
Integrative Transcriptomic Analysis Reveals Upregulated Apoptotic Signaling in Wound-Healing Pathway in Rat Liver Fibrosis Models
 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Model
2.2. Biochemical Analysis of Liver Enzymes
2.3. RNA Extraction and RNA Sequencing (RNA-seq)
2.4. Collection of Public Transcriptome Data for Liver Fibrosis
2.5. Preprocessing of RNA Sequencing Data
2.6. Differential Expression and Functional Enrichment Analysis
2.7. Western Blot
2.8. Statistical Analysis
3. Results
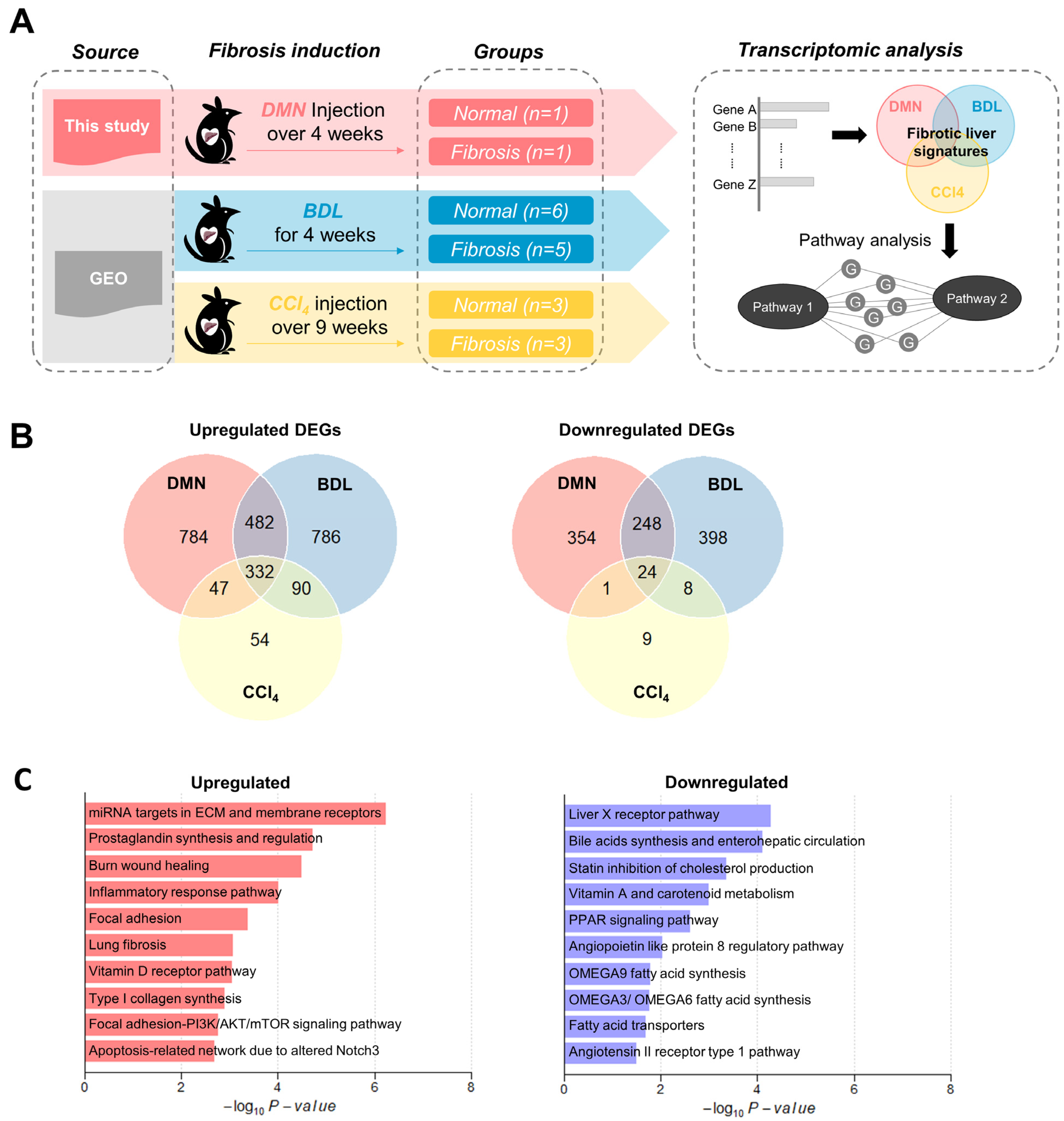
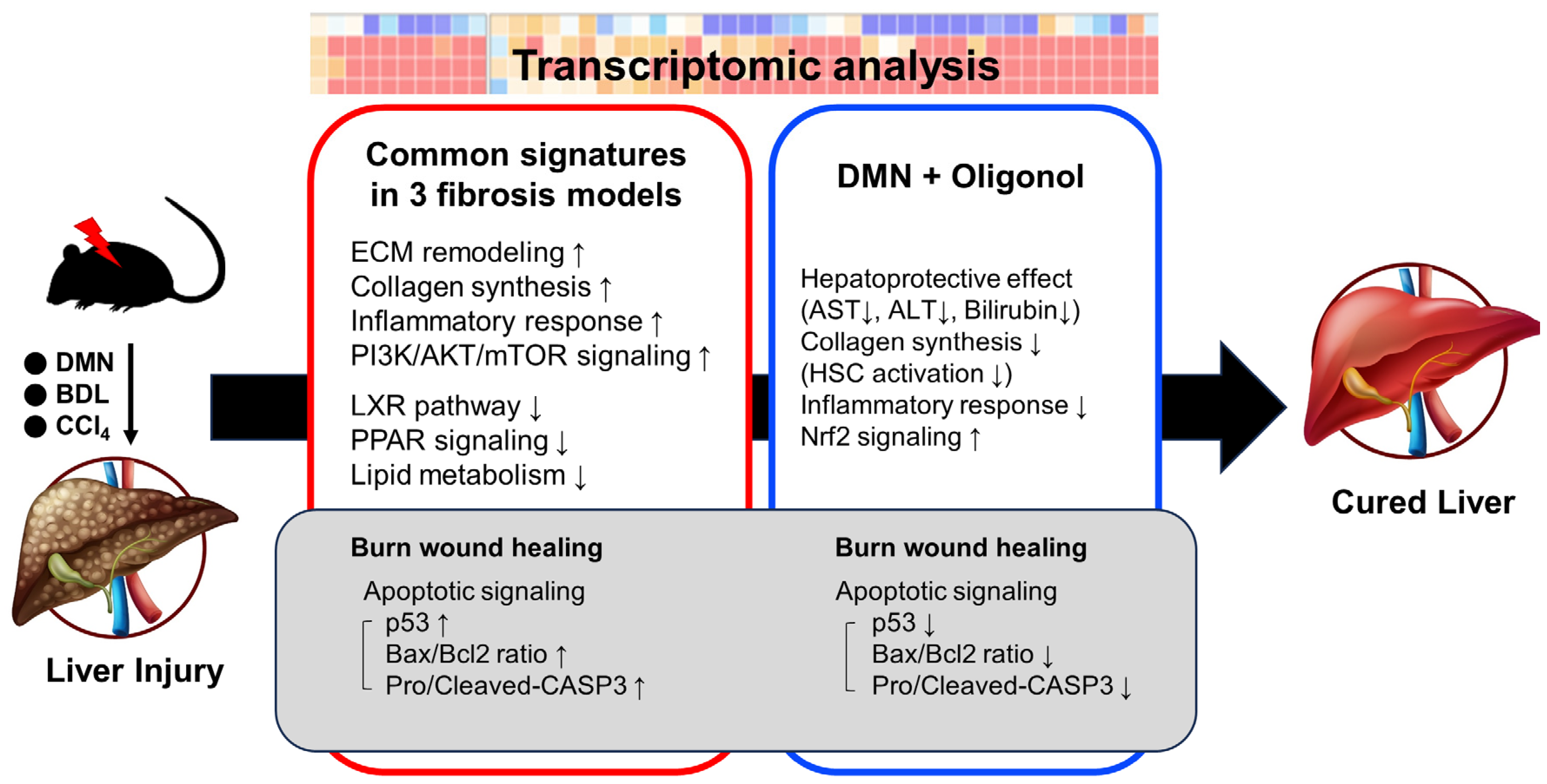
3.1. Common Transcriptomic Signatures of Multiple Liver Fibrosis Models Reveal Known Pathogenic Features of Fibrosis
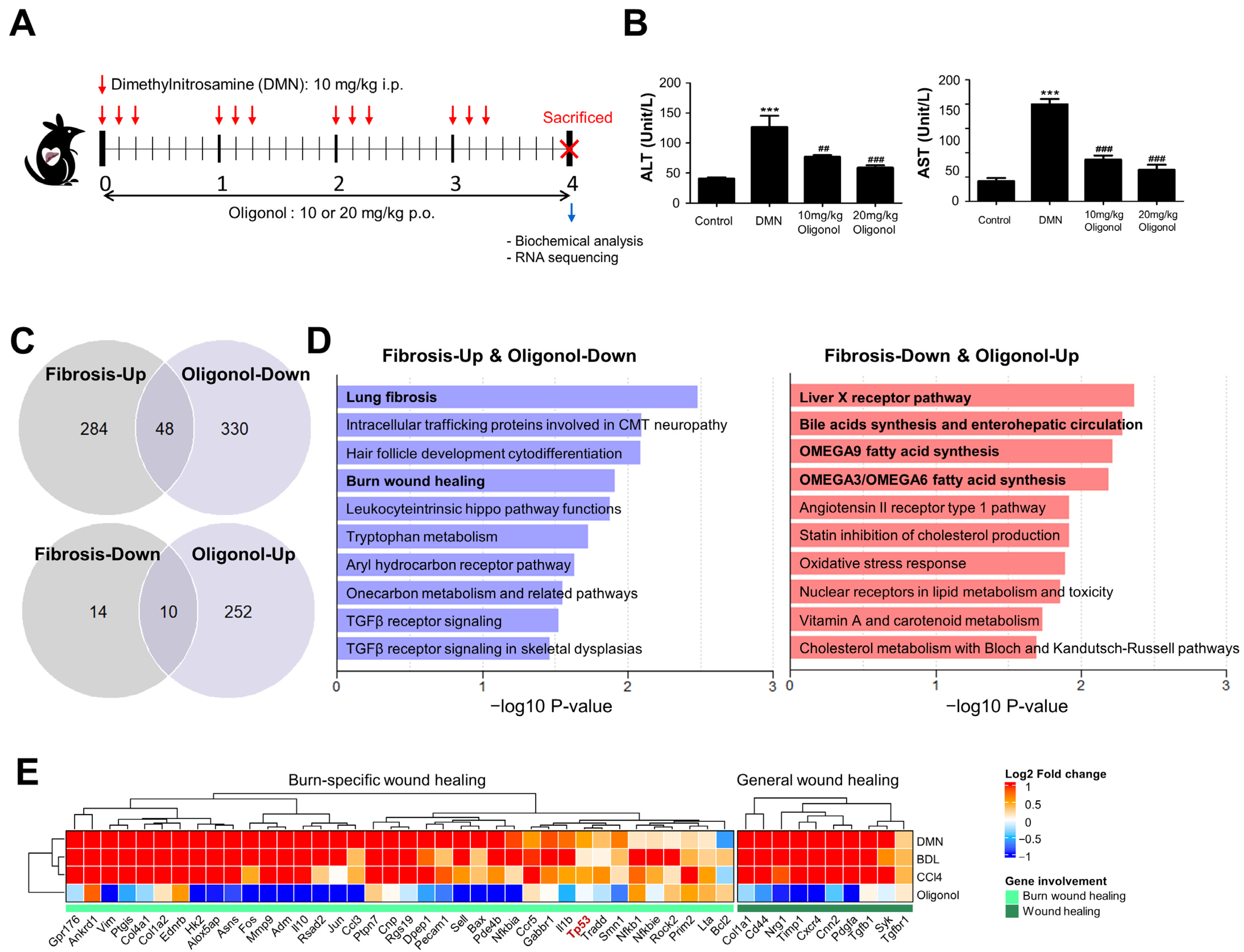
3.2. Transcriptomic Signatures Indicate the Impact of Oligonol Treatment on the Burn-Wound-Healing Pathway in Liver Fibrosis Models
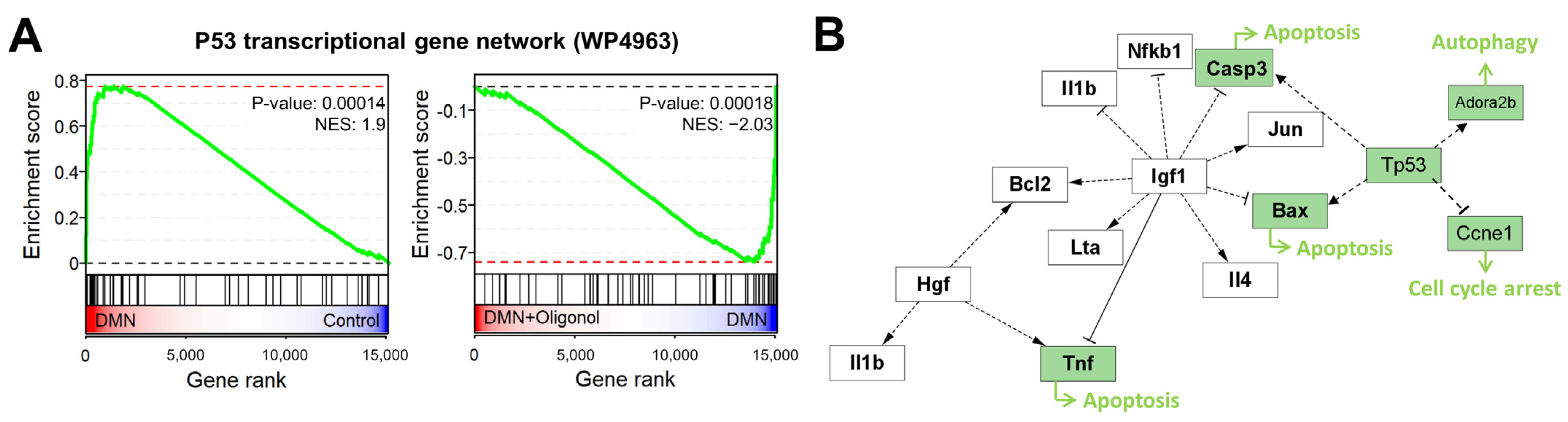
3.3. Expression of p53 Targets Was Overall Elevated in Liver Fibrosis Models
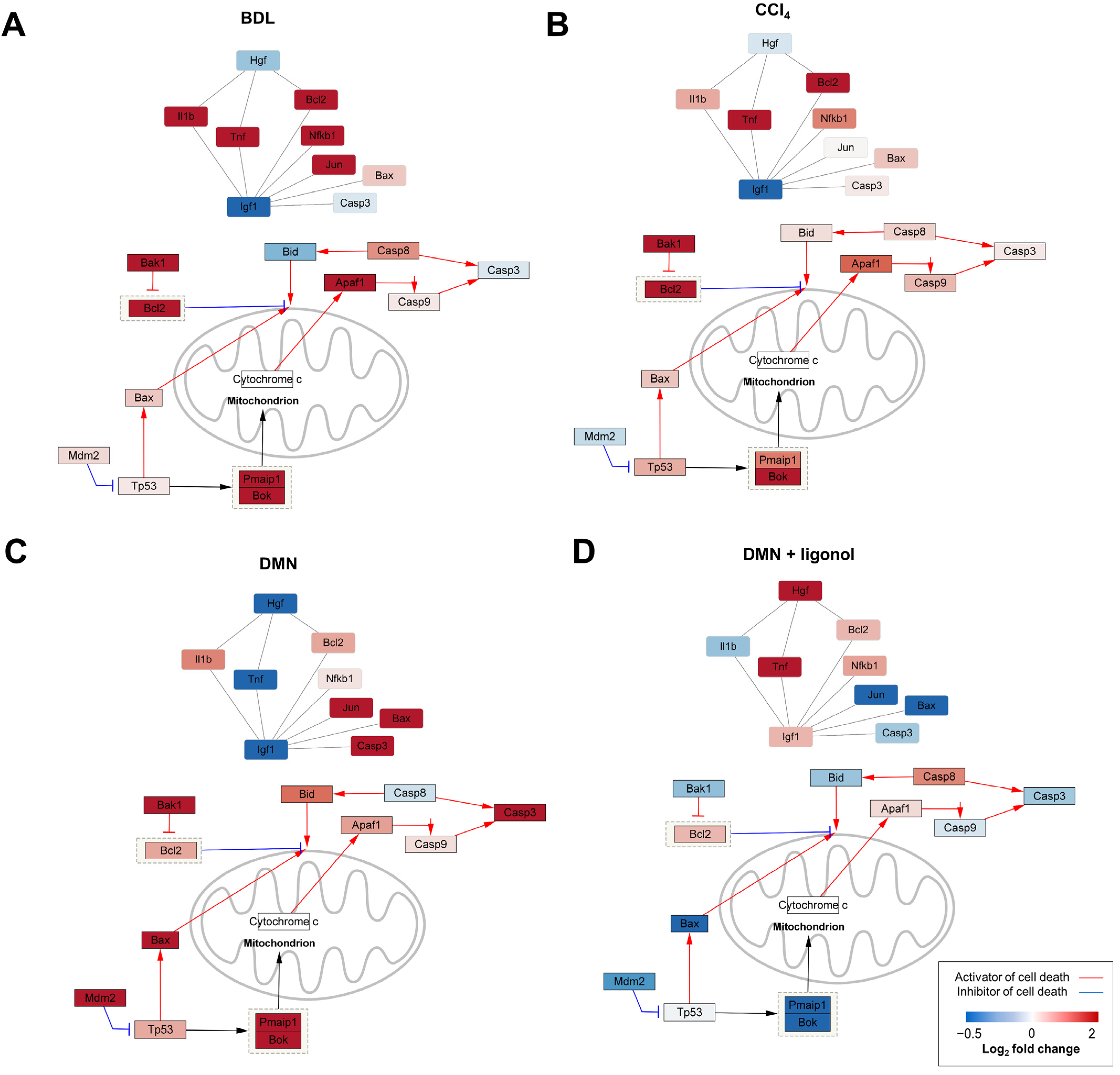
3.4. p53-Associated Apoptotic Genes May Be a Plausible Mechanism in Liver Fibrosis
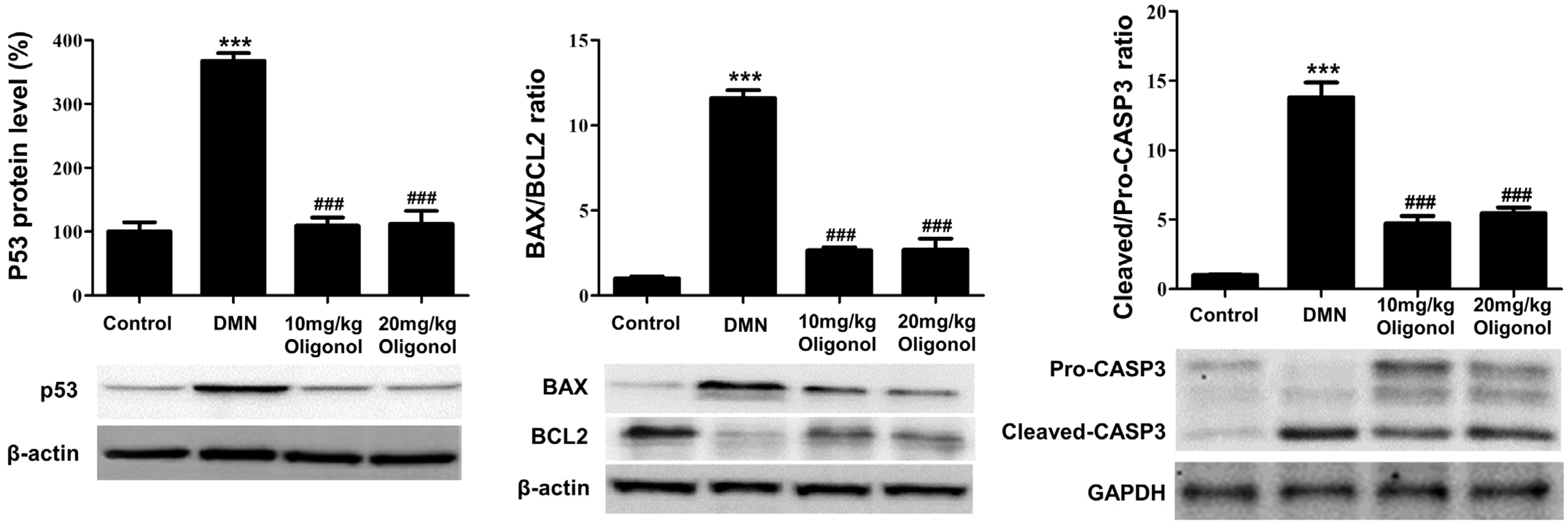
3.5. Apoptotic Genes Affected by DMN Exposure Are Restored with Oligonol Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.X.; Torok, N.J. Liver injury and the activation of the hepatic myofibroblasts. Curr. Pathobiol. Rep. 2013, 1, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.; Francis, M.; DiPietro, L.A. Differential apoptosis in mucosal and dermal wound healing. Adv. Wound Care (New Rochelle) 2014, 3, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Herrera, J.; Henke, C.A.; Bitterman, P.B. Extracellular matrix as a driver of progressive fibrosis. J. Clin. Investig. 2018, 128, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Kisseleva, T.; Cong, M.; Paik, Y.; Scholten, D.; Jiang, C.; Benner, C.; Iwaisako, K.; Moore-Morris, T.; Scott, B.; Tsukamoto, H.; et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 9448–9453. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Conde de la Rosa, L.; Goicoechea, L.; Torres, S.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Role of oxidative stress in liver disorders. Livers 2022, 2, 283–314. [Google Scholar] [CrossRef]
- Guicciardi, M.E.; Gores, G.J. Apoptosis: A mechanism of acute and chronic liver injury. Gut 2005, 54, 1024–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popov, Y.; Schuppan, D. Targeting liver fibrosis: Strategies for development and validation of antifibrotic therapies. Hepatology 2009, 50, 1294–1306. [Google Scholar] [CrossRef] [PubMed]
- Delire, B.; Starkel, P.; Leclercq, I. Animal models for fibrotic liver diseases: What we have, what we need, and what is under development. J. Clin. Transl. Hepatol. 2015, 3, 53–66. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, Z.; Mo, Q.; Zhou, G.; Wang, Y. Gallic acid attenuates dimethylnitrosamine-induced liver fibrosis by alteration of SMAD phosphoisoform signaling in rats. Biomed Res. Int. 2018, 2018, 1682743. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, C.; Luedde, T.; Sauerbruch, T.; Scholten, D.; Streetz, K.; Tacke, F.; Tolba, R.; Trautwein, C.; Trebicka, J.; Weiskirchen, R. Experimental liver fibrosis research: Update on animal models, legal issues and translational aspects. Fibrogenesis Tissue Repair. 2013, 6, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tag, C.G.; Sauer-Lehnen, S.; Weiskirchen, S.; Borkham-Kamphorst, E.; Tolba, R.H.; Tacke, F.; Weiskirchen, R. Bile duct ligation in mice: Induction of inflammatory liver injury and fibrosis by obstructive cholestasis. J. Vis. Exp. 2015, 96, 52438. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Liu, X.; Koyama, Y.; Wang, P.; Lan, T.; Kim, I.G.; Kim, I.H.; Ma, H.Y.; Kisseleva, T. The types of hepatic myofibroblasts contributing to liver fibrosis of different etiologies. Front. Pharmacol. 2014, 5, 167. [Google Scholar] [CrossRef] [Green Version]
- Weiskirchen, R. Hepatoprotective and anti-fibrotic agents: It’s time to take the next step. Front. Pharmacol. 2015, 6, 303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Bak, J.; Yoon, S.; Moon, J.O. Protective effect of oligonol on dimethylnitrosamine-induced liver fibrosis in rats via the JNK/NF-kappaB and PI3K/Art/Nrf2 signaling pathways. Antioxidants 2021, 10, 366. [Google Scholar] [CrossRef]
- Noh, J.S.; Park, C.H.; Yokozawa, T. Treatment with oligonol, a low-molecular polyphenol derived from lychee fruit, attenuates diabetes-induced hepatic damage through regulation of oxidative stress and lipid metabolism. Br. J. Nutr. 2011, 106, 1013–1022. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Park, W.H.; Lee, S.; Suh, D.H.; Kim, K.; No, J.H.; Kim, Y.B. Oligonol, a low molecular weight polyphenol, enhances apoptotic cell death in ovarian cancer cells via suppressing NF-kappaB activation. Nutr. Cancer 2019, 71, 141–148. [Google Scholar] [CrossRef]
- Park, C.H.; Park, K.H.; Hong, S.G.; Lee, J.S.; Baek, J.H.; Lee, G.I.; Heo, J.W.; Yokozawa, T. Oligonol, a low-molecular-weight polyphenol derived from lychee peel, attenuates diabetes-induced pancreatic damage by inhibiting inflammatory responses via oxidative stress-dependent mitogen-activated protein kinase/nuclear factor-kappa B signaling. Phytother. Res. 2018, 32, 2541–2550. [Google Scholar] [CrossRef]
- Kundu, J.K.; Choi, K.-S.; Fujii, H.; Sun, B.; Surh, Y.-J. Oligonol, a lychee fruit-derived low molecular weight polyphenol formulation, inhibits UVB-induced cyclooxygenase-2 expression, and induces NAD(P)H:quinone oxidoreductase-1 expression in hairless mouse skin. J. Funct. Foods 2009, 1, 98–108. [Google Scholar] [CrossRef]
- Reitman, S.; Frankel, S. A colorimetric method for the determination of serum glutamic oxalacetic and glutamic pyruvic transaminases. Am. J. Clin. Pathol. 1957, 28, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Muise, E.S.; Han, S.; Kutchukian, P.S.; Costet, P.; Zhu, Y.; Kan, Y.; Zhou, H.; Shah, V.; Huang, Y.; et al. Molecular profiling reveals a common metabolic signature of tissue fibrosis. Cell Rep. Med. 2020, 1, 100056. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Wen, S.; Li, Y.; Wei, Y.; Fang, B.; Li, C.; Huang, Q.; Lin, X. Comprehensive analysis of transcriptomics and metabolomics to illustrate the underlying mechanism of helenalin against hepatic fibrosis. Eur. J. Pharmacol. 2022, 919, 174770. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Gene Ontology Consortium. Gene ontology consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Caligiuri, A.; Gentilini, A.; Pastore, M.; Gitto, S.; Marra, F. Cellular and molecular mechanisms underlying liver fibrosis regression. Cells 2021, 10, 2759. [Google Scholar] [CrossRef]
- Okunishi, K.; Sisson, T.H.; Huang, S.K.; Hogaboam, C.M.; Simon, R.H.; Peters-Golden, M. Plasmin overcomes resistance to prostaglandin E2 in fibrotic lung fibroblasts by reorganizing protein kinase A signaling. J. Biol. Chem. 2011, 286, 32231–32243. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Wettlaufer, S.H.; Hogaboam, C.; Aronoff, D.M.; Peters-Golden, M. Prostaglandin E(2) inhibits collagen expression and proliferation in patient-derived normal lung fibroblasts via E prostanoid 2 receptor and cAMP signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L405–L413. [Google Scholar] [CrossRef] [Green Version]
- Baum, B.J.; Moss, J.; Breul, S.D.; Berg, R.A.; Crystal, R.G. Effect of cyclic AMP on the intracellular degradation of newly synthesized collagen. J. Biol. Chem. 1980, 255, 2843–2847. [Google Scholar] [CrossRef]
- Tian, L.Y.; Smit, D.J.; Jucker, M. The role of PI3K/AKT/mTOR signaling in hepatocellular carcinoma metabolism. Int. J. Mol. Sci. 2023, 24, 2652. [Google Scholar] [CrossRef]
- Zhang, Q.; Xiang, S.; Liu, Q.; Gu, T.; Yao, Y.; Lu, X. PPARγ antagonizes hypoxia-induced activation of hepatic stellate cell through cross mediating PI3K/AKT and cGMP/PKG signaling. PPAR Res. 2018, 2018, 6970407. [Google Scholar] [CrossRef] [Green Version]
- Calkin, A.C.; Tontonoz, P. Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat. Rev. Mol. Cell Biol. 2012, 13, 213–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A-González, N.; Castrillo, A. Liver X receptors as regulators of macrophage inflammatory and metabolic pathways. Biochim. Biophys. Acta 2011, 1812, 982–994. [Google Scholar] [CrossRef]
- Becares, N.; Gage, M.C.; Voisin, M.; Shrestha, E.; Martin-Gutierrez, L.; Liang, N.; Louie, R.; Pourcet, B.; Pello, O.M.; Luong, T.V.; et al. Impaired LXRalpha phosphorylation attenuates progression of fatty liver disease. Cell Rep. 2019, 26, 984–995.e6. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as metabolic regulators in the liver: Lessons from liver-specific PPAR-null mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Wang, C.; Deng, X.; He, J.; Yang, L.; Yuan, G. ANGPTL8 in metabolic homeostasis: More friend than foe? Open Biol. 2021, 11, 210106. [Google Scholar] [CrossRef]
- Verloh, N.; Einspieler, I.; Utpatel, K.; Menhart, K.; Brunner, S.; Hofheinz, F.; van den Hoff, J.; Wiggermann, P.; Evert, M.; Stroszczynski, C.; et al. In vivo confirmation of altered hepatic glucose metabolism in patients with liver fibrosis/cirrhosis by (18)F-FDG PET/CT. EJNMMI Res. 2018, 8, 98. [Google Scholar] [CrossRef]
- Walter, A.S.; Volkmer, E.; Gauglitz, G.; Böcker, W.; Saller, M.M. Systematic review of molecular pathways in burn wound healing. Burns 2023, in press. [CrossRef]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Ji, G.; Zhang, L. The role of p53 in liver fibrosis. Front. Pharmacol. 2022, 13, 1057829. [Google Scholar] [CrossRef]
- Kodama, T.; Takehara, T.; Hikita, H.; Shimizu, S.; Shigekawa, M.; Tsunematsu, H.; Li, W.; Miyagi, T.; Hosui, A.; Tatsumi, T.; et al. Increases in p53 expression induce CTGF synthesis by mouse and human hepatocytes and result in liver fibrosis in mice. J. Clin. Investig. 2011, 121, 3343–3356. [Google Scholar] [CrossRef] [Green Version]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [Green Version]
- Khurana, A.; Sayed, N.; Allawadhi, P.; Weiskirchen, R. It’s all about the spaces between cells: Role of extracellular matrix in liver fibrosis. Ann. Transl. Med. 2021, 9, 728. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Jiang, Z.; Li, J.; Lin, H.; Xu, B.; Liao, X.; Fu, Z.; Ao, H.; Guo, G.; Liu, M. Metformin improves burn wound healing by modulating microenvironmental fibroblasts and macrophages. Cells 2022, 11, 4094. [Google Scholar] [CrossRef]
- Jiang, J.X.; Venugopal, S.; Serizawa, N.; Chen, X.; Scott, F.; Li, Y.; Adamson, R.; Devaraj, S.; Shah, V.; Gershwin, M.E.; et al. Reduced nicotinamide adenine dinucleotide phosphate oxidase 2 plays a key role in stellate cell activation and liver fibrogenesis in vivo. Gastroenterology 2010, 139, 1375–1384. [Google Scholar] [CrossRef] [Green Version]
- Takehara, T.; Tatsumi, T.; Suzuki, T.; Rucker, E.B., 3rd; Hennighausen, L.; Jinushi, M.; Miyagi, T.; Kanazawa, Y.; Hayashi, N. Hepatocyte-specific disruption of Bcl-xL leads to continuous hepatocyte apoptosis and liver fibrotic responses. Gastroenterology 2004, 127, 1189–1197. [Google Scholar] [CrossRef]
- Hirsova, P.; Bohm, F.; Dohnalkova, E.; Nozickova, B.; Heikenwalder, M.; Gores, G.J.; Weber, A. Hepatocyte apoptosis is tumor promoting in murine nonalcoholic steatohepatitis. Cell Death Dis. 2020, 11, 80. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.H.; Hu, S.J.; Yu, B.; Jiang, S.S.; Zhang, J.; Luo, D.; Yang, M.W.; Su, W.Y.; Shao, Y.L.; Deng, H.L.; et al. Apoptosis in the aging liver. Oncotarget 2017, 8, 102640–102652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.C.; Han, X.; Dou, J.Y.; Yuan, M.H.; Zhou, M.J.; Cui, Z.Y.; Lian, L.H.; Nan, J.X.; Zhang, X.; Wu, Y.L. Protective role of Siberian onions against toxin-induced liver dysfunction: An insight into health-promoting effects. Food Funct. 2022, 13, 4678–4690. [Google Scholar] [CrossRef] [PubMed]
- Chaitanya, G.V.; Steven, A.J.; Babu, P.P. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal 2010, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Aruoma, O.I.; Sun, B.; Fujii, H.; Neergheen, V.S.; Bahorun, T.; Kang, K.S.; Sung, M.K. Low molecular proanthocyanidin dietary biofactor oligonol: Its modulation of oxidative stress, bioefficacy, neuroprotection, food application and chemoprevention potentials. Biofactors 2006, 27, 245–265. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.Y.; Maeda, T.; Fujii, H.; Yokozawa, T.; Kim, H.Y.; Cho, E.J.; Shibamoto, T. Oligonol improves memory and cognition under an amyloid β25–35—induced Alzheimer’s mouse model. Nutr. Res. 2014, 34, 595–603. [Google Scholar] [CrossRef] [Green Version]
- Park, C.H.; Lee, J.Y.; Kim, M.Y.; Shin, S.H.; Roh, S.S.; Choi, J.S.; Chung, H.Y.; Song, Y.O.; Shin, Y.S.; Yokozawa, T. Oligonol, a low-molecular-weight polyphenol derived from lychee fruit, protects the pancreas from apoptosis and proliferation via oxidative stress in streptozotocin-induced diabetic rats. Food Funct. 2016, 7, 3056–3063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yum, H.W.; Zhong, X.; Park, J.; Na, H.K.; Kim, N.; Lee, H.S.; Surh, Y.J. Oligonol inhibits dextran sulfate sodium-induced colitis and colonic adenoma formation in mice. Antioxid. Redox Signal 2013, 19, 102–114. [Google Scholar] [CrossRef] [Green Version]
- Issa, R.; Williams, E.; Trim, N.; Kendall, T.; Arthur, M.J.; Reichen, J.; Benyon, R.C.; Iredale, J.P. Apoptosis of hepatic stellate cells: Involvement in resolution of biliary fibrosis and regulation by soluble growth factors. Gut 2001, 48, 548–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biological Samples | Source | Animal/Sex/Age | Sequencing Platform | No. of Samples (Per Group) |
|---|---|---|---|---|
| Livers from DMN Model | This study | SD rat/male/6~7 weeks | DNBSEQ-T7 | 3 (1 normal, 1 fibrosis, 1 oligonol) |
| Livers from BDL Model | GEO (GSE152250, [22]) | SD rat/male/6~8 weeks | HiSeq 4000 | 11 (6 normal, 5 fibrosis) |
| Livers from CCl4 Model | GEO (GSE189402, [23]) | SD rat/male/8 weeks | NovaSeq 6000 | 6 (3 normal, 3 fibrosis) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Lee, C.; Noh, S.G.; Kim, S.; Chung, H.Y.; Lee, H.; Moon, J.-O. Integrative Transcriptomic Analysis Reveals Upregulated Apoptotic Signaling in Wound-Healing Pathway in Rat Liver Fibrosis Models. Antioxidants 2023, 12, 1588. https://doi.org/10.3390/antiox12081588
Kim J, Lee C, Noh SG, Kim S, Chung HY, Lee H, Moon J-O. Integrative Transcriptomic Analysis Reveals Upregulated Apoptotic Signaling in Wound-Healing Pathway in Rat Liver Fibrosis Models. Antioxidants. 2023; 12(8):1588. https://doi.org/10.3390/antiox12081588
Chicago/Turabian StyleKim, Jihyun, Changyong Lee, Sang Gyun Noh, Seungwoo Kim, Hae Young Chung, Haeseung Lee, and Jeon-Ok Moon. 2023. "Integrative Transcriptomic Analysis Reveals Upregulated Apoptotic Signaling in Wound-Healing Pathway in Rat Liver Fibrosis Models" Antioxidants 12, no. 8: 1588. https://doi.org/10.3390/antiox12081588