
Novel NADPH Oxidase-2 Inhibitors as Potential Anti-Inflammatory and Neuroprotective Agents


Abstract
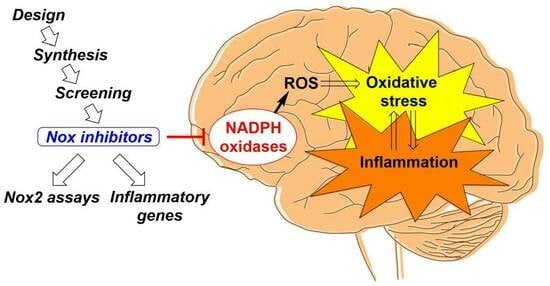
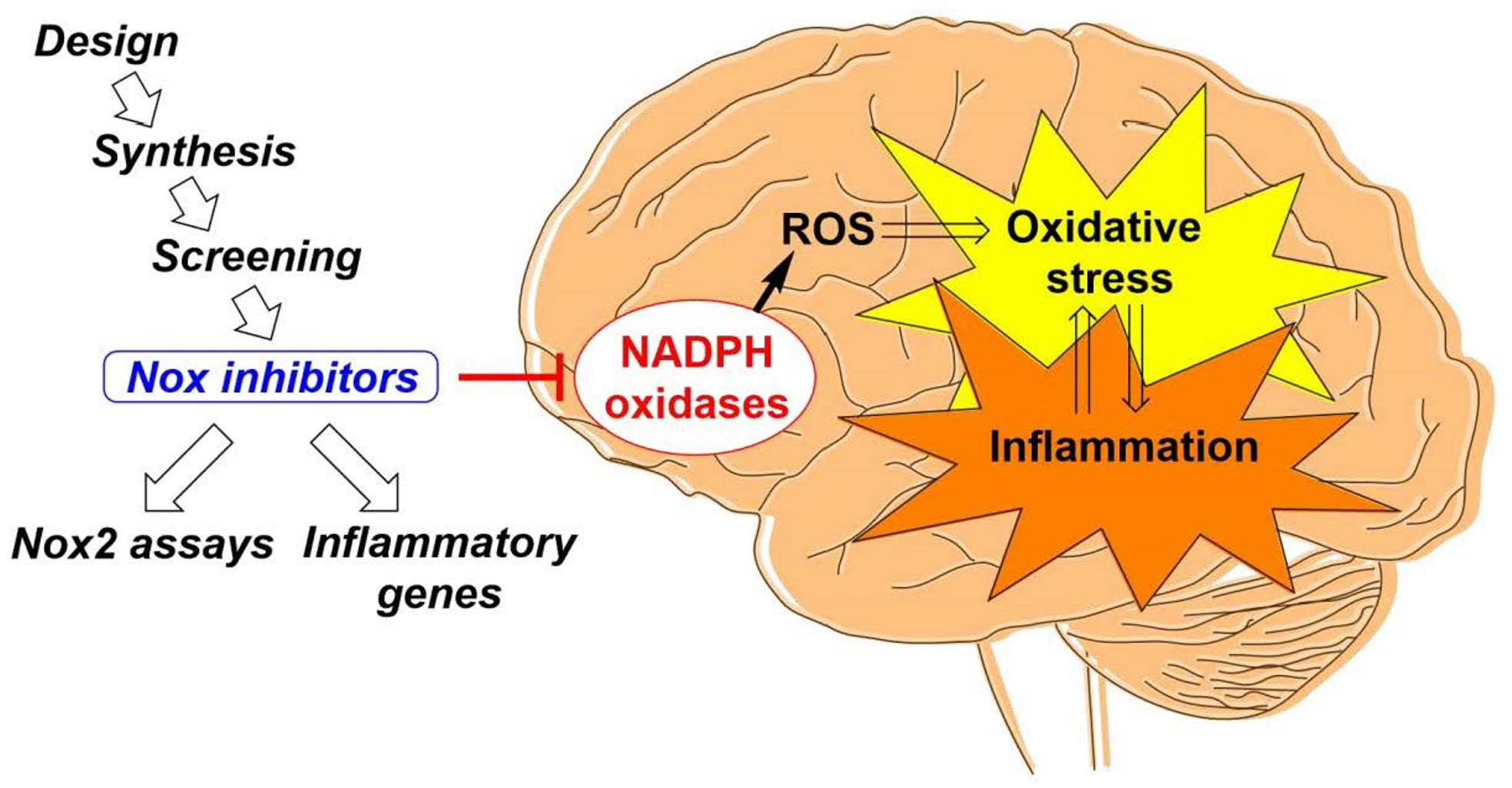
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
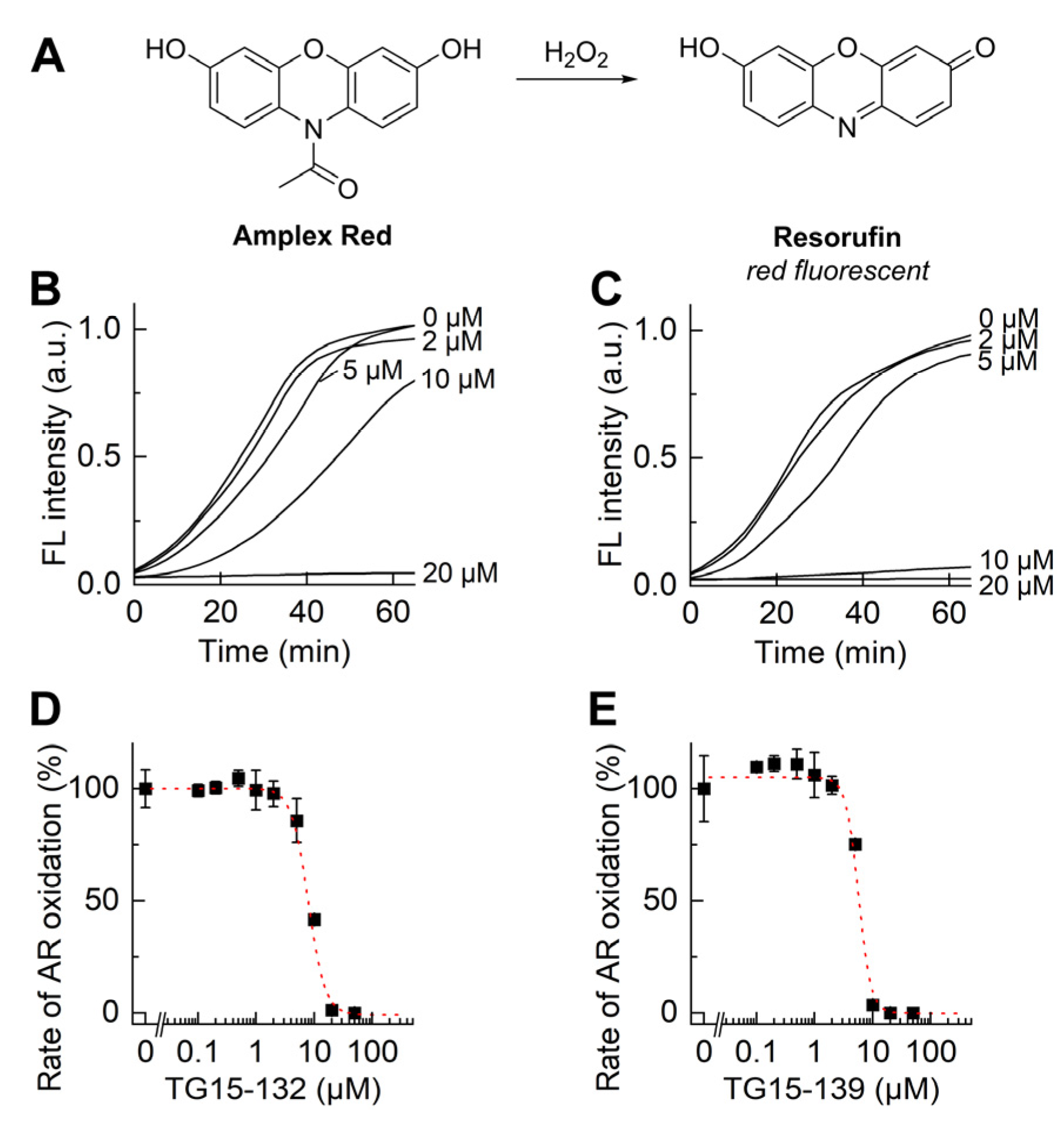
2.3. Plate Reader-Based Analyses of O2•− and H2O2 Production by Activated dHL60 Cells
2.4. Rapid HPLC Analyses of O2•− Production by Activated dHL60 Cells
2.5. Oxygen Consumption Measurements
2.6. Cytotoxicity
2.7. Gene Expression Analysis
2.8. Plasma and Brain In Vivo Pharmacokinetics Studies
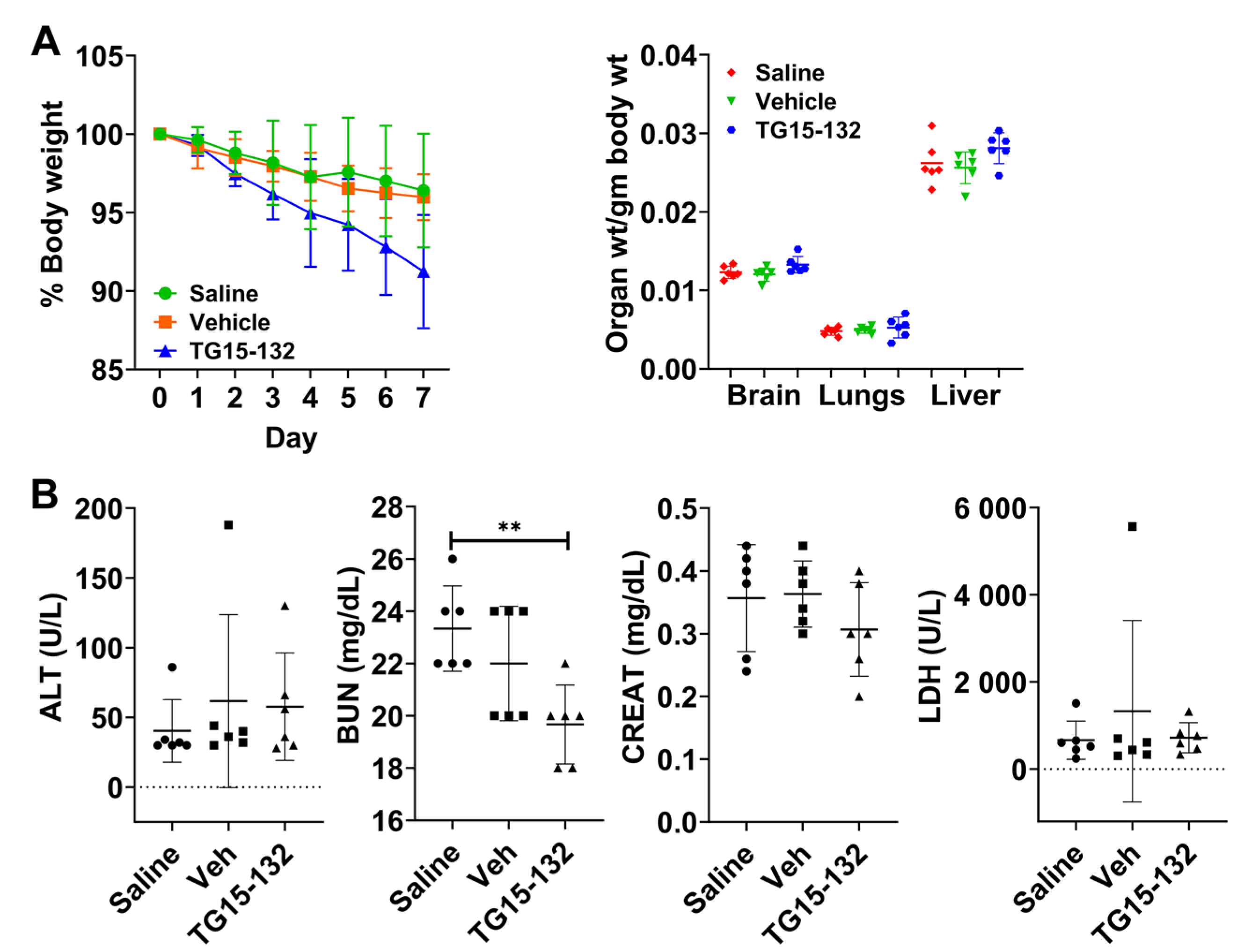
2.9. Short-Term In Vivo Toxicity
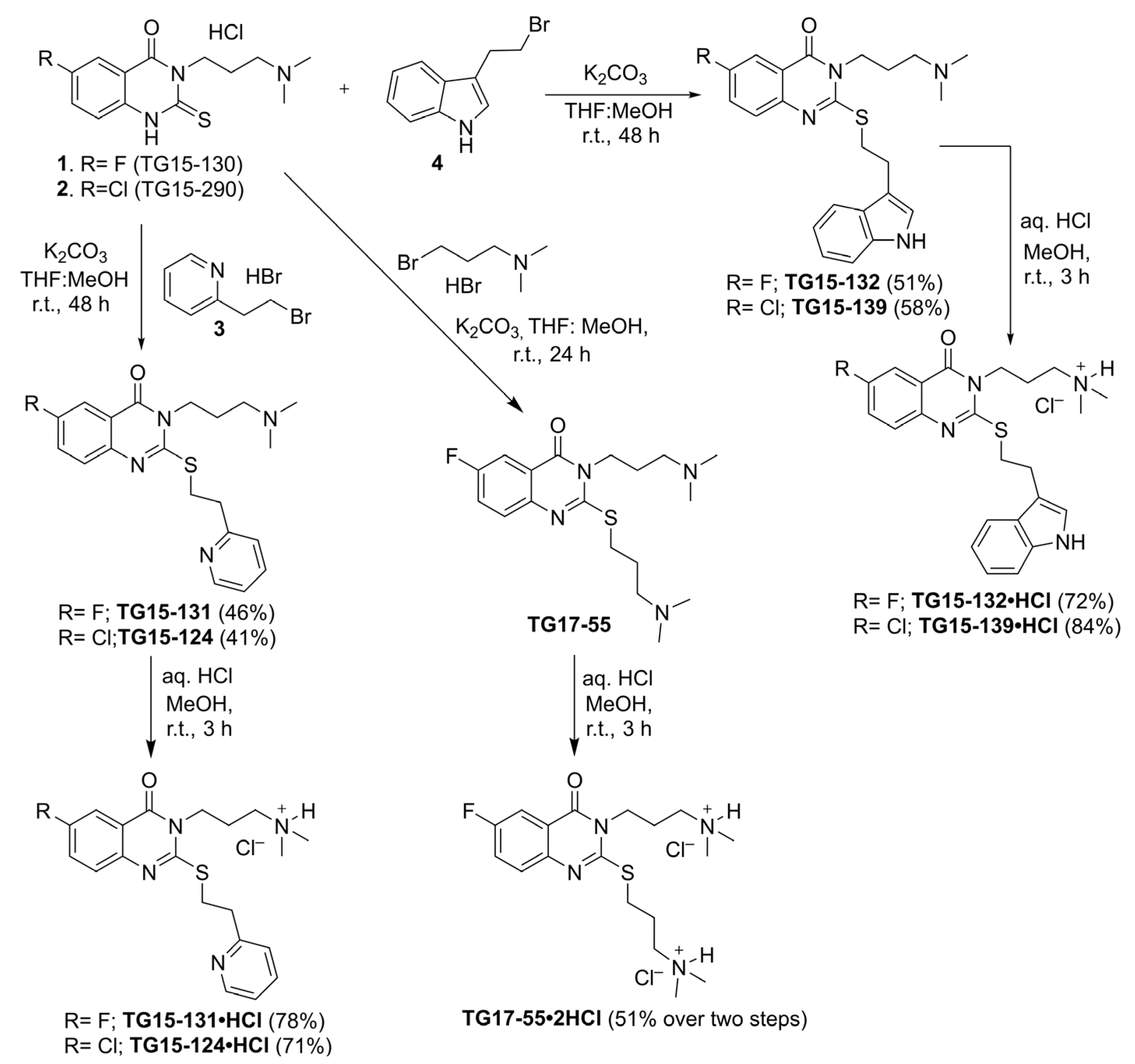
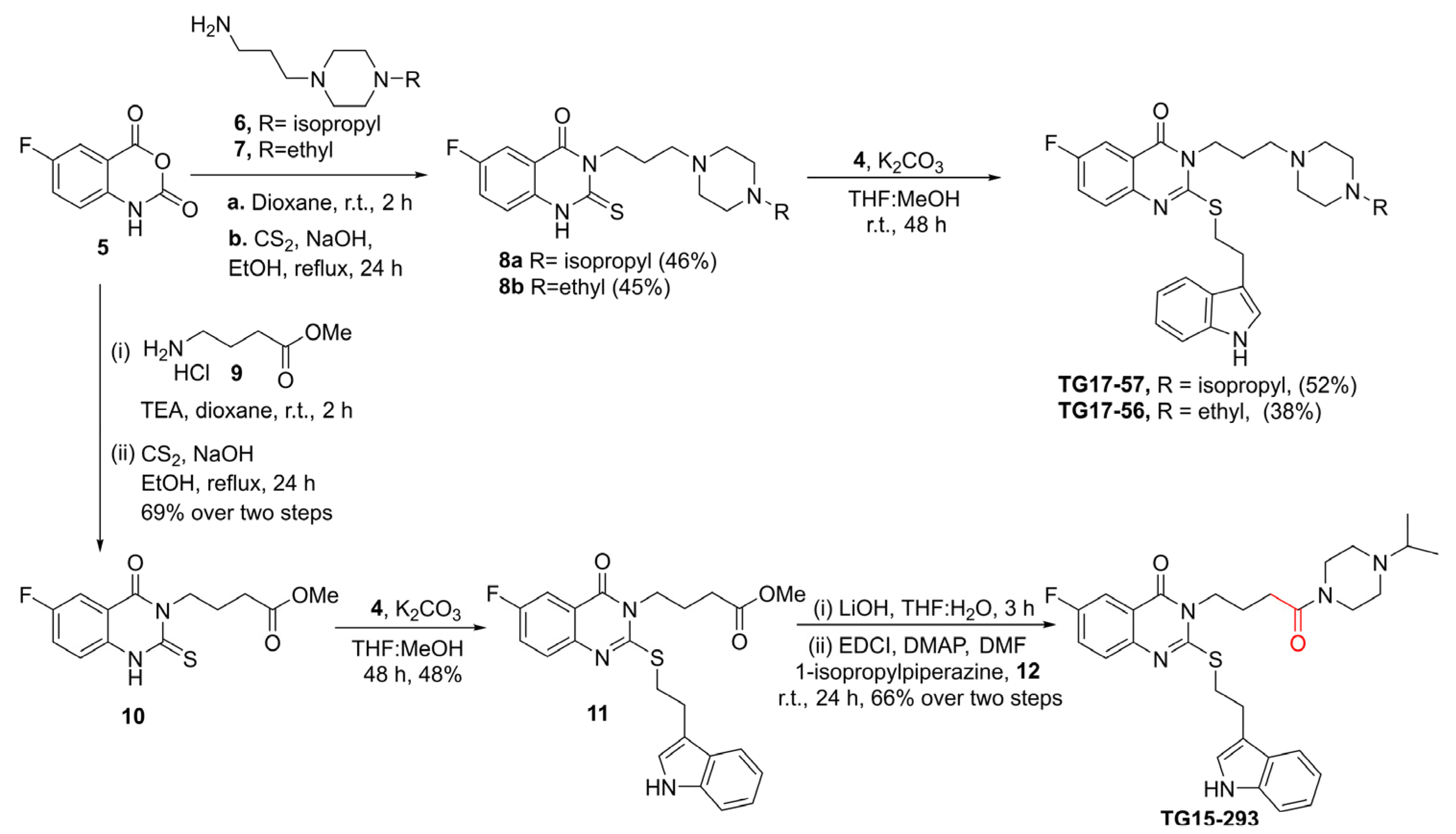
2.10. Synthesis of Nox2 Inhibitors TG15-132 and Derivatives
3. Results
3.1. Identification of Novel Nox2 Inhibitors
3.2. Characterization of Nox2 Inhibition in Differentiated HL60 Cells
3.3. Structure–Activity Relationship Studies
3.4. Effect of TG15-132 on Molecular Markers of Inflammation in THP-1 Cells
3.5. In Vivo Pharmacokinetic and Toxicity Study of Seven Day Dosing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.H.; Griffiths, H.R. The dual role of Reactive Oxygen Species in autoimmune and inflammatory diseases: Evidence from preclinical models. Free Radic. Biol. Med. 2018, 125, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Nocella, C.; D’Amico, A.; Cammisotto, V.; Bartimoccia, S.; Castellani, V.; Loffredo, L.; Marini, L.; Ferrara, G.; Testa, M.; Motta, G.; et al. Structure, Activation, and Regulation of NOX2: At the Crossroad between the Innate Immunity and Oxidative Stress-Mediated Pathologies. Antioxidants 2023, 12, 429. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Zhang, L.; Hong, J.S.; Zhang, D.; Zhao, J.; Wang, Q. Nicotinamide Adenine Dinucleotide Phosphate Oxidase and Neurodegenerative Diseases: Mechanisms and Therapy. Antioxid. Redox Signal. 2020, 33, 374–393. [Google Scholar] [CrossRef]
- Bonner, M.Y.; Arbiser, J.L. Targeting NADPH oxidases for the treatment of cancer and inflammation. Cell. Mol. Life Sci. 2012, 69, 2435–2442. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Espinosa, D.R.; Massieu, L.; Montiel, T.; Morán, J. Role of NADPH oxidase-2 in the progression of the inflammatory response secondary to striatum excitotoxic damage. J. Neuroinflamm. 2019, 16, 91. [Google Scholar] [CrossRef]
- Hu, C.-F.; Wu, S.-P.; Lin, G.-J.; Shieh, C.-C.; Hsu, C.-S.; Chen, J.-W.; Chen, S.-H.; Hong, J.-S.; Chen, S.-J. Microglial Nox2 Plays a Key Role in the Pathogenesis of Experimental Autoimmune Encephalomyelitis. Front. Immunol. 2021, 12, 638381. [Google Scholar] [CrossRef]
- Khayrullina, G.; Bermudez, S.; Hopkins, D.; Yauger, Y.; Byrnes, K.R. Differential effects of NOX2 and NOX4 inhibition after rodent spinal cord injury. PLoS ONE 2023, 18, e0281045. [Google Scholar] [CrossRef]
- Huang, W.-Y.; Liu, K.-H.; Lin, S.; Chen, T.-Y.; Tseng, C.-Y.; Chen, H.-Y.; Wu, H.-M.; Hsu, K.-S. NADPH oxidase 2 as a potential therapeutic target for protection against cognitive deficits following systemic inflammation in mice. Brain Behav. Immun. 2020, 84, 242–252. [Google Scholar] [CrossRef]
- Singel, K.L.; Segal, B.H. NOX2-dependent regulation of inflammation. Clin. Sci. 2016, 130, 479–490. [Google Scholar] [CrossRef]
- Steinhubl, S.R. Why have antioxidants failed in clinical trials? Am. J. Cardiol. 2008, 101, 14d–19d. [Google Scholar] [CrossRef] [PubMed]
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.H.; Stocker, R.; Vollbracht, C.; Paulsen, G.; Riley, D.; Daiber, A.; Cuadrado, A. Antioxidants in Translational Medicine. Antioxid. Redox Signal. 2015, 23, 1130–1143. [Google Scholar] [CrossRef] [PubMed]
- Sassetti, E.; Clausen, M.H.; Laraia, L. Small-Molecule Inhibitors of Reactive Oxygen Species Production. J. Med. Chem. 2021, 64, 5252–5275. [Google Scholar] [CrossRef] [PubMed]
- Diebold, B.A.; Smith, S.M.; Li, Y.; Lambeth, J.D. NOX2 As a Target for Drug Development: Indications, Possible Complications, and Progress. Antioxid. Redox Signal. 2015, 23, 375–405. [Google Scholar] [CrossRef]
- Eastman, C.L.; D’Ambrosio, R.; Ganesh, T. Modulating neuroinflammation and oxidative stress to prevent epilepsy and improve outcomes after traumatic brain injury. Neuropharmacology 2020, 172, 107907. [Google Scholar] [CrossRef]
- Cifuentes-Pagano, E.; Meijles, D.N.; Pagano, P.J. The quest for selective Nox inhibitors and therapeutics: Challenges, triumphs and pitfalls. Antioxid. Redox Signal. 2014, 20, 2741–2754. [Google Scholar] [CrossRef]
- Cifuentes-Pagano, E.; Csanyi, G.; Pagano, P.J. NADPH oxidase inhibitors: A decade of discovery from Nox2ds to HTS. Cell. Mol. Life Sci. 2012, 69, 2315–2325. [Google Scholar] [CrossRef]
- Altenhofer, S.; Radermacher, K.A.; Kleikers, P.W.; Wingler, K.; Schmidt, H.H. Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement. Antioxid. Redox Signal. 2015, 23, 406–427. [Google Scholar] [CrossRef]
- Augsburger, F.; Filippova, A.; Rasti, D.; Seredenina, T.; Lam, M.; Maghzal, G.; Mahiout, Z.; Jansen-Dürr, P.; Knaus, U.G.; Doroshow, J.; et al. Pharmacological characterization of the seven human NOX isoforms and their inhibitors. Redox Biol. 2019, 26, 101272. [Google Scholar] [CrossRef]
- Reis, J.; Massari, M.; Marchese, S.; Ceccon, M.; Aalbers, F.S.; Corana, F.; Valente, S.; Mai, A.; Magnani, F.; Mattevi, A. A closer look into NADPH oxidase inhibitors: Validation and insight into their mechanism of action. Redox Biol. 2020, 32, 101466. [Google Scholar] [CrossRef] [PubMed]
- Wind, S.; Beuerlein, K.; Eucker, T.; Muller, H.; Scheurer, P.; Armitage, M.E.; Ho, H.; Schmidt, H.H.; Wingler, K. Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. Br. J. Pharmacol. 2010, 161, 885–898. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ganesh, T.; Diebold, B.A.; Zhu, Y.; McCoy, J.W.; Smith, S.M.; Sun, A.; Lambeth, J.D. Thioxo-dihydroquinazolin-one Compounds as Novel Inhibitors of Myeloperoxidase. ACS Med. Chem. Lett. 2015, 6, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Cheng, G.; Zielonka, M.; Ganesh, T.; Sun, A.; Joseph, J.; Michalski, R.; O’Brien, W.J.; Lambeth, J.D.; Kalyanaraman, B. High-throughput assays for superoxide and hydrogen peroxide: Design of a screening workflow to identify inhibitors of NADPH oxidases. J. Biol. Chem. 2014, 289, 16176–16189. [Google Scholar] [CrossRef]
- Zielonka, J.; Zielonka, M.; VerPlank, L.; Cheng, G.; Hardy, M.; Ouari, O.; Ayhan, M.M.; Podsiadly, R.; Sikora, A.; Lambeth, J.D.; et al. Mitigation of NADPH Oxidase 2 Activity as a Strategy to Inhibit Peroxynitrite Formation. J. Biol. Chem. 2016, 291, 7029–7044. [Google Scholar] [CrossRef]
- Zielonka, J.; Hardy, M.; Michalski, R.; Sikora, A.; Zielonka, M.; Cheng, G.; Ouari, O.; Podsiadły, R.; Kalyanaraman, B. Recent Developments in the Probes and Assays for Measurement of the Activity of NADPH Oxidases. Cell Biochem. Biophys. 2017, 75, 335–349. [Google Scholar] [CrossRef]
- Seredenina, T.; Chiriano, G.; Filippova, A.; Nayernia, Z.; Mahiout, Z.; Fioraso-Cartier, L.; Plastre, O.; Scapozza, L.; Krause, K.H.; Jaquet, V. A subset of N-substituted phenothiazines inhibits NADPH oxidases. Free Radic. Biol. Med. 2015, 86, 239–249. [Google Scholar] [CrossRef]
- Seredenina, T.; Nayernia, Z.; Sorce, S.; Maghzal, G.J.; Filippova, A.; Ling, S.C.; Basset, O.; Plastre, O.; Daali, Y.; Rushing, E.J.; et al. Evaluation of NADPH oxidases as drug targets in a mouse model of familial amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2016, 97, 95–108. [Google Scholar] [CrossRef]
- Hirano, K.; Chen, W.S.; Chueng, A.L.; Dunne, A.A.; Seredenina, T.; Filippova, A.; Ramachandran, S.; Bridges, A.; Chaudry, L.; Pettman, G.; et al. Discovery of GSK2795039, a Novel Small Molecule NADPH Oxidase 2 Inhibitor. Antioxid. Redox Signal. 2015, 23, 358–374. [Google Scholar] [CrossRef]
- Mason, H.; Rai, G.; Kozyr, A.; De Jonge, N.; Gliniewicz, E.; Berg, L.J.; Wald, G.; Dorrier, C.; Henderson, M.J.; Zakharov, A.; et al. Development of an improved and specific inhibitor of NADPH oxidase 2 to treat traumatic brain injury. Redox Biol. 2023, 60, 102611. [Google Scholar] [CrossRef]
- Zielonka, J.; Sikora, A.; Joseph, J.; Kalyanaraman, B. Peroxynitrite is the major species formed from different flux ratios of co-generated nitric oxide and superoxide: Direct reaction with boronate-based fluorescent probe. J. Biol. Chem. 2010, 285, 14210–14216. [Google Scholar] [CrossRef]
- Zielonka, J.; Vasquez-Vivar, J.; Kalyanaraman, B. Detection of 2-hydroxyethidium in cellular systems: A unique marker product of superoxide and hydroethidine. Nat. Protoc. 2008, 3, 8–21. [Google Scholar] [CrossRef]
- Zielonka, J.; Hardy, M.; Kalyanaraman, B. HPLC study of oxidation products of hydroethidine in chemical and biological systems: Ramifications in superoxide measurements. Free Radic. Biol. Med. 2009, 46, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Dranka, B.P.; Gifford, A.; Ghosh, A.; Zielonka, J.; Joseph, J.; Kanthasamy, A.G.; Kalyanaraman, B. Diapocynin prevents early Parkinson’s disease symptoms in the leucine-rich repeat kinase 2 (LRRK2R¹⁴⁴¹G) transgenic mouse. Neurosci. Lett. 2013, 549, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Edwards, E.E.; Thomas, S.N. P-Selectin and ICAM-1 synergy in mediating THP-1 monocyte adhesion in hemodynamic flow is length dependent. Integr. Biol. 2017, 9, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Zielonka, M.; Cheng, G.; Hardy, M.; Kalyanaraman, B. High-Throughput Screening of NOX Inhibitors. Methods Mol. Biol. 2019, 1982, 429–446. [Google Scholar] [CrossRef]
- Zielonka, J.; Zielonka, M.; Kalyanaraman, B. HPLC-Based Monitoring of Oxidation of Hydroethidine for the Detection of NADPH Oxidase-Derived Superoxide Radical Anion. Methods Mol. Biol. 2019, 1982, 243–258. [Google Scholar] [CrossRef]
- Rawat, V.; Eastman, C.L.; Amaradhi, R.; Banik, A.; Fender, J.S.; Dingledine, R.J.; D’Ambrosio, R.; Ganesh, T. Temporal Expression of Neuroinflammatory and Oxidative Stress Markers and Prostaglandin E2 Receptor EP2 Antagonist Effect in a Rat Model of Epileptogenesis. ACS Pharmacol. Transl. Sci. 2023, 6, 128–138. [Google Scholar] [CrossRef]
- Rawat, V.; Goux, W.; Piechaczyk, M.; SR, D.M. c-Fos Protects Neurons Through a Noncanonical Mechanism Involving HDAC3 Interaction: Identification of a 21-Amino Acid Fragment with Neuroprotective Activity. Mol. Neurobiol. 2016, 53, 1165–1180. [Google Scholar] [CrossRef]
- Rawat, V.; Wang, S.; Sima, J.; Bar, R.; Liraz, O.; Gundimeda, U.; Parekh, T.; Chan, J.; Johansson, J.O.; Tang, C.; et al. ApoE4 Alters ABCA1 Membrane Trafficking in Astrocytes. J. Neurosci. 2019, 39, 9611–9622. [Google Scholar] [CrossRef]
- Rawat, V.; Banik, A.; Amaradhi, R.; Rojas, A.; Taval, S.; Nagy, T.; Dingledine, R.; Ganesh, T. Pharmacological antagonism of EP2 receptor does not modify basal cardiovascular and respiratory function, blood cell counts, and bone morphology in animal models. Biomed. Pharmacother. 2022, 147, 112646. [Google Scholar] [CrossRef]
- Smith, S.M.; Min, J.; Ganesh, T.; Diebold, B.; Kawahara, T.; Zhu, Y.; McCoy, J.; Sun, A.; Snyder, J.P.; Fu, H.; et al. Ebselen and congeners inhibit NADPH oxidase 2-dependent superoxide generation by interrupting the binding of regulatory subunits. Chem. Biol. 2012, 19, 752–763. [Google Scholar] [CrossRef]
- Zielonka, J.; Kalyanaraman, B. Hydroethidine- and MitoSOX-derived red fluorescence is not a reliable indicator of intracellular superoxide formation: Another inconvenient truth. Free Radic. Biol. Med. 2010, 48, 983–1001. [Google Scholar] [CrossRef] [PubMed]
- Michalski, R.; Thiebaut, D.; Michałowski, B.; Ayhan, M.M.; Hardy, M.; Ouari, O.; Rostkowski, M.; Smulik-Izydorczyk, R.; Artelska, A.; Marcinek, A.; et al. Oxidation of ethidium-based probes by biological radicals: Mechanism, kinetics and implications for the detection of superoxide. Sci. Rep. 2020, 10, 18626. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.K.; Lu, J.; Doroshow, J.H. Effects of Iodonium Analogs on Nadph Oxidase 1 in Human Colon Cancer Cells. Antioxidants 2021, 10, 1757. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, Z.; Zhuan, L.; Wang, T.; Guo, S.; Wang, S.; Liu, J.; Ye, Z. Effects of apocynin on oxidative stress and expression of apoptosis-related genes in testes of diabetic rats. Mol. Med. Rep. 2013, 7, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Teleanu, D.M.; Niculescu, A.-G.; Lungu, I.I.; Radu, C.I.; Vladâcenco, O.; Roza, E.; Costăchescu, B.; Grumezescu, A.M.; Teleanu, R.I. An Overview of Oxidative Stress, Neuroinflammation, and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 5938. [Google Scholar] [CrossRef]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef]
- Onose, G.; Anghelescu, A.; Blendea, D.; Ciobanu, V.; Daia, C.; Firan, F.C.; Oprea, M.; Spinu, A.; Popescu, C.; Ionescu, A.; et al. Cellular and Molecular Targets for Non-Invasive, Non-Pharmacological Therapeutic/Rehabilitative Interventions in Acute Ischemic Stroke. Int. J. Mol. Sci. 2022, 23, 907. [Google Scholar] [CrossRef]
- Moghaddam, M.H.; Bayat, A.H.; Eskandari, N.; Abdollahifar, M.A.; Fotouhi, F.; Forouzannia, A.; Rafiei, R.; Hatari, S.; Seraj, A.; Shahidi, A.; et al. Elderberry diet ameliorates motor function and prevents oxidative stress-induced cell death in rat models of Huntington disease. Brain Res. 2021, 1762, 147444. [Google Scholar] [CrossRef]
- Obrador, E.; Salvador, R.; Lopez-Blanch, R.; Jihad-Jebbar, A.; Valles, S.L.; Estrela, J.M. Oxidative Stress, Neuroinflammation and Mitochondria in the Pathophysiology of Amyotrophic Lateral Sclerosis. Antioxidants 2020, 9, 901. [Google Scholar] [CrossRef] [PubMed]
- Solleiro-Villavicencio, H.; Rivas-Arancibia, S. Effect of Chronic Oxidative Stress on Neuroinflammatory Response Mediated by CD4+T Cells in Neurodegenerative Diseases. Front. Cell. Neurosci. 2018, 12, 114. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Probe | Species Monitored | Excitation Filter | Emission Filter |
|---|---|---|---|
| HE a | O2•− | 485 nm | 574 nm |
| HE b | O2•− | 370 nm | 565 nm |
| CBA | H2O2 | 355 nm | 460 nm |
| Amplex Red | H2O2 | 535 nm | 595 nm |
| Gene | Accession No. | Primer Sequences |
|---|---|---|
| NOX2 | NM_000397 | F: GGTCCCATGTTTCTGTATCTC R: CCCTTCTTCTTCATCTGTAGC |
| p47phox | NM_000265 | F: GCTGGTGGGTCATCAGGAAA R: GCCCTGACTTTTGCAGGTACA |
| p67phox | NM_000433 | F: CCTGCAACTACCTTGAACCAGTT R: GGACTGCGGAGAGCTTTCC |
| Tnfα | NM_000594 | F: CTCTTCTGCCTGCTGCACTTTG R: ATGGGCTACAGGCTTGTCACTC |
| IL1β | NM_000576 | F: CCACAGACCTTCCAGGAGAATG R: GTGCAGTTCAGTGATCGTACAGG |
| β-Actin | NM_001101 | F: GGTGACAGCAGTCGGTTGGAG R: AGTGGGGTGGCTTTTAGGATGG |
| GAPDH | NM_001256799 | F: TGCACCACCAACTGCTTAGC R: GGCATGGACTGTGGTCATGAG |
| Compound | CBA Assay | Amplex Red Assay | Viability Assay a |
|---|---|---|---|
| TG15-132 | 4.3 ± 1.3 b | 7.9 ± 3.0 b | 34.5 ± 6.0 |
| TG15-139 | 3.1 ± 1.4 b | 4.9 ± 2.3 b | 14.1 ± 1.7 |
| TG15-293 | 23.6 ± 18.1 | 67.8 ± 2.6 | 38.3 ± 7.5 |
| TG17-56 | 5.3 ± 0.6 | 7.8 ± 4.0 | 16.7 ± 2.5 |
| TG17-57 | 7.0 ± 2.8 | 9.2 ± 0.4 | 20.7 ± 5.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juric, M.; Rawat, V.; Amaradhi, R.; Zielonka, J.; Ganesh, T. Novel NADPH Oxidase-2 Inhibitors as Potential Anti-Inflammatory and Neuroprotective Agents. Antioxidants 2023, 12, 1660. https://doi.org/10.3390/antiox12091660
Juric M, Rawat V, Amaradhi R, Zielonka J, Ganesh T. Novel NADPH Oxidase-2 Inhibitors as Potential Anti-Inflammatory and Neuroprotective Agents. Antioxidants. 2023; 12(9):1660. https://doi.org/10.3390/antiox12091660
Chicago/Turabian StyleJuric, Matea, Varun Rawat, Radhika Amaradhi, Jacek Zielonka, and Thota Ganesh. 2023. "Novel NADPH Oxidase-2 Inhibitors as Potential Anti-Inflammatory and Neuroprotective Agents" Antioxidants 12, no. 9: 1660. https://doi.org/10.3390/antiox12091660