
Advances in Understanding the Role of NRF2 in Liver Pathophysiology and Its Relationship with Hepatic-Specific Cyclooxygenase-2 Expression
 , ,
, ,
Abstract
:1. Introduction
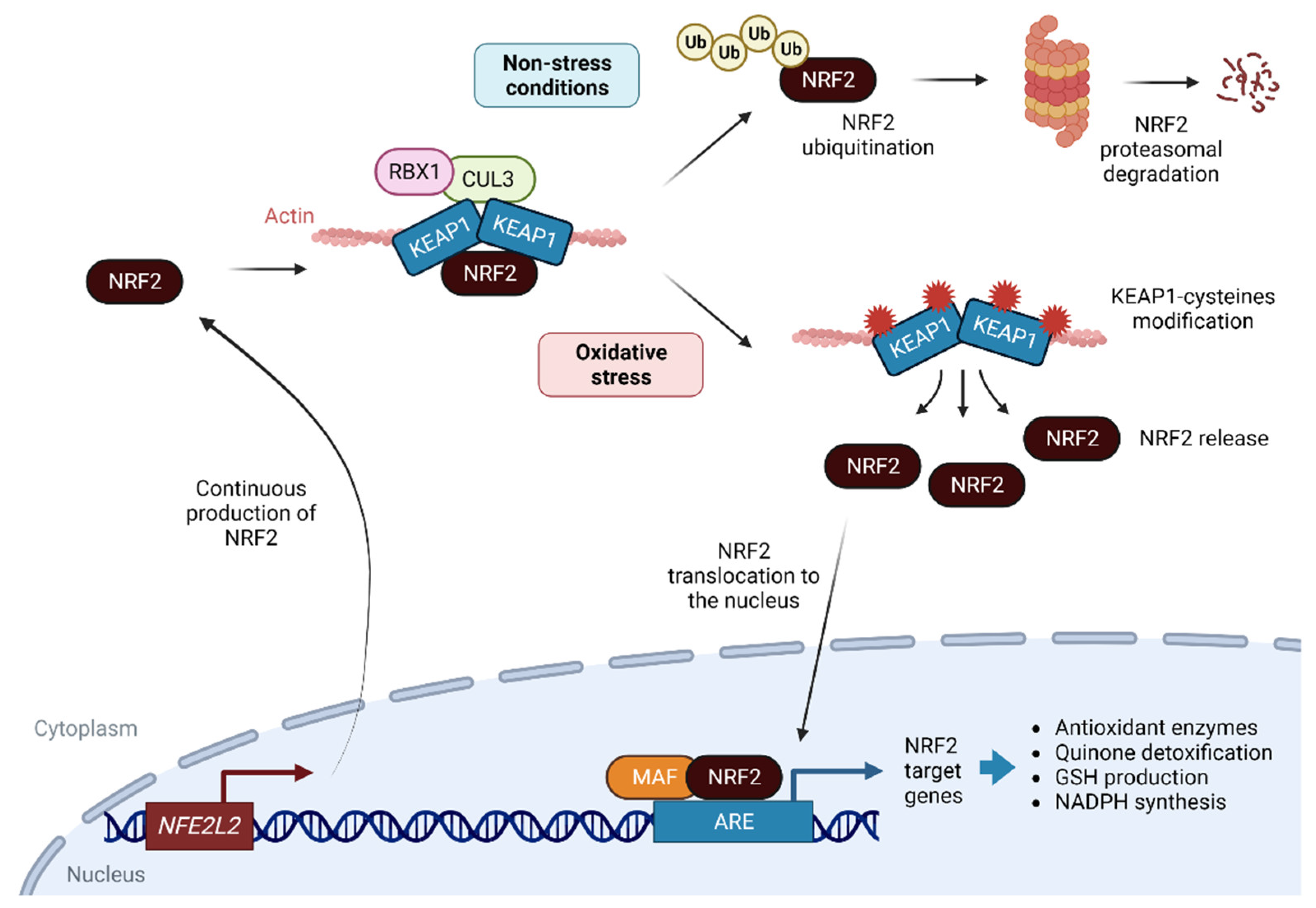
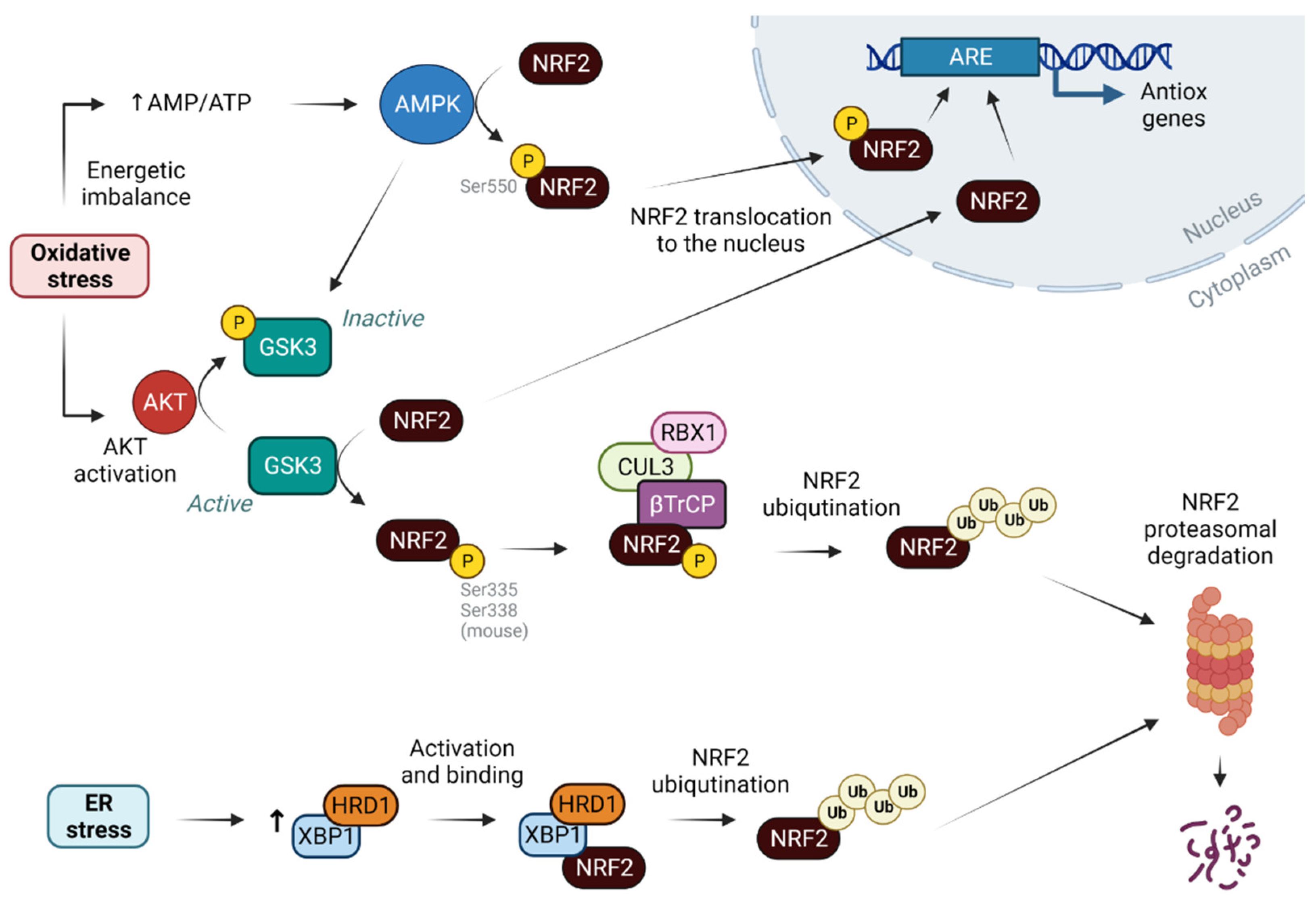
2. NRF2 as a Sensor of Cellular Redox State
3. NRF2 and Its Antioxidant Role
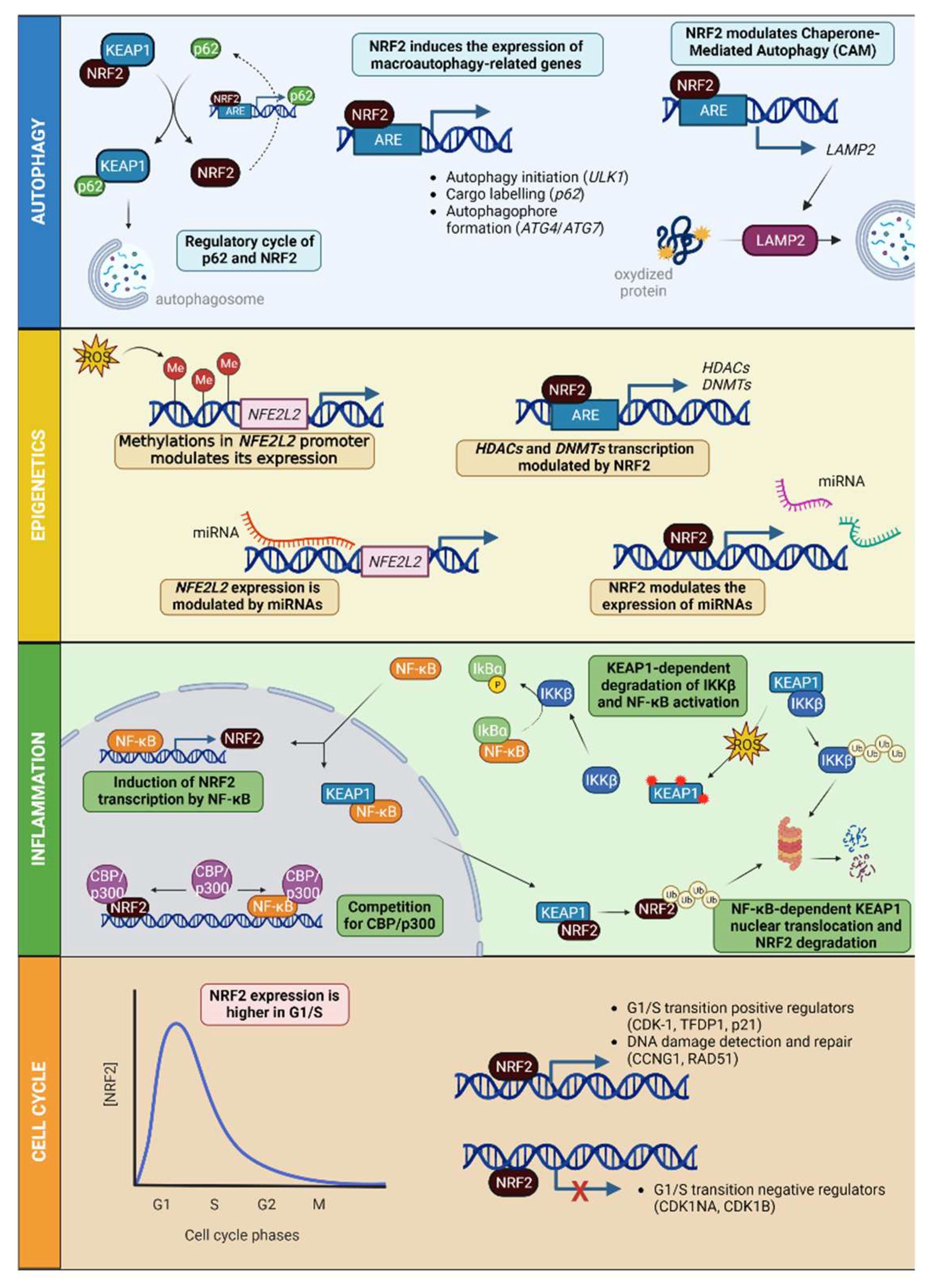
4. NRF2 beyond Its Antioxidant Role
4.1. NRF2 in Autophagy and Protein Degradation
4.2. NRF2, Epigenetics and miRNAs
4.3. NRF2 Interaction with NF-κB and Inflammation
4.4. NRF2 in Cell Cycle
5. NRF2 in Liver Pathology
5.1. NRF2 in Liver Inflammation
5.2. NRF2 and Insulin Resistance
5.3. NRF2 and Liver Regeneration
5.4. NRF2 in Acute Liver Injury
5.5. NRF2 in MAFLD/NASH
5.6. NRF2 in ALD
5.7. NRF2 in Hepatocellular Carcinoma (HCC)
5.8. NRF2 in Ischemia-Reperfusion Injury

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Liver Pathology | NRF2 Regulation | Consequence of NRF2 Regulation | Reference |
|---|---|---|---|
| Inflammation | Blocking of the recruitment of RNA Pol II | Inhibition of pro-inflammatory cytokines | [72] |
| Insulin resistance | Increase the phosphorylation of AMPK in the liver and the insulin signaling in skeletal muscle | Improvement of glucose tolerance | [75] |
| Acute liver injury | Absence of NRF2 in APAP treatment activates CYP4204E1, decreases GSH levels and accumulates ROS. | Enhancement of oxidative stress | [92] |
| Absence of NRF2 enhances oxidative stress when treated with both APAP and ConA | The liver is unable to detoxify xenobiotics | [93,94] | |
| KEAP1-dependent release of NRF2 by natural components | Reduce cell death and tissue damage by increasing hepatic detoxification capacity | [97] | |
| MAFLD/NASH | Inhibition of lipogenesis and increase fatty acid oxidation through the activation of ARE-containing transcription factors | Improvement of steatosis | [9] |
| Suppression of key enzymes involved in lipid synthesis and reduction of hepatic lipid storage | [105] | ||
| Inhibition of LXRα activity, LXRα-dependent liver steatosis and SREBP-1c and lipogenic genes | [106] | ||
| Inhibition of NOX-2 activation | Enhancement of hepatocellular antioxidant capacity and improvement of mitochondrial dysfunction | [108] | |
| Inhibition of TGFβ signaling in stellate cells | Reduction of liver fibrosis | [112] | |
| ALD | Upregulation of hepatic very low-density lipoprotein receptor levels | Contribution to the pathogenesis of ALD | [119] |
| CYP2E1 detoxifying enzyme increase NRF2 expression | Increase HMOX1 expression | [120] | |
| HCC | Reduction of apoptosis and downregulation of genes involved in cell cycle regulation and DNA replication | Reduction of fibrogenesis, lower tumor incidence, reduced tumor number and size | [74] |
| Increased NRF2 levels may promote cancer cell proliferation | Contribution to pro-oncogenic pathways and therefore to HCC | [126] | |
| Elevated NRF2 levels could promote cancer cells to be less sensitive to chemotherapy and ionizing radiation | [127,128] | ||
| I/R | Regulation of the key genes in NADPH synthesis (G6PD, PGD, MEI, IDH) and activation of NQO1 and NQO2 | Control of NADPH levels, glutathione metabolism, and lipid biosynthesis | [133] |
| Upregulation of anti-apoptotic BCL-2 proteins and reduction of BAX, cytochrome c release and caspase activation | Prevention of apoptosis | [134,135,136] | |
| Activation of yes-associated protein 1 (YAP) and genes involved in regeneration (CTGF, CYR61, and ANKRD1) or phase II enzymes (MnSOD and CAT). Reduction of ROS production, and infiltration of CD68+7-Ly6G+ neutrophils | Protection against I/R damage | [138] |
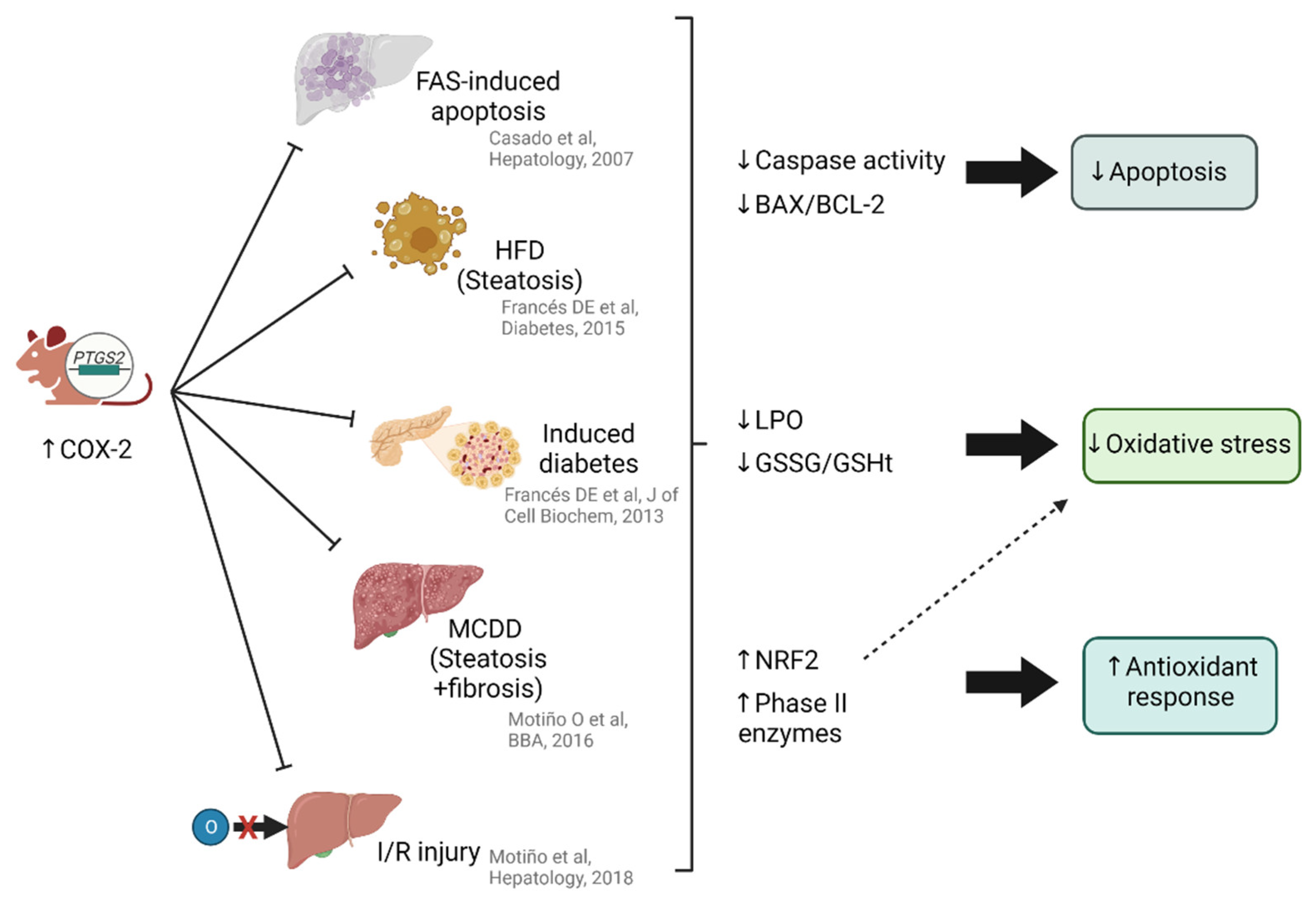
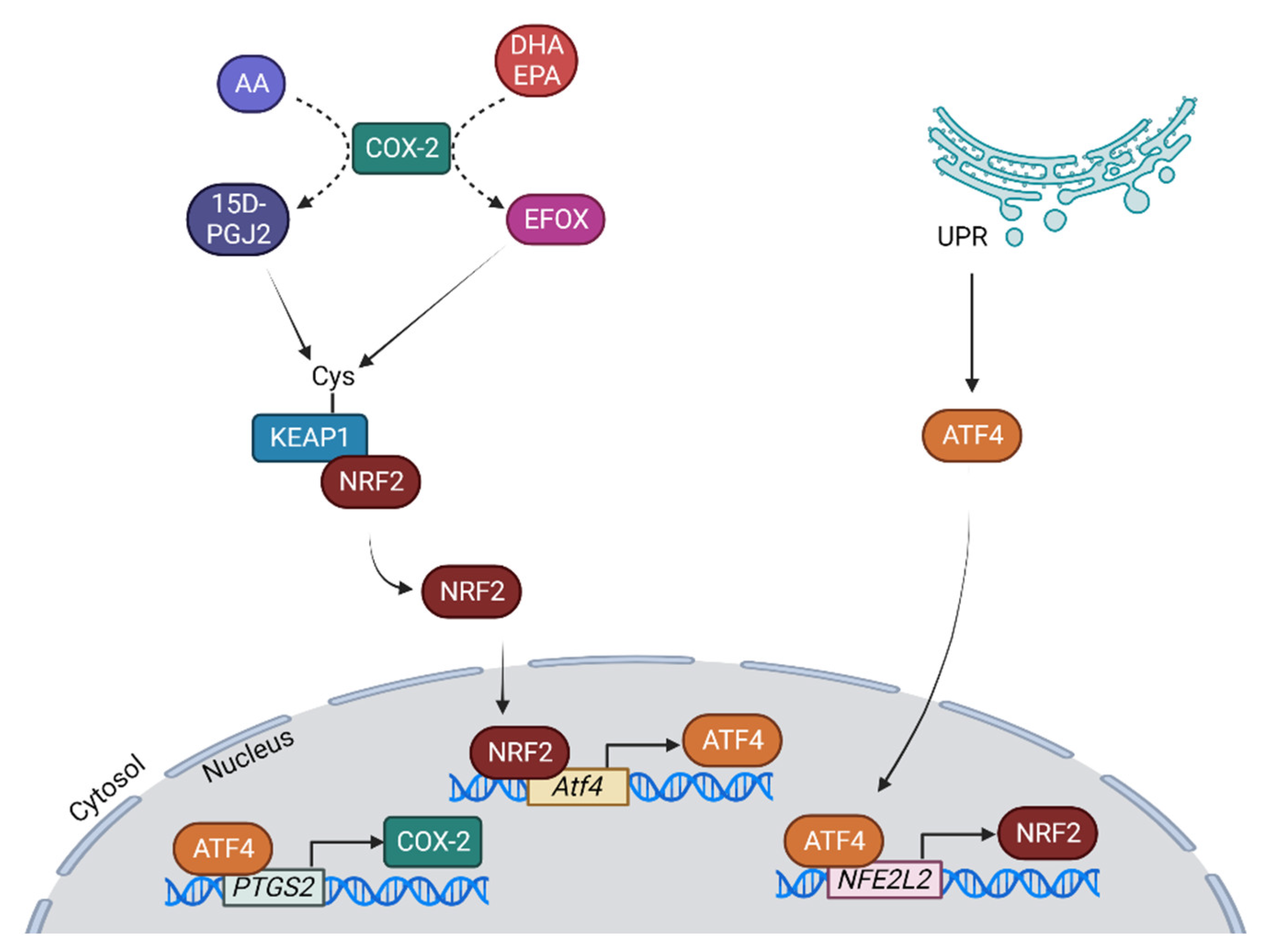
6. The Relationship between NRF2 and Cyclooxygenase 2 (COX-2) in Liver Pathology
7. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Harman, D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Del Río, L.A.; Lopez-Huertas, E. ROS Generation in Peroxisomes and its Role in Cell Signaling. Plant Cell Physiol. 2016, 57, pcw076. [Google Scholar] [CrossRef]
- Cederbaum, A.I. Cytochrome P450 2E1-dependent oxidant stress and upregulation of anti-oxidant defense in liver cells. J. Gastroenterol. Hepatol. 2006, 21 (Suppl. S3), S22–S25. [Google Scholar] [CrossRef]
- Rada, B.; Leto, T.L. Oxidative Innate Immune Defenses by Nox/Duox Family NADPH Oxidases. Contrib. Microbiol. 2008, 15, 164–187. [Google Scholar] [CrossRef] [Green Version]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Checa, J.; Aran, J.M. Reactive Oxygen Species: Drivers of Physiological and Pathological Processes. J. Inflamm. Res. 2020, 13, 1057–1073. [Google Scholar] [CrossRef]
- Brieger, K.; Schiavone, S.; Miller, F.J., Jr.; Krause, K.-H. Reactive oxygen species: From health to disease. Swiss Med. Wkly. 2012, 142, w13659. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Callejas, N.A.; Boscá, L.; Williams, C.S.; DuBois, R.N.; Martín-Sanz, P. Regulation of cyclooxygenase 2 expression in hepatocytes by CCAAT/enhancer-binding proteins. Gastroenterology 2000, 119, 493–501. [Google Scholar] [CrossRef]
- Francés, D.E.; Motiño, O.; Agrá, N.; González-Rodríguez, Á.; Fernández-Álvarez, A.; Cucarella, C.; Mayoral, R.; Castro-Sánchez, L.; García-Casarrubios, E.; Boscá, L.; et al. Hepatic Cyclooxygenase-2 Expression Protects Against Diet-Induced Steatosis, Obesity, and Insulin Resistance. Diabetes 2015, 64, 1522–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motiño, O.; Agra, N.; Contreras, R.B.; Domínguez-Moreno, M.; García-Monzón, C.; Vargas-Castrillón, J.; Carnovale, C.E.; Boscá, L.; Casado, M.; Mayoral, R.; et al. Cyclooxygenase-2 expression in hepatocytes attenuates non-alcoholic steatohepatitis and liver fibrosis in mice. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2016, 1862, 1710–1723. [Google Scholar] [CrossRef] [PubMed]
- Motiño, O.; Francés, D.E.; Casanova, N.; Fuertes-Agudo, M.; Cucarella, C.; Flores, J.M.; Vallejo-Cremades, M.T.; Olmedilla, L.; Peña, J.P.; Bañares, R.; et al. Protective Role of Hepatocyte Cyclooxygenase-2 Expression Against Liver Ischemia–Reperfusion Injury in Mice. Hepatology 2019, 70, 650–665. [Google Scholar] [CrossRef]
- Fuertes-Agudo, M.; Luque-Tévar, M.; Cucarella, C.; Brea, R.; Boscá, L.; Quintana-Cabrera, R.; Martín-Sanz, P.; Casado, M. COX-2 Expression in Hepatocytes Improves Mitochondrial Function after Hepatic Ischemia-Reperfusion Injury. Antioxidants 2022, 11, 1724. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.-L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [Green Version]
- Tong, K.I.; Padmanabhan, B.; Kobayashi, A.; Shang, C.; Hirotsu, Y.; Yokoyama, S.; Yamamoto, M. Different Electrostatic Potentials Define ETGE and DLG Motifs as Hinge and Latch in Oxidative Stress Response. Mol. Cell. Biol. 2007, 27, 7511–7521. [Google Scholar] [CrossRef] [Green Version]
- Motohashi, H.; Yamamoto, M. Nrf2–Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef]
- Rushmore, T.H.; Morton, M.R.; Pickett, C.B. The Antioxidant Responsive Element. Activation by Oxidative Stress and Identification of the DNA Consensus Sequence Required for Functional Activity. J. Biol. Chem. 1991, 266, 11632–11639. [Google Scholar] [CrossRef]
- Kopacz, A.; Kloska, D.; Forman, H.J.; Jozkowicz, A.; Grochot-Przeczek, A. Beyond repression of Nrf2: An update on Keap1. Free. Radic. Biol. Med. 2020, 157, 63–74. [Google Scholar] [CrossRef]
- Cuadrado, A. Structural and functional characterization of Nrf2 degradation by glycogen synthase kinase 3/β-TrCP. Free. Radic. Biol. Med. 2015, 88, 147–157. [Google Scholar] [CrossRef]
- Reichard, J.F.; Motz, G.T.; Puga, A. Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1. Nucleic Acids Res. 2007, 35, 7074–7086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.; Xu, C.; Pan, Z.; Keum, Y.-S.; Kim, J.-H.; Shen, G.; Yu, S.; Oo, K.T.; Ma, J.; Kong, A.-N.T. Butylated hydroxyanisole regulates ARE-mediated gene expression via Nrf2 coupled with ERK and JNK signaling pathway in HepG2 cells. Mol. Carcinog. 2006, 45, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP Augments Promoter-Specific DNA Binding of Nrf2 during the Antioxidant Response. Mol. Cell. Biol. 2009, 29, 2658–2672. [Google Scholar] [CrossRef] [Green Version]
- Kawai, Y.; Garduño, L.; Theodore, M.; Yang, J.; Arinze, I.J. Acetylation-Deacetylation of the Transcription Factor Nrf2 (Nuclear Factor Erythroid 2-related Factor 2) Regulates Its Transcriptional Activity and Nucleocytoplasmic Localization. J. Biol. Chem. 2011, 286, 7629–7640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mobasher, M.A.; González-Rodriguez, A.; Santamaría, B.; Ramos, S.; Martín, M.Á.; Goya, L.; Rada, P.; Letzig, L.; James, L.P.; Cuadrado, A.; et al. Protein tyrosine phosphatase 1B modulates GSK3β/Nrf2 and IGFIR signaling pathways in acetaminophen-induced hepatotoxicity. Cell Death Dis. 2013, 4, e626. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, K.; Hirano, I.; Itoh, T.; Tanaka, M.; Miyajima, A.; Suzuki, A.; Motohashi, H.; Yamamoto, M. Nrf2 Enhances Cholangiocyte Expansion in Pten-Deficient Livers. Mol. Cell. Biol. 2014, 34, 900–913. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.; Scott, J.W.; Pan, D.A.; Hudson, E.R. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003, 546, 113–120. [Google Scholar] [CrossRef]
- Joo, M.S.; Kim, W.D.; Lee, K.Y.; Kim, J.H.; Koo, J.H.; Kim, S.G. AMPK Facilitates Nuclear Accumulation of Nrf2 by Phosphorylating at Serine 550. Mol. Cell. Biol. 2016, 36, 1931–1942. [Google Scholar] [CrossRef] [Green Version]
- Rada, P.; Rojo, A.I.; Offergeld, A.; Feng, G.J.; Velasco-Martín, J.P.; González-Sancho, J.M.; Valverde, Á.M.; Dale, T.; Regadera, J.; Cuadrado, A.; et al. WNT-3A Regulates an Axin1/NRF2 Complex That Regulates Antioxidant Metabolism in Hepatocytes. Antioxid. Redox Signal. 2015, 22, 555–571. [Google Scholar] [CrossRef]
- Wu, T.; Zhao, F.; Gao, B.; Tan, C.; Yagishita, N.; Nakajima, T.; Wong, P.K.; Chapman, E.; Fang, D.; Zhang, D.D. Hrd1 Suppresses Nrf2-Mediated Cellular Protection during Liver Cirrhosis. Genes Dev. 2014, 28, 708–722. [Google Scholar] [CrossRef] [Green Version]
- Silva-Llanes, I.; Shin, C.H.; Jiménez-Villegas, J.; Gorospe, M.; Lastres-Becker, I. The Transcription Factor NRF2 Has Epigenetic Regulatory Functions Modulating HDACs, DNMTs, and miRNA Biogenesis. Antioxidants 2023, 12, 641. [Google Scholar] [CrossRef]
- Kovac, S.; Angelova, P.R.; Holmström, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2015, 1850, 794–801. [Google Scholar] [CrossRef] [Green Version]
- Erkens, R.; Suvorava, T.; Sutton, T.R.; Fernandez, B.O.; Mikus-Lelinska, M.; Barbarino, F.; Flögel, U.; Kelm, M.; Feelisch, M.; Cortese-Krott, M.M. Nrf2 Deficiency Unmasks the Significance of Nitric Oxide Synthase Activity for Cardioprotection. Oxidative Med. Cell. Longev. 2018, 2018, 8309698. [Google Scholar] [CrossRef] [Green Version]
- Kopacz, A.; Klóska, D.; Proniewski, B.; Cysewski, D.; Personnic, N.; Piechota-Polańczyk, A.; Kaczara, P.; Zakrzewska, A.; Forman, H.J.; Dulak, J.; et al. Keap1 controls protein S-nitrosation and apoptosis-senescence switch in endothelial cells. Redox Biol. 2020, 28, 101304. [Google Scholar] [CrossRef]
- Rosa, A.C.; Corsi, D.; Cavi, N.; Bruni, N.; Dosio, F. Superoxide Dismutase Administration: A Review of Proposed Human Uses. Molecules 2021, 26, 1844. [Google Scholar] [CrossRef]
- Trist, B.G.; Hilton, J.B.; Hare, D.J.; Crouch, P.J.; Double, K.L. Superoxide Dismutase 1 in Health and Disease: How a Frontline Antioxidant Becomes Neurotoxic. Angew. Chem. Int. Ed. 2021, 60, 9215–9246. [Google Scholar] [CrossRef]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.H.; Tran, G.-B.; Nguyen, C.T. Anti-oxidative effects of superoxide dismutase 3 on inflammatory diseases. J. Mol. Med. 2020, 98, 59–69. [Google Scholar] [CrossRef]
- Zeeshan, H.M.A.; Lee, G.H.; Kim, H.-R.; Chae, H.-J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Zhang, Y.; Dusting, G.J. NADPH Oxidase-Mediated Redox Signaling: Roles in Cellular Stress Response, Stress Tolerance, and Tissue Repair. Pharmacol. Rev. 2011, 63, 218–242. [Google Scholar] [CrossRef] [Green Version]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 Is a Direct PERK Substrate and Effector of PERK-Dependent Cell Survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarcinelli, C.; Dragic, H.; Piecyk, M.; Barbet, V.; Duret, C.; Barthelaix, A.; Ferraro-Peyret, C.; Fauvre, J.; Renno, T.; Chaveroux, C.; et al. ATF4-Dependent NRF2 Transcriptional Regulation Promotes Antioxidant Protection during Endoplasmic Reticulum Stress. Cancers 2020, 12, 569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [Green Version]
- Pajares, M.; Jiménez-Moreno, N.; García-Yagüe, Á.J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rábano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef] [Green Version]
- Milan, G.; Romanello, V.; Pescatore, F.; Armani, A.; Paik, J.-H.; Frasson, L.; Seydel, A.; Zhao, J.; Abraham, R.; Goldberg, A.L.; et al. Regulation of Autophagy and the Ubiquitin-Proteasome System by the FoxO Transcriptional Network during Muscle Atrophy. Nat. Commun. 2015, 6, 6670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajares, M.; Rojo, A.I.; Arias, E.; Díaz-Carretero, A.; Cuervo, A.M.; Cuadrado, A. Transcription factor NFE2L2/NRF2 modulates chaperone-mediated autophagy through the regulation of LAMP2A. Autophagy 2018, 14, 1310–1322. [Google Scholar] [CrossRef] [Green Version]
- Hedman, Å.K.; Zilmer, M.; Sundström, J.; Lind, L.; Ingelsson, E. DNA methylation patterns associated with oxidative stress in an ageing population. BMC Med. Genom. 2016, 9, 72. [Google Scholar] [CrossRef] [Green Version]
- Bu, H.; Wedel, S.; Cavinato, M.; Jansen-Dürr, P. MicroRNA Regulation of Oxidative Stress-Induced Cellular Senescence. Oxidative Med. Cell. Longev. 2017, 2017, 2398696. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Khor, T.O.; Cheung, K.-L.; Li, W.; Wu, T.-Y.; Huang, Y.; Foster, B.A.; Kan, Y.W.; Kong, A.-N. Nrf2 Expression Is Regulated by Epigenetic Mechanisms in Prostate Cancer of TRAMP Mice. PLoS ONE 2010, 5, e8579. [Google Scholar] [CrossRef] [Green Version]
- Taheri, Z.; Aghdaei, H.A.; Irani, S.; Modarressi, M.H.; Noormohammadi, Z. Evaluation of the Epigenetic Demethylation of NRF2, a Master Transcription Factor for Antioxidant Enzymes, in Colorectal Cancer. Rep. Biochem. Mol. Biol. 2020, 9, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.M.; Rushworth, S.A.; Murray, M.Y.; Bowles, K.M.; MacEwan, D.J. Understanding the role of NRF2-regulated miRNAs in human malignancies. Oncotarget 2013, 4, 1130–1142. [Google Scholar] [CrossRef] [Green Version]
- Quiles, J.M.; Pepin, M.E.; Sunny, S.; Shelar, S.B.; Challa, A.K.; Dalley, B.; Hoidal, J.R.; Pogwizd, S.M.; Wende, A.R.; Rajasekaran, N.S. Identification of Nrf2-responsive microRNA networks as putative mediators of myocardial reductive stress. Sci. Rep. 2021, 11, 11977. [Google Scholar] [CrossRef] [PubMed]
- Kaundal, R.K.; Datusalia, A.K.; Sharma, S.S. Posttranscriptional regulation of Nrf2 through miRNAs and their role in Alzheimer’s disease. Pharmacol. Res. 2022, 175, 106018. [Google Scholar] [CrossRef]
- Cheng, X.; Ku, C.-H.; Siow, R.C. Regulation of the Nrf2 antioxidant pathway by microRNAs: New players in micromanaging redox homeostasis. Free Radic. Biol. Med. 2013, 64, 4–11. [Google Scholar] [CrossRef]
- Xu, W.; Li, F.; Liu, Z.; Xu, Z.; Sun, B.; Cao, J.; Liu, Y. MicroRNA-27b inhibition promotes Nrf2/ARE pathway activation and alleviates intracerebral hemorrhage-induced brain injury. Oncotarget 2017, 8, 70669–70684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Mata, M.; Gaidatzis, D.; Vitanescu, M.; Stadler, M.B.; Wentzel, C.; Scheiffele, P.; Filipowicz, W.; Großhans, H. Potent degradation of neuronal mi RNA s induced by highly complementary targets. EMBO Rep. 2015, 16, 500–511. [Google Scholar] [CrossRef] [Green Version]
- Karin, M.; Ben-Neriah, Y. Phosphorylation Meets Ubiquitination: The Control of NF-κB Activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef]
- Lawrence, T. The Nuclear Factor NF-kappa B Pathway in Inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.J.; Liu, Z.-G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.-F.; Kuo, H.-P.; Liu, M.; Chou, C.-K.; Xia, W.; Du, Y.; Shen, J.; Chen, C.-T.; Huo, L.; Hsu, M.-C.; et al. KEAP1 E3 Ligase-Mediated Downregulation of NF-κB Signaling by Targeting IKKβ. Mol. Cell 2009, 36, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, N.; Slocum, S.L.; Skoko, J.J.; Shin, S.; Kensler, T.W.; Chiou, Y.-S.; Huang, Q.; Ho, C.-T.; Wang, Y.-J.; Pan, M.-H.; et al. When NRF2 Talks, Who’s Listening? Antioxid. Redox Signal. 2010, 13, 1649–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-κB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Li, H.; Liu, Q.; Liu, F.; Tang, L.; Li, C.; Yuan, Y.; Zhan, Y.; Xu, W.; Li, W.; et al. Nuclear factor p65 interacts with Keap1 to repress the Nrf2-ARE pathway. Cell. Signal. 2011, 23, 883–892. [Google Scholar] [CrossRef]
- Lastra, D.; Escoll, M.; Cuadrado, A. Transcription Factor NRF2 Participates in Cell Cycle Progression at the Level of G1/S and Mitotic Checkpoints. Antioxidants 2022, 11, 946. [Google Scholar] [CrossRef]
- Hu, M.; Zou, Y.; Nambiar, S.M.; Lee, J.; Yang, Y.; Dai, G. Keap1 modulates the redox cycle and hepatocyte cell cycle in regenerating liver. Cell Cycle 2014, 13, 2349–2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariza, J.; González-Reyes, J.A.; Jódar, L.; Díaz-Ruiz, A.; de Cabo, R.; Villalba, J.M. Mitochondrial permeabilization without caspase activation mediates the increase of basal apoptosis in cells lacking Nrf2. Free Radic. Biol. Med. 2016, 95, 82–95. [Google Scholar] [CrossRef] [Green Version]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/Small Maf Heterodimer Mediates the Induction of Phase II Detoxifying Enzyme Genes through Antioxidant Response Elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Hurtado-Navarro, L.; Angosto-Bazarra, D.; Pelegrín, P.; Baroja-Mazo, A.; Cuevas, S. NLRP3 Inflammasome and Pyroptosis in Liver Pathophysiology: The Emerging Relevance of Nrf2 Inducers. Antioxidants 2022, 11, 870. [Google Scholar] [CrossRef]
- Jin, C.; Flavell, R.A. Molecular Mechanism of NLRP3 Inflammasome Activation. J. Clin. Immunol. 2010, 30, 628–631. [Google Scholar] [CrossRef]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Mohs, A.; Otto, T.; Schneider, K.M.; Peltzer, M.; Boekschoten, M.; Holland, C.H.; Hudert, C.A.; Kalveram, L.; Wiegand, S.; Saez-Rodriguez, J.; et al. Hepatocyte-specific NRF2 activation controls fibrogenesis and carcinogenesis in steatohepatitis. J. Hepatol. 2021, 74, 638–648. [Google Scholar] [CrossRef]
- Xu, J.; Donepudi, A.C.; Moscovitz, J.E.; Slitt, A.L. Keap1-Knockdown Decreases Fasting-Induced Fatty Liver via Altered Lipid Metabolism and Decreased Fatty Acid Mobilization from Adipose Tissue. PLoS ONE 2013, 8, e79841. [Google Scholar] [CrossRef] [PubMed]
- Meakin, P.J.; Chowdhry, S.; Sharma, R.S.; Ashford, F.B.; Walsh, S.V.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; Dillon, J.F.; Hayes, J.D.; Ashford, M.L.J. Susceptibility of Nrf2-Null Mice to Steatohepatitis and Cirrhosis upon Consumption of a High-Fat Diet Is Associated with Oxidative Stress, Perturbation of the Unfolded Protein Response, and Disturbance in the Expression of Metabolic Enzymes but Not with Insulin Resistance. Mol. Cell. Biol. 2014, 34, 3305–3320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Y.; Hu, M.; Lee, J.; Nambiar, S.M.; Garcia, V.; Bao, Q.; Chan, J.Y.; Dai, G. Nrf2 is essential for timely M phase entry of replicating hepatocytes during liver regeneration. Am. J. Physiol. Liver Physiol. 2015, 308, G262–G268. [Google Scholar] [CrossRef] [Green Version]
- Beyer, T.A.; Xu, W.; Teupser, D.; Keller, U.A.D.; Bugnon, P.; Hildt, E.; Thiery, J.; Kan, Y.W.; Werner, S. Impaired liver regeneration in Nrf2 knockout mice: Role of ROS-mediated insulin/IGF-1 resistance. EMBO J. 2008, 27, 212–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayoub, R.; Vogel, A.; Schuett, J.; Lupke, M.; Spieker, S.M.; Kettern, N.; Hildt, E.; Melter, M.; Weiss, T.S. Nrf2 Activates Augmenter of Liver Regeneration (ALR) via Antioxidant Response Element and Links Oxidative Stress to Liver Regeneration. Mol. Med. 2013, 19, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Cordero-Espinoza, L.; Huch, M. The balancing act of the liver: Tissue regeneration versus fibrosis. J. Clin. Investig. 2018, 128, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Kaminsky-Kolesnikov, Y.; Rauchbach, E.; Abu-Halaka, D.; Hahn, M.; García-Ruiz, C.; Fernandez-Checa, J.C.; Madar, Z.; Tirosh, O. Cholesterol Induces Nrf-2- and HIF-1α-Dependent Hepatocyte Proliferation and Liver Regeneration to Ameliorate Bile Acid Toxicity in Mouse Models of NASH and Fibrosis. Oxidative Med. Cell. Longev. 2020, 2020, 5393761. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Dorko, K.; Antoine, D.J.; Clarke, J.I.; Gholami, P.; Li, F.; Kumer, S.C.; Schmitt, T.M.; Forster, J.; Fan, F.; et al. Bile acid-induced necrosis in primary human hepatocytes and in patients with obstructive cholestasis. Toxicol. Appl. Pharmacol. 2015, 283, 168–177. [Google Scholar] [CrossRef]
- Zhang, D.Y.; Friedman, S.L. Fibrosis-dependent mechanisms of hepatocarcinogenesis. Hepatology 2012, 56, 769–775. [Google Scholar] [CrossRef] [Green Version]
- Udompap, P.; Kim, D.; Kim, W.R. Current and Future Burden of Chronic Nonmalignant Liver Disease. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2015, 13, 2031–2041. [Google Scholar] [CrossRef] [Green Version]
- Tiegs, G.; Hentschel, J.; Wendel, A. A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J. Clin. Investig. 1992, 90, 196–203. [Google Scholar] [CrossRef]
- Osburn, W.O.; Yates, M.S.; Dolan, P.D.; Chen, S.; Liby, K.T.; Sporn, M.B.; Taguchi, K.; Yamamoto, M.; Kensler, T.W. Genetic or Pharmacologic Amplification of Nrf2 Signaling Inhibits Acute Inflammatory Liver Injury in Mice. Toxicol. Sci. 2008, 104, 218–227. [Google Scholar] [CrossRef]
- Li, S.; Hong, M.; Tan, H.-Y.; Wang, N.; Feng, Y. Insights into the Role and Interdependence of Oxidative Stress and Inflammation in Liver Diseases. Oxidative Med. Cell. Longev. 2016, 2016, 4234061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.H.; Son, J.Y.; Kim, K.S.; Park, Y.J.; Kim, H.R.; Park, J.H.; Kim, K.-B.; Lee, K.Y.; Kang, K.W.; Kim, I.S.; et al. Estrogen Deficiency Potentiates Thioacetamide-Induced Hepatic Fibrosis in Sprague-Dawley Rats. Int. J. Mol. Sci. 2019, 20, 3709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussein, R.M.; Sawy, D.M.; Kandeil, M.A.; Farghaly, H.S. Chlorogenic acid, quercetin, coenzyme Q10 and silymarin modulate Keap1-Nrf2/heme oxygenase-1 signaling in thioacetamide-induced acute liver toxicity. Life Sci. 2021, 277, 119460. [Google Scholar] [CrossRef]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell Death and Cell Death Responses in Liver Disease: Mechanisms and Clinical Relevance. Gastroenterology 2014, 147, 765–783.e4. [Google Scholar] [CrossRef] [Green Version]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Hinson, J.A.; Reid, A.B.; McCullough, S.S.; James, L.P. Acetaminophen-Induced Hepatotoxicity: Role of Metabolic Activation, Reactive Oxygen/Nitrogen Species, and Mitochondrial Permeability Transition. Drug Metab. Rev. 2004, 36, 805–822. [Google Scholar] [CrossRef] [PubMed]
- González-Rodríguez, Á.; Reibert, B.; Amann, T.; Constien, R.; Rondinone, C.M.; Valverde, Á.M. In vivo siRNA delivery of Keap1 modulates death and survival signaling pathways and attenuates Concanavalin A-induced acute liver injury in mice. Dis. Model. Mech. 2014, 7, 1093–1100. [Google Scholar] [CrossRef]
- Enomoto, A. High Sensitivity of Nrf2 Knockout Mice to Acetaminophen Hepatotoxicity Associated with Decreased Expression of ARE-Regulated Drug Metabolizing Enzymes and Antioxidant Genes. Toxicol. Sci. 2001, 59, 169–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reisman, S.A.; Csanaky, I.L.; Aleksunes, L.M.; Klaassen, C.D. Altered Disposition of Acetaminophen in Nrf2-null and Keap1-knockdown Mice. Toxicol. Sci. 2009, 109, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Lv, H.; Hong, L.; Tian, Y.; Yin, C.; Zhu, C.; Feng, H. Corilagin alleviates acetaminophen-induced hepatotoxicity via enhancing the AMPK/GSK3β-Nrf2 signaling pathway. Cell Commun. Signal. 2019, 17, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Liu, C.-Y.; Wang, T.; Yu, H.-M.; Ouyang, S.-H.; Wu, Y.-P.; Gong, H.-B.; Ma, X.-H.; Jiao, G.-L.; Fu, L.-L.; et al. (+)-Clausenamide protects against drug-induced liver injury by inhibiting hepatocyte ferroptosis. Cell Death Dis. 2020, 11, 781. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, H.; Chen, Q.; Jiao, F.; Shi, C.; Pei, M.; Lv, J.; Zhang, H.; Wang, L.; Gong, Z. TNF-α/HMGB1 inflammation signalling pathway regulates pyroptosis during liver failure and acute kidney injury. Cell Prolif. 2020, 53, e12829. [Google Scholar] [CrossRef]
- Tomasi, M.L.; Ryoo, M.; Yang, H.; Ara, A.I.; Ko, K.S.; Lu, S.C. Molecular mechanisms of lipopolysaccharide-mediated inhibition of glutathione synthesis in mice. Free Radic. Biol. Med. 2014, 68, 148–158. [Google Scholar] [CrossRef] [Green Version]
- Chalasani, N.; Younossi, Z.; LaVine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [Green Version]
- Tanase, D.M.; Gosav, E.M.; Costea, C.F.; Ciocoiu, M.; Lacatusu, C.M.; Maranduca, M.A.; Ouatu, A.; Floria, M. The Intricate Relationship between Type 2 Diabetes Mellitus (T2DM), Insulin Resistance (IR), and Nonalcoholic Fatty Liver Disease (NAFLD). J. Diabetes Res. 2020, 2020, 3920196. [Google Scholar] [CrossRef] [PubMed]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2012, 52, 59–69. [Google Scholar] [CrossRef]
- Serviddio, G.; Bellanti, F.; Vendemiale, G. Free radical biology for medicine: Learning from nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2013, 65, 952–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, B.W.; Adams, L.A. Non-alcoholic fatty liver disease. Crit. Rev. Clin. Lab. Sci. 2011, 48, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Yates, M.S.; Tran, Q.T.; Dolan, P.M.; Osburn, W.O.; Shin, S.; McCulloch, C.C.; Silkworth, J.B.; Taguchi, K.; Yamamoto, M.; Williams, C.R.; et al. Genetic versus chemoprotective activation of Nrf2 signaling: Overlapping yet distinct gene expression profiles between Keap1 knockout and triterpenoid-treated mice. Carcinogenesis 2009, 30, 1024–1031. [Google Scholar] [CrossRef] [Green Version]
- Kay, H.Y.; Kim, W.D.; Hwang, S.J.; Choi, H.-S.; Gilroy, R.K.; Wan, Y.-J.Y.; Kim, S.G.; Wang, X.; Hai, C.; Chen, Y.-J.; et al. Nrf2 Inhibits LXRα-Dependent Hepatic Lipogenesis by Competing with FXR for Acetylase Binding. Antioxid. Redox Signal. 2011, 15, 2135–2146. [Google Scholar] [CrossRef]
- Liao, Z.; Zhang, J.; Liu, B.; Yan, T.; Xu, F.; Xiao, F.; Wu, B.; Bi, K.; Jia, Y. Polysaccharide from Okra (Abelmoschus esculentus (L.) Moench) Improves Antioxidant Capacity via PI3K/AKT Pathways and Nrf2 Translocation in a Type 2 Diabetes Model. Molecules 2019, 24, 1906. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, T.; Liu, P.; Yang, F.; Wang, X.; Zheng, W.; Sun, W. Hesperetin ameliorates hepatic oxidative stress and inflammation via the PI3K/AKT-Nrf2-ARE pathway in oleic acid-induced HepG2 cells and a rat model of high-fat diet-induced NAFLD. Food Funct. 2021, 12, 3898–3918. [Google Scholar] [CrossRef]
- Zhang, C.-Y.; Yuan, W.-G.; He, P.; Lei, J.-H.; Wang, C.-X. Liver fibrosis and hepatic stellate cells: Etiology, pathological hallmarks and therapeutic targets. World J. Gastroenterol. 2016, 22, 10512–10522. [Google Scholar] [CrossRef]
- Lu, C.; Xu, W.; Shao, J.; Zhang, F.; Chen, A.; Zheng, S. Nrf2 induces lipocyte phenotype via a SOCS3-dependent negative feedback loop on JAK2/STAT3 signaling in hepatic stellate cells. Int. Immunopharmacol. 2017, 49, 203–211. [Google Scholar] [CrossRef]
- Lyu, H.; Wang, H.; Li, L.; Zhu, J.; Chen, F.; Chen, Y.; Liu, C.; Fu, J.; Yang, B.; Zhang, Q.; et al. Hepatocyte-specific deficiency of Nrf2 exacerbates carbon tetrachloride-induced liver fibrosis via aggravated hepatocyte injury and subsequent inflammatory and fibrogenic responses. Free Radic. Biol. Med. 2020, 150, 136–147. [Google Scholar] [CrossRef]
- Oh, C.J.; Kim, J.-Y.; Min, A.-K.; Park, K.-G.; Harris, R.A.; Kim, H.-J.; Lee, I.-K. Sulforaphane attenuates hepatic fibrosis via NF-E2-related factor 2-mediated inhibition of transforming growth factor-β/Smad signaling. Free Radic. Biol. Med. 2012, 52, 671–682. [Google Scholar] [CrossRef]
- Thorgeirsson, S.S. The central role of the c-Met pathway in rebuilding the liver. Gut 2012, 61, 1105–1106. [Google Scholar] [CrossRef] [PubMed]
- Kroy, D.C.; Schumacher, F.; Ramadori, P.; Hatting, M.; Bergheim, I.; Gassler, N.; Boekschoten, M.V.; Müller, M.; Streetz, K.L.; Trautwein, C. Hepatocyte specific deletion of c-Met leads to the development of severe non-alcoholic steatohepatitis in mice. J. Hepatol. 2014, 61, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Ramadori, P.; Drescher, H.; Erschfeld, S.; Fragoulis, A.; Kensler, T.W.; Wruck, C.J.; Cubero, F.J.; Trautwein, C.; Streetz, K.L.; Kroy, D.C. Genetic Nrf2 Overactivation Inhibits the Deleterious Effects Induced by Hepatocyte-Specific c-met Deletion during the Progression of NASH. Oxidative Med. Cell. Longev. 2017, 2017, 3420286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramadori, P.; Drescher, H.; Erschfeld, S.; Schumacher, F.; Berger, C.; Fragoulis, A.; Schenkel, J.; Kensler, T.W.; Wruck, C.J.; Trautwein, C.; et al. Hepatocyte-specific Keap1 deletion reduces liver steatosis but not inflammation during non-alcoholic steatohepatitis development. Free Radic. Biol. Med. 2016, 91, 114–126. [Google Scholar] [CrossRef]
- Gao, B.; Bataller, R. Alcoholic Liver Disease: Pathogenesis and New Therapeutic Targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [Green Version]
- Dey, A.; Cederbaum, A.I. Alcohol and oxidative liver injury. Hepatology 2006, 43, S63–S74. [Google Scholar] [CrossRef]
- Wang, Y.; Kou, Y.; Wang, X.; Cederbaum, A.; Wang, R. Multifactorial Comparative Proteomic Study of Cytochrome P450 2E1 Function in Chronic Alcohol Administration. PLoS ONE 2014, 9, e92504. [Google Scholar] [CrossRef]
- Gong, P.; Cederbaum, A.I. Nrf2 is increased by CYP2E1 in rodent liver and HepG2 cells and protects against oxidative stress caused by CYP2E1. Hepatology 2006, 43, 144–153. [Google Scholar] [CrossRef]
- Lamlé, J.; Marhenke, S.; Borlak, J.; von Wasielewski, R.; Eriksson, C.P.; Geffers, R.; Manns, M.P.; Yamamoto, M.; Vogel, A. Nuclear Factor-Eythroid 2–Related Factor 2 Prevents Alcohol-Induced Fulminant Liver Injury. Gastroenterology 2008, 134, 1159–1168. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Dou, X.; Li, S.; Zhang, X.; Sun, X.; Zhou, Z.; Song, Z. Nuclear factor (erythroid-derived 2)-like 2 activation-induced hepatic very-low-density lipoprotein receptor overexpression in response to oxidative stress contributes to alcoholic liver disease in mice. Hepatology 2014, 59, 1381–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.C.; Liu, J.; Klaassen, C.D. Role of Nrf2 in preventing ethanol-induced oxidative stress and lipid accumulation. Toxicol. Appl. Pharmacol. 2012, 262, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Hong, Z.; Shao, S.; Li, L.; Yang, B.; Hou, Y.; Wang, H.; Xu, Y.; Zhang, Q.; Pi, J.; et al. Liver-specific Nrf2 deficiency accelerates ethanol-induced lethality and hepatic injury in vivo. Toxicol. Appl. Pharmacol. 2021, 426, 115617. [Google Scholar] [CrossRef]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [Green Version]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Lister, A.; Nedjadi, T.; Kitteringham, N.R.; Campbell, F.; Costello, E.; Lloyd, B.; Copple, I.M.; Williams, S.; Owen, A.; Neoptolemos, J.P.; et al. Nrf2 is overexpressed in pancreatic cancer: Implications for cell proliferation and therapy. Mol. Cancer 2011, 10, 37. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Bodas, M.; Wakabayashi, N.; Bunz, F.; Biswal, S.; Lee, S.B.; Sellers, B.N.; DeNicola, G.M.; Kerimi, A.; Williamson, G.; et al. Gain of Nrf2 Function in Non-Small-Cell Lung Cancer Cells Confers Radioresistance. Antioxid. Redox Signal. 2010, 13, 1627–1637. [Google Scholar] [CrossRef] [Green Version]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell Biology of Ischemia/Reperfusion Injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, O.; Xu, M.; Zhou, F.; Wein, A.N.; Upadhya, G.A.; Ye, L.; Wong, B.W.; Lin, Y.; O’farrelly, C.; Chapman, W.C. NRF2 assessment in discarded liver allografts: A role in allograft function and salvage. Am. J. Transplant. 2022, 22, 58–70. [Google Scholar] [CrossRef]
- Banan, B.; Watson, R.; Xu, M.; Lin, Y.; Chapman, W. Development of a normothermic extracorporeal liver perfusion system toward improving viability and function of human extended criteria donor livers. Liver Transplant. 2016, 22, 979–993. [Google Scholar] [CrossRef] [Green Version]
- Bardallo, R.G.; Company-Marin, I.; Folch-Puy, E.; Roselló-Catafau, J.; Panisello-Rosello, A.; Carbonell, T. PEG35 and Glutathione Improve Mitochondrial Function and Reduce Oxidative Stress in Cold Fatty Liver Graft Preservation. Antioxidants 2022, 11, 158. [Google Scholar] [CrossRef]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial Role of Nrf2 in Regulating NADPH Generation and Consumption. Toxicol. Sci. 2011, 123, 590–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niture, S.K.; Jaiswal, A.K. Nrf2 Protein Up-regulates Antiapoptotic Protein Bcl-2 and Prevents Cellular Apoptosis. J. Biol. Chem. 2012, 287, 9873–9886. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.Z.; Huang, Y.P.; Wang, X.; Wang, H.P.; Ren, F.; Tian, R.F.; Cheng, X.; Cai, J.; Zhang, Y.; Zhu, X.Y.; et al. Integrated Omics Reveals Tollip as an Regulator and Therapeutic Target for Hepatic Ischemia-Reperfusion Injury in Mice. Hepatology 2019, 70, 1750–1769. [Google Scholar] [CrossRef] [PubMed]
- Duarte, S.; Shen, X.-D.; Fondevila, C.; Busuttil, R.W.; Coito, A.J. Fibronectin-α4β1 Interactions in Hepatic Cold Ischemia and Reperfusion Injury: Regulation of MMP-9 and MT1-MMP via the p38 MAPK Pathway. Am. J. Transplant. 2012, 12, 2689–2699. [Google Scholar] [CrossRef] [Green Version]
- Niture, S.K.; Jaiswal, A.K. Nrf2-induced antiapoptotic Bcl-xL protein enhances cell survival and drug resistance. Free Radic. Biol. Med. 2013, 57, 119–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Lu, T.; Zhang, C.; Xu, J.; Xue, Z.; Busuttil, R.W.; Xu, N.; Xia, Q.; Kupiec-Weglinski, J.W.; Ji, H. Activation of YAP attenuates hepatic damage and fibrosis in liver ischemia-reperfusion injury. J. Hepatol. 2019, 71, 719–730. [Google Scholar] [CrossRef]
- Smith, J.B.; Willis, A.L. Aspirin Selectively Inhibits Prostaglandin Production in Human Platelets. Nat. New Biol. 1971, 231, 235–237. [Google Scholar] [CrossRef]
- DuBois, R.N.; Abramson, S.B.; Crofford, L.; Gupta, R.A.; Simon, L.S.; Putte, L.B.A.; Lipsky, P.E.; Werner, M.; Jordan, P.M.; Romp, E.; et al. Cyclooxygenase in biology and disease. FASEB J. 1998, 12, 1063–1073. [Google Scholar] [CrossRef] [Green Version]
- Casado, M.; Callejas, N.A.; Rodrigo, J.; Zhao, X.; Dey, S.K.; Boscá, L.; Martín-Sanz, P. Contribution of cyclooxygenase-2 to liver regeneration after partial hepatectomy. FASEB J. 2001, 15, 2016–2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishizawa, N.; Ito, Y.; Eshima, K.; Ohkubo, H.; Kojo, K.; Inoue, T.; Raouf, J.; Jakobsson, P.-J.; Uematsu, S.; Akira, S.; et al. Inhibition of microsomal prostaglandin E synthase-1 facilitates liver repair after hepatic injury in mice. J. Hepatol. 2018, 69, 110–120. [Google Scholar] [CrossRef]
- Hamada, T.; Tsuchihashi, S.; Avanesyan, A.; Duarte, S.; Moore, C.; Busuttil, R.W.; Coito, A.J. Cyclooxygenase-2 Deficiency Enhances Th2 Immune Responses and Impairs Neutrophil Recruitment in Hepatic Ischemia/Reperfusion Injury. J. Immunol. 2008, 180, 1843–1853. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Li, Y.M.; Li, X.; Shi, B.; He, M.Y.; Zhu, X.L.; Zhou, W.C.; Wachtel, M.S.; Frezza, E. COX-2 Inhibition Improves Immune System Homeostasis and Decreases Liver Damage in Septic Rats. J. Surg. Res. 2009, 157, 43–47. [Google Scholar] [CrossRef]
- Du, Y.; Chen, J.; Liu, D.; Bai, Q.; Song, J.; Guan, J.; Gao, J.; Liu, B.; Ma, X. Celecoxib attenuates liver steatosis and inflammation in non-alcoholic steatohepatitis induced by high-fat diet in rats. Mol. Med. Rep. 2011, 4, 811–816. [Google Scholar] [CrossRef]
- Morteau, O.; Morham, S.G.; Sellon, R.; Dieleman, L.A.; Langenbach, R.; Smithies, O.; Sartor, R.B. Impaired mucosal defense to acute colonic injury in mice lacking cyclooxygenase-1 or cyclooxygenase-2. J. Clin. Investig. 2000, 105, 469–478. [Google Scholar] [CrossRef] [Green Version]
- Duarte, S.; Kato, H.; Kuriyama, N.; Suko, K.; Ishikawa, T.-O.; Busuttil, R.W.; Herschman, H.R.; Coito, A.J. Hepatic Ischemia and Reperfusion Injury in the Absence of Myeloid Cell-Derived COX-2 in Mice. PLoS ONE 2014, 9, e96913. [Google Scholar] [CrossRef] [PubMed]
- Casado, M.; Mollá, B.; Roy, R.; Fernández-Martínez, A.; Cucarella, C.; Mayoral, R.; Boscá, L.; Martín-Sanz, P. Protection against Fas-induced liver apoptosis in transgenic mice expressing cyclooxygenase 2 in hepatocytes. Hepatology 2007, 45, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Francés, D.E.; Ingaramo, P.I.; Mayoral, R.; Través, P.; Casado, M.; Valverde, Á.M.; Martín-Sanz, P.; Carnovale, C.E. Cyclooxygenase-2 over-expression inhibits liver apoptosis induced by hyperglycemia. J. Cell. Biochem. 2013, 114, 669–680. [Google Scholar] [CrossRef]
- Masuda, Y.; Vaziri, N.D.; Takasu, C.; Li, S.; Robles, L.; Pham, C.; Le, A.; Vo, K.; Farzaneh, S.H.; Stamos, M.J.; et al. Salutary effect of pre-treatment with an Nrf2 inducer on ischemia reperfusion injury in the rat liver. Gastroenterol. Hepatol. 2014, 1, 1–7. [Google Scholar] [CrossRef]
- Chen, M.; Sun, X.; Wei, W.; Cucarella, C.; Martín-Sanz, P.; Casado, M.; Pi, L.; Ren, B.; Cao, Q. Hepatic COX-2 expression protects mice from an alcohol-high fat diet-induced metabolic disorder by involving protein acetylation related energy metabolism. Alcohol 2021, 92, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Ke, B.; Shen, X.-D.; Zhang, Y.; Ji, H.; Gao, F.; Yue, S.; Kamo, N.; Zhai, Y.; Yamamoto, M.; Busuttil, R.W.; et al. KEAP1-NRF2 complex in ischemia-induced hepatocellular damage of mouse liver transplants. J. Hepatol. 2013, 59, 1200–1207. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Li, L.; Iwamoto, N.; Nakajima-Takagi, Y.; Kaneko, H.; Nakayama, Y.; Eguchi, M.; Wada, Y.; Kumagai, Y.; Yamamoto, M. The Antioxidant Defense System Keap1-Nrf2 Comprises a Multiple Sensing Mechanism for Responding to a Wide Range of Chemical Compounds. Mol. Cell. Biol. 2009, 29, 493–502. [Google Scholar] [CrossRef] [Green Version]
- Kansanen, E.; Kivelä, A.M.; Levonen, A.-L. Regulation of Nrf2-dependent gene expression by 15-deoxy-Δ12,14-prostaglandin J2. Free Radic. Biol. Med. 2009, 47, 1310–1317. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Antonucci, L.; Karin, M. NRF2 as a regulator of cell metabolism and inflammation in cancer. Carcinogenesis 2020, 41, 405–416. [Google Scholar] [CrossRef]
- Ishii, T. Close teamwork between Nrf2 and peroxiredoxins 1 and 6 for the regulation of prostaglandin D2 and E2 production in macrophages in acute inflammation. Free Radic. Biol. Med. 2015, 88, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Liong, E.C.; Huang, H.; On Tse, W.; Lau, K.S.; Pan, J.; Nanji, A.A.; Fung, M.L.; Xing, F.; Tipoe, G.L. Cyclooxygenase-1 Serves a Vital Hepato-Protective Function in Chemically Induced Acute Liver Injury. Toxicol. Sci. 2015, 143, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Groeger, A.L.; Cipollina, C.; Cole, M.P.; Woodcock, S.R.; Bonacci, G.; Rudolph, T.K.; Rudolph, V.; Freeman, B.A.; Schopfer, F.J. Cyclooxygenase-2 generates anti-inflammatory mediators from omega-3 fatty acids. Nat. Chem. Biol. 2010, 6, 433–441. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Urgard, E.; Vooder, T.; Metspalu, A. The role of COX-2 and Nrf2/ARE in anti-inflammation and antioxidative stress: Aging and anti-aging. Med. Hypotheses 2011, 77, 174–178. [Google Scholar] [CrossRef]
- Zdanov, S.; Toussaint, O.; Debacq-Chainiaux, F. p53 and ATF-2 partly mediate the overexpression of COX-2 in H2O2-induced premature senescence of human fibroblasts. Biogerontology 2009, 10, 291–298. [Google Scholar] [CrossRef]
- Jessen, C.; Kreß, J.K.C.; Baluapuri, A.; Hufnagel, A.; Schmitz, W.; Kneitz, S.; Roth, S.; Marquardt, A.; Appenzeller, S.; Ade, C.P.; et al. The transcription factor NRF2 enhances melanoma malignancy by blocking differentiation and inducing COX2 expression. Oncogene 2020, 39, 6841–6855. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Zhao, L.; Zou, W.; Shen, W.; Zhang, H.; He, Q. Activation of Cyclooxygenase-2 by ATF4 during Endoplasmic Reticulum Stress Regulates Kidney Podocyte Autophagy Induced by Lupus Nephritis. Cell. Physiol. Biochem. 2018, 48, 753–764. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuertes-Agudo, M.; Luque-Tévar, M.; Cucarella, C.; Martín-Sanz, P.; Casado, M. Advances in Understanding the Role of NRF2 in Liver Pathophysiology and Its Relationship with Hepatic-Specific Cyclooxygenase-2 Expression. Antioxidants 2023, 12, 1491. https://doi.org/10.3390/antiox12081491
Fuertes-Agudo M, Luque-Tévar M, Cucarella C, Martín-Sanz P, Casado M. Advances in Understanding the Role of NRF2 in Liver Pathophysiology and Its Relationship with Hepatic-Specific Cyclooxygenase-2 Expression. Antioxidants. 2023; 12(8):1491. https://doi.org/10.3390/antiox12081491
Chicago/Turabian StyleFuertes-Agudo, Marina, María Luque-Tévar, Carme Cucarella, Paloma Martín-Sanz, and Marta Casado. 2023. "Advances in Understanding the Role of NRF2 in Liver Pathophysiology and Its Relationship with Hepatic-Specific Cyclooxygenase-2 Expression" Antioxidants 12, no. 8: 1491. https://doi.org/10.3390/antiox12081491