T.A.; Karamalakis, A.P.; Evason, K.J.; Brown, K.K.; Cox, A.G. The KEAP1–NRF2 pathway regulates TFEB/TFE3-dependent lysosomal biogenesis. Proc. Natl. Acad. Sci. USA 2023, 120, e2217425120. [Google Scholar] [CrossRef]
- Gumeni, S.; Papanagnou, E.-D.; Manola, M.S.; Trougakos, I.P. Nrf2 activation induces mitophagy and reverses Parkin/Pink1 knock down-mediated neuronal and muscle degeneration phenotypes. Cell Death Dis. 2021, 12, 671. [Google Scholar] [CrossRef]
- Pajares, M.; Jiménez-Moreno, N.; García-Yagüe, J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rábano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef]
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Wang, Z.; Fu, Z.; Ma, H.; Jiang, M.; Xu, A.; Zhang, W. p62/SQSTM1 protects against cisplatin-induced oxidative stress in kidneys by mediating the cross talk between autophagy and the Keap1-Nrf2 signalling pathway. Free. Radic. Res. 2019, 53, 800–814. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Kimura, T.; Takabatake, Y.; Namba, T.; Kaimori, J.; Kitamura, H.; Matsui, I.; Niimura, F.; Matsusaka, T.; Fujita, N.; et al. Autophagy guards against cisplatin-induced acute kidney injury. Am. J. Pathol. 2012, 180, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, R.S.; Lokhandwala, M.F.; Banday, A.A. Age-Related Mitochondrial Impairment and Renal Injury Is Ameliorated by Sulforaphane via Activation of Transcription Factor NRF2. Antioxidants 2022, 11, 156. [Google Scholar] [CrossRef]
- Sasaki, A.; Koike, N.; Murakami, T.; Suzuki, K. Dimethyl fumarate ameliorates cisplatin-induced renal tubulointerstitial lesions. J. Toxicol. Pathol. 2019, 32, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, Y.; Imoto, A.; Kawakami, F.; Ouchi, M.; Morita, A.; Yokoba, M.; Takenaka, T.; Ichikawa, T.; Katagiri, M.; Nielsen, R.; et al. In vitro study on effect of bardoxolone methyl on cisplatin-induced cellular senescence in human proximal tubular cells. Mol. Cell Biochem. 2022, 477, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Racke, M.K.; Nicholas, J.A.; Boster, A.L.; Imitola, J.; O’Connell, C. Design of oral agents for the management of multiple sclerosis: Benefit and risk assessment for dimethyl fumarate. Drug Des. Dev. Ther. 2014, 8, 897–908. [Google Scholar] [CrossRef]
- Atilano-Roque, A.; Aleksunes, L.M.; Joy, M.S. Bardoxolone methyl modulates efflux transporter and detoxifying enzyme expression in cisplatin-induced kidney cell injury. Toxicol. Lett. 2016, 259, 52–59. [Google Scholar] [CrossRef]
- Huang, S.; You, J.; Wang, K.; Li, Y.; Zhang, Y.; Wei, H.; Liang, X.; Liu, Y. N-Acetylcysteine Attenuates Cisplatin-Induced Acute Kidney Injury by Inhibiting the C5a Receptor. BioMed Res. Int. 2019, 2019, 4805853. [Google Scholar] [CrossRef]
- Güntürk, I.; Yazici, C.; Köse, K.; Dağli, F.; Yücel, B.; Yay, A. The effect of N-acetylcysteine on inflammation and oxidative stress in cisplatin induced nephrotoxicity: A rat model. Turk. J. Med. Sci. 2019, 49, 1789–1799. [Google Scholar] [CrossRef]
- Lee, J.; Jung, S.-Y.; Yang, K.-J.; Kim, Y.; Lee, D.; Lee, M.H.; Kim, D.-K. α-Lipoic acid prevents against cisplatin cytotoxicity via activation of the NRF2/HO-1 antioxidant pathway. PLoS ONE 2019, 14, e0226769. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Gonzalez, P.D.; Lopez-Hernandez, F.J.; Perez-Barriocanal, F.; Morales, A.I.; Lopez-Novoa, J.M. Quercetin reduces cisplatin nephrotoxicity in rats without compromising its anti-tumour activity. Nephrol. Dial. Transplant. 2011, 26, 3484–3495. [Google Scholar] [CrossRef] [PubMed]
- Hao, Q.; Xiao, X.; Zhen, J.; Feng, J.; Song, C.; Jiang, B.; Hu, Z. Resveratrol attenuates acute kidney injury by inhibiting death receptor-mediated apoptotic pathways in a cisplatin-induced rat model. Mol. Med. Rep. 2016, 14, 3683–3689. [Google Scholar] [CrossRef] [PubMed]
- Soetikno, V.; Sari, S.D.P.; Maknun, L.U.; Sumbung, N.K.; Rahmi, D.N.I.; Pandhita, B.A.W.; Louisa, M.; Estuningtyas, A. Pre-Treatment with Curcumin Ameliorates Cisplatin-Induced Kidney Damage by Suppressing Kidney Inflammation and Apoptosis in Rats. Drug Res. 2019, 69, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-H.; Tsai, K.-L.; Chou, W.-C.; Cheng, H.-C.; Huang, Y.-T.; Ou, H.-C.; Chang, Y.-C. Quercetin Mitigates Cisplatin-Induced Oxidative Damage and Apoptosis in Cardiomyocytes through Nrf2/HO-1 Signaling Pathway. Am. J. Chin. Med. 2022, 50, 1281–1298. [Google Scholar] [CrossRef] [PubMed]
- Boots, A.W.; Haenen, G.R.; Bast, A. Health effects of quercetin: From antioxidant to nutraceutical. Eur. J. Pharmacol. 2008, 585, 325–337. [Google Scholar] [CrossRef]
- Wang, X.-J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Jeddi, F.; Soozangar, N.; Sadeghi, M.R.; Somi, M.H.; Samadi, N. Contradictory roles of Nrf2/Keap1 signaling pathway in cancer prevention/promotion and chemoresistance. DNA Repair 2017, 54, 13–21. [Google Scholar] [CrossRef]
- Solis, L.M.; Behrens, C.; Dong, W.; Suraokar, M.; Ozburn, N.C.; Moran, C.A.; Corvalan, A.H.; Biswal, S.; Swisher, S.G.; Bekele, B.N.; et al. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin. Cancer Res. 2010, 16, 3743–3753. [Google Scholar] [CrossRef]
- Yoo, N.J.; Kim, H.R.; Kim, Y.R.; An, C.H.; Lee, S.H. Somatic mutations of the KEAP1 gene in common solid cancers. Histopathology 2012, 60, 943–952. [Google Scholar] [CrossRef]
- Goldstein, L.D.; Lee, J.; Gnad, F.; Klijn, C.; Schaub, A.; Reeder, J.; Daemen, A.; Bakalarski, C.E.; Holcomb, T.; Shames, D.S.; et al. Recurrent Loss of NFE2L2 Exon 2 Is a Mechanism for Nrf2 Pathway Activation in Human Cancers. Cell Rep. 2016, 16, 2605–2617. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Hanada, N.; Takahata, T.; Zhou, Q.; Ye, X.; Sun, R.; Itoh, J.; Ishiguro, A.; Kijima, H.; Mimura, J.; Itoh, K.; et al. Methylation of the KEAP1 gene promoter region in human colorectal cancer. BMC Cancer 2012, 12, 66. [Google Scholar] [CrossRef] [PubMed]
- Köhler, U.A.; Kurinna, S.; Schwitter, D.; Marti, A.; Schäfer, M.; Hellerbrand, C.; Speicher, T.; Werner, S. Activated Nrf2 impairs liver regeneration in mice by activation of genes involved in cell-cycle control and apoptosis. Hepatology 2014, 60, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; de la Vega, M.R.; Chapman, E.; Ooi, A.; Zhang, D.D. The effects of NRF2 modulation on the initiation and progression of chemically and genetically induced lung cancer. Mol. Carcinog. 2018, 57, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Rush, B.M.; Bondi, C.D.; Stocker, S.D.; Barry, K.M.; Small, S.A.; Ong, J.; Jobbagy, S.; Stolz, D.B.; Bastacky, S.I.; Chartoumpekis, D.V.; et al. Genetic or pharmacologic Nrf2 activation increases proteinuria in chronic kidney disease in mice. Kidney Int. 2021, 99, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef]
- Hong, D.S.; Kurzrock, R.; Supko, J.G.; He, X.; Naing, A.; Wheler, J.; Lawrence, D.; Eder, J.P.; Meyer, C.J.; Ferguson, D.A.; et al. A phase I first-in-human trial of bardoxolone methyl in patients with advanced solid tumors and lymphomas. Clin. Cancer Res. 2012, 18, 3396–3406. [Google Scholar] [CrossRef]
- Atwell, L.L.; Zhang, Z.; Mori, M.; Farris, P.E.; Vetto, J.T.; Naik, A.M.; Oh, K.Y.; Thuillier, P.; Ho, E.; Shannon, J. Sulforaphane Bioavailability and Chemopreventive Activity in Women Scheduled for Breast Biopsy. Cancer Prev. Res. 2015, 8, 1184–1191. [Google Scholar] [CrossRef]
- Traka, M.H.; Melchini, A.; Coode-Bate, J.; Al Kadhi, O.; Saha, S.; Defernez, M.; Troncoso-Rey, P.; Kibblewhite, H.; O’Neill, C.M.; Bernuzzi, F.; et al. Transcriptional changes in prostate of men on active surveillance after a 12-mo glucoraphanin-rich broccoli intervention—Results from the Effect of Sulforaphane on prostate CAncer PrEvention (ESCAPE) randomized controlled trial. Am. J. Clin. Nutr. 2019, 109, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Tahata, S.; Singh, S.V.; Lin, Y.; Hahm, E.-R.; Beumer, J.H.; Christner, S.M.; Rao, U.N.; Sander, C.; Tarhini, A.A.; Tawbi, H.; et al. Evaluation of Biodistribution of Sulforaphane after Administration of Oral Broccoli Sprout Extract in Melanoma Patients with Multiple Atypical Nevi. Cancer Prev. Res. 2018, 11, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Tschöp, M.H.; Stumvoll, M.; Ristow, M. Opposing Effects of Antidiabetic Interventions on Malignant Growth and Metastasis. Cell Metab. 2016, 23, 959–960. [Google Scholar] [CrossRef] [PubMed]
- Ashabi, G.; Khalaj, L.; Khodagholi, F.; Goudarzvand, M.; Sarkaki, A. Pre-treatment with metformin activates Nrf2 antioxidant pathways and inhibits inflammatory responses through induction of AMPK after transient global cerebral ischemia. Metab. Brain Dis. 2015, 30, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Jin, X.; Zhang, J.; Li, L.; Zhao, J. Metformin suppresses Nrf2-mediated chemoresistance in hepatocellular carcinoma cells by increasing glycolysis. Aging 2020, 12, 17582–17600. [Google Scholar] [CrossRef]
- Bai, M.; Yang, L.; Liao, H.; Liang, X.; Xie, B.; Xiong, J.; Tao, X.; Chen, X.; Cheng, Y.; Chen, X.; et al. Metformin sensitizes endometrial cancer cells to chemotherapy through IDH1-induced Nrf2 expression via an epigenetic mechanism. Oncogene 2018, 37, 5666–5681. [Google Scholar] [CrossRef]
- Hsu, W.-H.; Hsiao, P.-J.; Lin, P.-C.; Chen, S.-C.; Lee, M.-Y.; Shin, S.-J. Effect of metformin on kidney function in patients with type 2 diabetes mellitus and moderate chronic kidney disease. Oncotarget 2018, 9, 5416–5423. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mapuskar, K.A.; Pulliam, C.F.; Zepeda-Orozco, D.; Griffin, B.R.; Furqan, M.; Spitz, D.R.; Allen, B.G. Redox Regulation of Nrf2 in Cisplatin-Induced Kidney Injury. Antioxidants 2023, 12, 1728. https://doi.org/10.3390/antiox12091728
Mapuskar KA, Pulliam CF, Zepeda-Orozco D, Griffin BR, Furqan M, Spitz DR, Allen BG. Redox Regulation of Nrf2 in Cisplatin-Induced Kidney Injury. Antioxidants. 2023; 12(9):1728. https://doi.org/10.3390/antiox12091728
Chicago/Turabian StyleMapuskar, Kranti A., Casey F. Pulliam, Diana Zepeda-Orozco, Benjamin R. Griffin, Muhammad Furqan, Douglas R. Spitz, and Bryan G. Allen. 2023. "Redox Regulation of Nrf2 in Cisplatin-Induced Kidney Injury" Antioxidants 12, no. 9: 1728. https://doi.org/10.3390/antiox12091728