

Targeting the Cysteine Redox Proteome in Parkinson’s Disease: The Role of Glutathione Precursors and Beyond

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
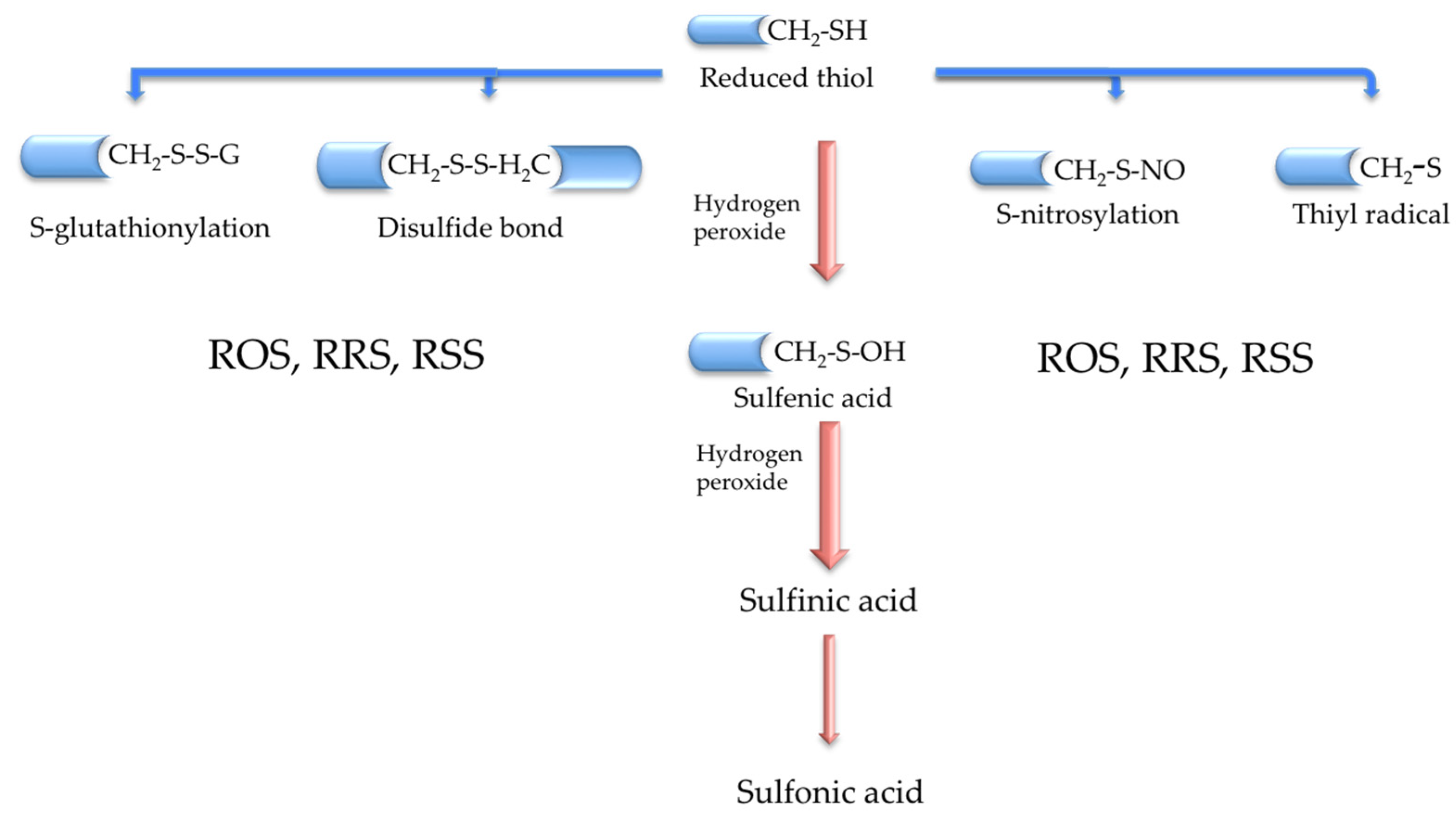
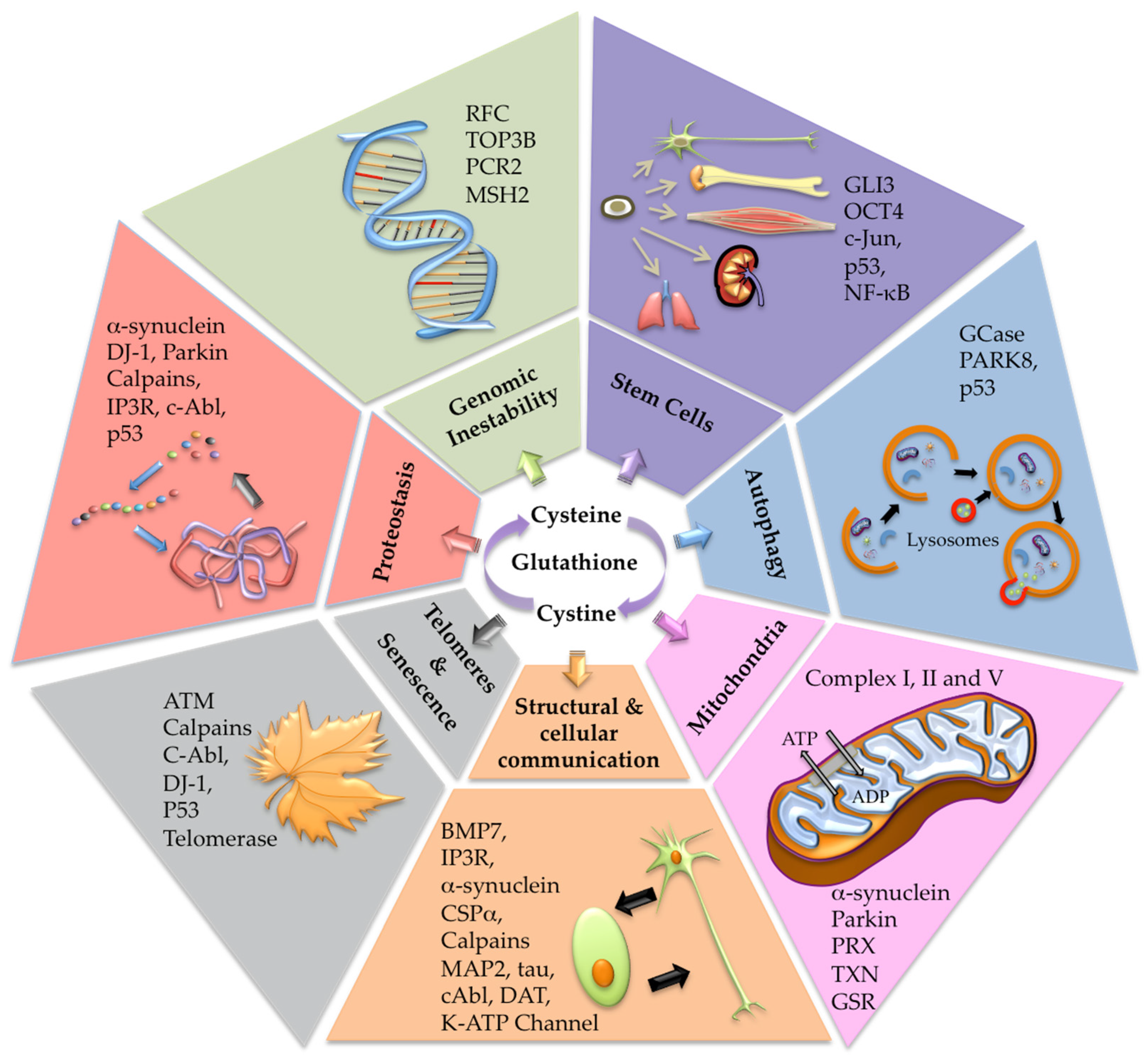
2. The Cysteine Redox Proteome Hub (Cysteinome and Cysteinet)
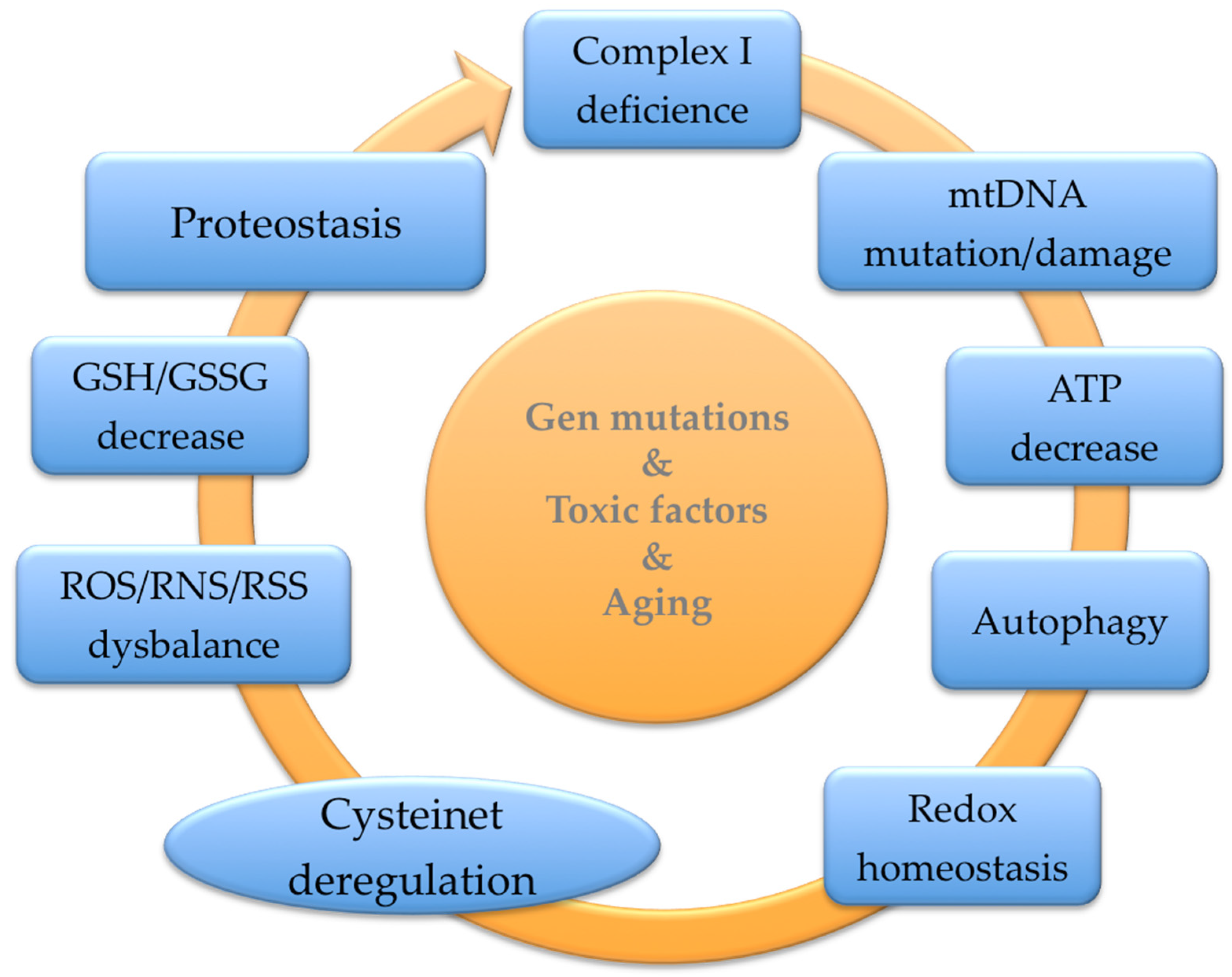
3. The Cysteinet Deregulation in Aging
4. Cysteinet Deregulation in PD
4.1. Extracellular Vesicles
4.2. DnaJ Homolog C (DNAJC)
4.3. Glucocerebrosidase
4.4. Leucine-Rich Repeat Kinase 2
4.5. Cellular-Abelson Tyrosine Kinase (c-Abl)
4.6. Parkin
4.7. Dopamine Transporter (DAT)
4.8. Protein Deglycase DJ-1
4.9. ATP-Sensitive Potassium Channel (K-ATP Channel)
4.10. Antioxidant Enzymatic System
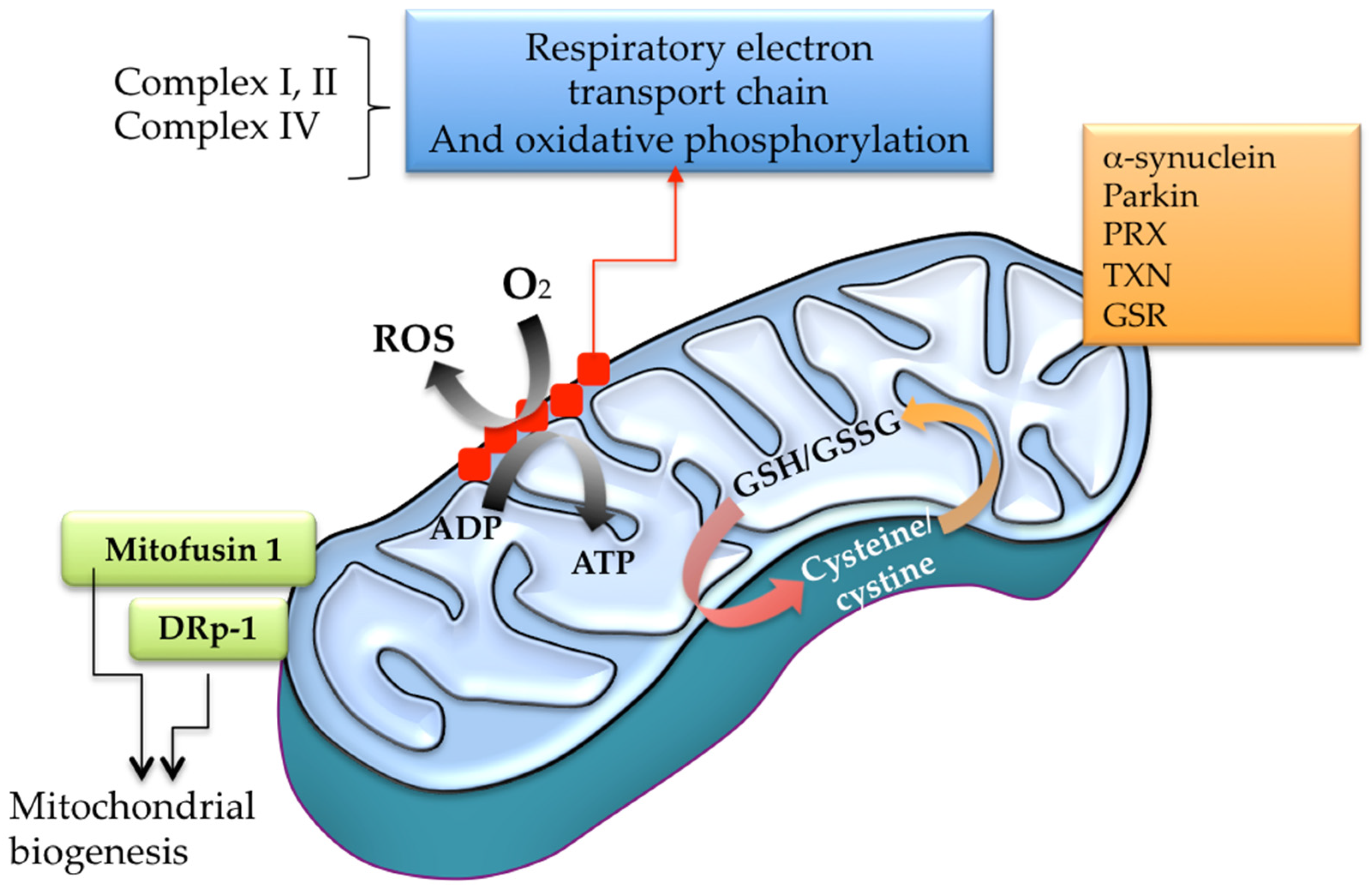
4.11. Mitochondrial Respiratory Chain and Oxidative Phosphorylation
4.12. Microtubule-Associated Protein Tau and MAP2
4.13. α-Synuclein and the Ubiquitin–Proteasome System
4.14. Pluripotency and Neural Stem Cells
4.15. Cysteinet Deregulation and Genomic Instability
4.16. Ataxia-Telangiectasia Mutated (ATM) and Apoptosis
4.17. Bone Morphogenetic Protein 7 and TGF-β
4.18. Telomerase and Senescence
5. Role of Glutathione Precursors in Parkinson’s Disease
5.1. Nutrients and GSH Status
5.1.1. Amino Acids, Peptides, and Proteins
5.1.2. Vitamins
5.1.3. Flavonoids and Thiol-Rich Compounds
5.1.4. Michael Acceptor Molecules (MAMs)
5.2. CoQ10-Related Compounds
5.3. N-Acetyl-Cysteine (NAC)
5.3.1. NAC Bioavailability and Safety
5.3.2. NAC in Cysteinet Regulation
5.3.3. NAC in Protein Misfolding
5.3.4. NAC in Cellular Vesicle Regulation and Signaling
5.3.5. NAC and Mitochondria
5.3.6. NAC and Protein Kinases
5.3.7. NAC and Telomerase
5.3.8. NAC-GSH and Nanotechnology
6. Preclinical and Clinical Studies of NAC in PD
7. Conclusions and Future Perspectives
- large-scale clinical trials;
- including PD patients, healthy people, and age-matched patients with comorbidities (e.g., diabetes, hypertension, obesity, and cardiovascular disorders);
- supplementing the diet with cysteine-rich food or low doses of NAC (between 1800 and 3000 mg/week);
- monitoring GSH and albumin redox status.
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Dorsey, E.R.; Elbaz, A.; Nichols, E.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.-Y.J.; Collado-Mateo, D.; et al. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Veldman, B.A.; Wijn, A.M.; Knoers, N.; Praamstra, P.; Horstinka, M.W.I.M. Genetic and environmental risk factors in Parkinson’s Disease. Clin. Neurol. Neurosurg. 1998, 100, 15–26. [Google Scholar] [CrossRef]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, X.; Bandres-Ciga, S.; Blauwendraat, C.; Cookson, M.R. The role of monogenic genes in idiopathic Parkinson’s disease. Neurobiol. Dis. 2019, 124, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Youdim, M.B. Understanding Parkinson’s disease. Sci. Am. 1997, 276, 52–59. [Google Scholar] [CrossRef]
- Forno, L.S. Neuropathology of Parkinson’s disease. J. Neuropathol. Exp. Neurol. 1996, 55, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Gelb, D.J.; Oliver, E.; Gilman, S. Diagnostic criteria for Parkinson disease. Arch. Neurol. 1999, 56, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Schulz-Schaeffer, W.J. The synaptic pathology of a-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010, 120, 131–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz-Schaeffer, W.J. Is cell death primary or secondary in the pathophysiology of idiopathic Parkinson’s disease? Biomolecules 2015, 5, 1467–1479. [Google Scholar] [CrossRef] [Green Version]
- Jellinger, K.A. A critical reappraisal of current staging of Lewy related pathology in human brain. Acta Neuropathol. 2008, 116, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Perl, D.P.; DeMartino, G.N.; McNaught, K.S. Lewy body formation is an aggresome-related process: A hypothesis. Lancet Neurol. 2004, 3, 496–503. [Google Scholar] [CrossRef]
- Fahn, S.; Cohen, G. The oxidant stress hypothesis in Parkinson’s disease: Evidence supporting it. Ann. Neurol. 1992, 32, 804–812. [Google Scholar] [CrossRef]
- Jenner, P. Oxidative stress in Parkinson’s disease. Ann. Neurol. 2003, 53, S26–S38. [Google Scholar] [CrossRef]
- Martinez, T.N.; Greenamyre, J.T. Toxin models of mitocondrial dysfunction in Parkinson’s disease. Antioxid. Redox Sign. 2012, 16, 920–924. [Google Scholar] [CrossRef] [Green Version]
- Go, Y.-M.; Chandler, J.D.; Jones, D.P. The cysteine proteome. Free Radic. Biol. Med. 2015, 84, 227–745. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Banaclocha, M. Cellular cysteine network (Cysteinet): Pharmacological intervention in brain aging and neurodegenerative diseases. In Frontiers in Clinical Drug Research-Central Nervous System; Atta-ur-Rahman, Ed.; Bentham Science Publishers: Al Sharjah, United Arab Emirates, 2016; Volume 2, pp. 105–172. [Google Scholar]
- Xiao, H.; Jedrychowski, M.P.; Schweppe, D.K.; Huttlin, E.L.; Yu, Q.; Heppner, D.E.; Li, J.; Long, J.; Mills, E.L.; Szpyt, J.; et al. A quantitative tissue-specific landscape of protein redox regulation during aging. Cell 2020, 180, 968–983. [Google Scholar] [CrossRef]
- Meng, J.; Fu, L.; Liu, K.; Tian, C.; Wu, Z.; Jung, Y.; Ferreira, R.B.; Carroll, K.S.; Blackwell, T.K.; Yang, J. Global profiling of distinct cysteine redox forms reveals wide-ranging redox regulation in C. elegans. Nat. Commun. 2021, 12, 1415. [Google Scholar] [CrossRef]
- Martinez-Banaclocha, M. N-Acetyl-Cysteine: Modulating the cysteine redox proteome in neurodegenerative diseases. Antioxidants 2022, 11, 416. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Banaclocha, M. Cysteine network (CYSTEINET) dysregulation in Parkinson’s disease: Role of N-acetylcysteine. Curr. Drug Metab. 2016, 17, 368–385. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Banaclocha, M. N-acetylcysteine: A natural antidote for Alzheimer’s disease. Alzheimers Dis. Dement. 2016, 1, 4–15. [Google Scholar]
- Martinez-Banaclocha, M. Potential role of N-acetyl-cysteine in the cysteine proteome in Parkinson’s sisease? Clin. Pharmacol. Ther. 2020, 107, 1055. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Banaclocha, M. Proteomic complexity in Parkinson’s disease: A redox signaling perspective of the pathophysiology and progression. Neuroscience 2021, 453, 287–300. [Google Scholar] [CrossRef]
- Weerapana, E.; Wang, C.; Simon, G.M.; Richter, F.; Khare, S.; Dillon, M.B.; Bachovchin, D.A.; Mowen, K.; Baker, D.; Cravatt, B.F. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 2010, 468, 790–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radzinski, M.; Oppenheim, T.; Metanis, N.; Reichmann, D. The cys sense: Thiol redox switches mediate life cycles of celular proteins. Biomolecules 2021, 11, 469. [Google Scholar] [CrossRef] [PubMed]
- Sitia, R.; Braakman, I. Quality control in the endoplasmic reticulum protein factory. Nature 2003, 426, 891–894. [Google Scholar] [CrossRef] [Green Version]
- Feleciano, D.R.; Kirstein, J. Collapse of redox homeostasis during aging and stress. Mol. Cell. Oncol. 2015, 3, e1091060. [Google Scholar] [CrossRef] [Green Version]
- Paulsen, C.E.; Carroll, K.S. Cysteine-mediated redox signaling: Chemistry, biology, and tools for discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.J. The oxidative environment and protein damage. Biochim. Biophys. Acta 2005, 1703, 93–109. [Google Scholar] [CrossRef]
- Corcoran, A.; Cotter, T.G. Redox regulation of protein kinases. FEBS J. 2013, 280, 1944–1965. [Google Scholar] [CrossRef]
- Meister, A. Glutathione metabolism and its selective modification. J. Biol. Chem. 1988, 263, 17205–17208. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Torner, M.; Carrero, D.; Pérez-Silva, J.G.; Álvarez-Puente, D.; Roiz-Valle, D.; Bretones, G.; Rodríguez, D.; Maeso, D.; Mateo-González, E.; Español, Y.; et al. Comparative genomics of mortal and immortal cnidarians unveils novel keys behind rejuvenation. Proc. Natl. Acad. Sci. USA 2022, 119, e2118763119. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, B.; Sakellariou, G.K.; Smith, N.T.; Brownridge, P.; Jackson, M.J. Differential cysteine labelling and global label-free proteomics reveals an altered metabolic state in skeletal muscle aging. J. Proteome Res. 2014, 13, 5008–5021. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.P.; Mody, V.C., Jr.; Carlson, J.L.; Lynn, M.J.; Sternberg, P., Jr. Redox analysis of human plasma allows separation of pro-oxidant events of aging from decline in antioxidant defenses. Free Radic. Biol. Med. 2002, 33, 1290–1300. [Google Scholar] [CrossRef]
- Nuttall, S.L.; Martin, U.; Sinclair, A.J.; Kendall, M.J. Glutathione: In sickness and in health. Lancet 1998, 351, 645–646. [Google Scholar] [CrossRef]
- Liedhegner, E.A.S.; Gao, X.H.; Mieyal, J.J. Mechanisms of altered redox regulation in neurodegenerative diseases—Focus on S-glutathionylation. Antioxid. Redox Signal. 2012, 16, 543–566. [Google Scholar] [CrossRef] [Green Version]
- Ansari, M.A.; Scheff, S.W. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exp. Neurol. 2010, 69, 155–167. [Google Scholar] [CrossRef] [Green Version]
- Miquel, J.; Ferrándiz, M.L.; De Juan, E.; Sevilla, I.; Martinez-Banaclocha, M. N-acetylcysteine protecs against age-related decline of oxidative phosphorylation in liver mitochondria. Eur. J. Pharmacol. 1995, 292, 333–335. [Google Scholar]
- Knuesting, J.; Scheibe, R. Small molecules govern thiol redox switches. Trends Plant. Sci. 2018, 23, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Levine, C.G.; Mitra, D.; Sharma, A.; Smith, C.L.; Hegde, R.S. The efficiency of protein compartmentalization into the secretory pathway. Mol. Biol. Cell 2005, 16, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Haas, A.L.; Rose, I.A. The mechanism of ubiquitin activating enzyme. J. Biol. Chem. 1982, 257, 10329–10337. [Google Scholar] [CrossRef]
- Shang, F.; Taylor, A. Ubiquitin-proteasome pathway and cellular responses to oxidative stress. Free Radic. Biol. Med. 2011, 51, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.D.; Hannink, M. Distinct cysteine cesidues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Muramatsu, A.; Saito, R.; Iso, T.; Shibata, T.; Kuwata, K.; Kawaguchi, S.; Iwawaki, T.; Adachi, S.; Suda, H.; et al. Molecular mechanism of cellular oxidative stress sensing by Keap1. Cell Rep. 2019, 28, 746–758.e4. [Google Scholar] [CrossRef] [Green Version]
- Hourihan, J.M.; Moronetti Mazzeo, L.E.; Fernandez-Cardenas, L.P.; Blackwell, T.K. Cysteine sulfenylation directs IRE-1 to actívate the SKN-1/Nrf2 antioxidant response. Mol. Cell 2016, 63, 553–566. [Google Scholar] [CrossRef] [Green Version]
- Pajares, M.; Cuadrado, A.; Rojo, A.I. Modulation of proteostasis by transcription factor NRF2 and impact in neurodegenerative diseases. Redox Biol. 2017, 11, 543–553. [Google Scholar] [CrossRef] [Green Version]
- Kirstein, J.; Morito, D.; Kakihana, T.; Sugihara, M.; Minnen, A.; Hipp, M.S.; Nussbaum-Krammer, C.; Hartl, F.U.; Nagata, K.; Morimoto, R.I. Proteotoxic stress and ageing triggers the loss of redox homeostasis across cellular compartments. EMBO J. 2015, 34, 2334–2349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzman, J.N.; Sanchez-Padilla, J.; Wokosin, D.; Kondapalli, J.; Ilijic, E.; Schumacker, P.T.; Surmeier, D.J. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 2010, 468, 696–700. [Google Scholar] [CrossRef] [Green Version]
- Horowitz, M.P.; Milanese, C.; Di Maio, R.; Hu, X.; Montero, L.M.; Sanders, L.H.; Tapias, V.; Sepe, S.; van Cappellen, W.A.; Burton, E.A.; et al. Single-cell redox imaging demonstrates a distinctive response of dopaminergic neurons to oxidative insults. Antioxid. Redox Signal. 2011, 15, 855–871. [Google Scholar] [CrossRef] [Green Version]
- Chinta, S.; Andersen, J. Redox imbalance in Parkinson’s disease. Biochim. Biophys. Acta 2008, 1780, 1362–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Garcia, A.; Zavala-Flores, L.; Rodriguez-Rocha, H.; Franco, R. Thiol-redox signaling, dopaminergic cell death, and Parkinson’s disease. Antioxid. Redox Signal. 2012, 17, 1764–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Redox mechanisms in neurodegeneration: From disease outcomes to therapeutic opportunities. Antioxid. Redox Signal. 2019, 30, 1450–1499. [Google Scholar] [CrossRef] [PubMed]
- Milanese, C.; Payán-Gómez, C.; Mastroberardino, P.G. Cysteine oxidation and redox signaling in dopaminergic neurons physiology and in Parkinson’s disease. Curr. Opin. Physiol. 2019, 9, 73–78. [Google Scholar] [CrossRef]
- Benedikter, B.J.; Weseler, A.R.; Wouters, E.F.M.; Savelkoul, P.H.M.; Rohde, G.G.U.; Stassen, F.R.M. Redox-dependent thiol modifications: Implications for the release of extracellular vesicles. Cell. Mol. Life Sci. 2018, 75, 2321–2337. [Google Scholar] [CrossRef] [Green Version]
- Bodega, G.; Alique, M.; Puebla, L.; Carracedo, J.; Ramírez, R.M. Microvesicles: ROS scavengers and ROS producers. J. Extracell. Vesicles 2019, 8, 1626654. [Google Scholar] [CrossRef] [Green Version]
- Mullen, L.; Hanschmann, E.M.; Lillig, C.H.; Herzenberg, L.A.; Ghezzi, P. Cysteine oxidation targets peroxiredoxins 1 and 2 for exosomal release through a novel mechanism of redox-dependent secretion. Mol. Med. 2015, 21, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Zima, A.V.; Blatter, L.A. Redox regulation of cardiac calcium channels and transporters. Cardiovasc. Res. 2006, 71, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Bánsághi, S.; Golenár, T.; Madesh, M.; Csordás, G.; RamachandraRao, S.; Sharma, K.; Yule, D.I.; Joseph, S.K.; Hajnóczky, G. Isoform- and species-specific control of inositol 1,4,5-trisphosphate (IP3) receptors by reactive oxygen species. J. Biol. Chem. 2014, 289, 8170–8181. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.A.; Rossi, A.M.; Riley, A.M.; Potter, B.V.; Taylor, C.W. Subtype-selective regulation of IP3 receptors by thimerosal via cysteine residues within the IP3-binding core and suppressor domain. Biochem. J. 2013, 451, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Pasquet, J.M.; Dachary-Prigent, J.; Nurden, A.T. Calcium influx is a determining factor of calpain activation and microparticleformation in platelets. Eur. J. Biochem. 1996, 239, 647–654. [Google Scholar] [CrossRef]
- Liu, J.; Liu, M.C.; Wang, K.K. Calpain in the CNS: From synaptic function to neurotoxicity. Sci. Signal. 2008, 1, re1. [Google Scholar] [CrossRef]
- Mouatt-Prigent, A.; Karlsson, J.O.; Agid, Y.; Hirsch, E.C. Increased M-calpain expression in the mesencephalon of patients with Parkinson’s disease but not in other neurodegenerative disorders involving the mesencephalon: A role in nerve cell death? Neuroscience 1996, 73, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Suk, J.E.; Bae, E.J.; Lee, J.H.; Paik, S.R.; Lee, S.J. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int. J. Biochem. Cell Biol. 2008, 40, 1835–1849. [Google Scholar] [CrossRef] [PubMed]
- Freundt, E.C.; Maynard, N.; Clancy, E.K.; Roy, S.; Bousset, L.; Sourigues, Y.; Covert, M.; Melki, R.; Kirkegaard, K.; Brahic, M. Neuron-to-neuron transmission of alpha-synuclein fibrils through axonal transport. Ann. Neurol. 2012, 72, 517–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roosen, D.A.; Blauwendraat, C.; Cookson, M.R.; Lewis, P.A. DNAJC proteins and pathways to parkinsonism. FEBS J. 2019, 286, 3080–3094. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, T.; Yoshida, S.; Sugeno, N.; Kobayashi, J.; Aoki, M. DnaJ/Hsp40 family and Parkinson’s disease. Front. Neurosci. 2018, 11, 743. [Google Scholar] [CrossRef]
- Chandra, S.; Gallardo, G.; Fernández-Chacón, R.; Schlüter, O.M.; Südhof, T.C. α-synuclein cooperates with CSP in preventing neurodegeneration. Cell 2005, 123, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Zarouchlioti, C.; Parfitt, D.A.; Li, W.; Gittings, L.M.; Cheetham, M.E. DNAJ proteins in neurodegeneration: Essential and protective factors. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20160534. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Wang, B.; Li, X.; Fu, C.; Wang, C.; Kang, X. α-Synuclein: A multifunctional player in exocytosis, endocytosis, and vesicle recycling. Front. Neurosci. 2019, 13, 28. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Koutras, C.; Donnelier, J.; Alshehri, M.; Fotouhi, M.; Girard, M.; Casha, S.; McPherson, P.S.; Robbins, S.M.; Braun, J.E.A. Neurons export extracellular vesicles enriched in cysteine string protein and misfolded protein cargo. Sci. Rep. 2017, 7, 956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, D.; Garcia-Garcia, E.; Royo, F.; Falcon-Perez, J.M.; Avila, J. Proteostasis of tau. Tau overexpression results in its secretion via membrane vesicles. FEBS Lett. 2012, 586, 47–54. [Google Scholar] [CrossRef]
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Krüger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E.; et al. The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 2017, 12, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Kovac, A.; Korff, A.; Cook, T.J.; Ginghina, C.; Bullock, K.M.; Yang, L.; Stewart, T.; Zheng, D.; Aro, P.; et al. CNS tau efflux via exosomes is likely increased in Parkinson’s disease but not in Alzheimer’s disease. Alzheimer’s Dement. 2016, 12, 1125–1131. [Google Scholar] [CrossRef] [Green Version]
- Gegg, M.E.; Schapira, A.H.V. The role of glucocerebrosidase in Parkinson disease pathogenesis. FEBS J. 2018, 285, 3591–3603. [Google Scholar] [CrossRef] [Green Version]
- Do, J.; McKinney, C.; Sharma, P.; Sidransky, E. Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener. 2019, 14, 36. [Google Scholar] [CrossRef] [Green Version]
- Burbulla, L.F.; Jeon, S.; Zheng, J.; Song, P.; Silverman, R.B.; Krainc, D.A. modulator of wild-type glucocerebrosidase improves pathogenic phenotypes in dopaminergic neuronal models of Parkinson’s disease. Sci. Transl. Med. 2019, 11, 514. [Google Scholar] [CrossRef]
- Baden, P.; Perez, M.J.; Raji, H.; Bertoli, F.; Kalb, S.; Illescas, M.; Spanos, F.; Giuliano, C.; Calogero, A.M.; Oldrati, M.; et al. Glucocerebrosidase is imported into mitochondria and preserves complex I integrity and energy metabolism. Nat. Commun. 2023, 14, 1930. [Google Scholar] [CrossRef]
- Liu, M.; Kang, S.; Ray, S.; Jackson, J.; Zaitsev, A.D.; Gerber, S.A.; Cuny, G.D.; Glicksman, M.A. Kinetic, mechanistic, and structural modeling studies of truncated wild-type leucine-rich repeat kinase 2 and the G2019S mutant. Biochemistry 2011, 50, 9399–9408. [Google Scholar] [CrossRef] [Green Version]
- Dvir, H.; Harel, M.; McCarthy, A.A.; Toker, L.; Silman, I.; Futerman, A.H.; Sussman, J.L. X-ray structure of human acid-b-glucosidase, the defective enzyme in Gaucher disease. EMBO Rep. 2003, 4, 704–709. [Google Scholar] [CrossRef] [Green Version]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, S.H.; Knape, M.J.; Boassa, D.; Mumdey, N.; Kornev, A.P.; Ellisman, M.H.; Taylor, S.S.; Herberg, F.W. The dynamic switchmechanism that leads to activation of LRRK2 is embedded in the DFG motif in the kinase domain. Proc. Natl. Acad. Sci. USA 2019, 116, 14979–14988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Maio, R.; Hoffman, E.K.; Rocha, E.M.; Keeney, M.T.; Sanders, L.H.; De Miranda, B.R.; Zharikov, A.; Van Laar, A.; Stepan, A.F.; Lanz, T.A.; et al. LRRK2 activation in idiopathic Parkinson’s disease. Sci. Transl. Med. 2018, 10, 451. [Google Scholar] [CrossRef] [Green Version]
- Aasly, J.O.; Sæther, O.; Johansen, K.K.; Bathen, T.F.; Giskeødegård, G.F.; White, L.R. Changes to intermediarymetabolites in sporadic and LRRK2 Parkinson’s disease demonstrated by protonmagnetic resonance spectroscopy. Parkinson Dis. 2015, 2015, 264896. [Google Scholar]
- Brahmachari, S.; Karuppagounder, S.S.; Ge, P.; Lee, S.; Dawson, V.L.; Dawson, T.M.; Ko, H.S. c-Abl and Parkinson’s disease: Mechanisms and therapeutic potential. J. Parkinson Dis. 2017, 7, 589–601. [Google Scholar] [CrossRef] [Green Version]
- Leonberg, A.K.; Chai, Y.C. The functional role of cysteine residues for c-Abl kinase activity. Mol. Cell. Biochem. 2007, 304, 207–212. [Google Scholar] [CrossRef]
- Kharbanda, S.; Kumar, V.; Dhar, S.; Pandey, P.; Chen, C.; Majumder, P.; Yuan, Z.M.; Whang, Y.; Strauss, W.; Pandita, T.K.; et al. Regulation of the hTERT telomerase catalytic subunit by the c-Abl tyrosine kinase. Curr. Biol. 2000, 10, 568–575. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Kim, S.; Park, Y.J.; Yun, S.P.; Kwon, S.H.; Kim, D.; Kim, D.Y.; Shin, J.S.; Cho, D.J.; Lee, G.Y.; et al. The c-Abl inhibitor, Radotinib HCl, is neuroprotective in a preclinical Parkinson’s disease mouse model. Hum. Mol. Genet. 2018, 27, 2344–2356. [Google Scholar] [CrossRef] [Green Version]
- Joselin, A.P.; Hewitt, S.J.; Callaghan, S.M.; Kim, R.H.; Chung, Y.H.; Mak, T.W.; Shen, J.; Slack, R.S.; Park, D.S. ROS-dependent regulation of parkin and DJ-1 localization during oxidative stress in neurons. Hum. Mol. Genet. 2012, 21, 4888–4903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshii, S.R.; Kishi, C.; Ishihara, N.; Mizushima, N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem. 2011, 286, 19630–19640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seirafi, M.; Kozlov, G.; Gehring, K. Parkin structure and function. FEBS J. 2015, 282, 2076–2088. [Google Scholar] [CrossRef] [Green Version]
- Yao, D.; Gu, Z.; Nakamura, T.; Shi, Z.Q.; Ma, Y.; Gaston, B.; Palmer, L.A.; Rockenstein, E.M.; Zhang, Z.; Masliah, E.; et al. Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. USA 2004, 101, 10810–10814. [Google Scholar] [CrossRef] [Green Version]
- Ozawa, K.; Komatsubara, A.T.; Nishimura, Y.; Sawada, T.; Kawafune, H.; Tsumoto, H.; Tsuji, Y.; Zhao, J.; Kyotani, Y.; Tanaka, T.; et al. S-nitrosylation regulates mitochondrial quality control via activation of parkin. Sci. Rep. 2013, 3, 2202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, F.; Yao, D.; Shi, Y.; Kabakoff, J.; Wu, W.; Reicher, J.; Ma, Y.; Moosmann, B.; Masliah, E.; Lipton, S.A.; et al. Oxidation of the cysteine-rich regions of parkin perturbs its E3 ligase activity and contributes to protein aggregation. Mol. Neurodegener. 2011, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- Dawson, T.M.; Dawson, V.L. The role of parkin in familial and sporadic Parkinson’s disease. Mov. Disord. 2014, 25, S32–S39. [Google Scholar] [CrossRef] [Green Version]
- Canet-Avilés, R.M.; Wilson, M.A.; Miller, D.W.; Ahmad, R.; McLendon, C.; Bandyopadhyay, S.; Baptista, M.J.; Ringe, D.; Petsko, G.A.; Cookson, M.R. The Parkinson’s disease DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitocondrial localization. Proc. Natl. Acad. Sci. USA 2004, 101, 9103–9108. [Google Scholar] [CrossRef] [Green Version]
- Torres, G.E.; Gainetdinov, R.R.; Caron, M.G. Plasma membrane monoamine transporters: Structure, regulation and function. Nat. Rev. Neurosci. 2003, 4, 13–25. [Google Scholar] [CrossRef]
- Chen, N.; Ferrer, J.V.; Javitch, J.A.; Justice, J.B., Jr. Transport-dependent accessibility of a cytoplasmic loop cysteine in the human dopamine transporter. J. Biol. Chem. 2000, 275, 1608–1614. [Google Scholar] [CrossRef] [Green Version]
- Rastedt, D.E.; Vaughan, R.A.; Foster, J.D. Palmitoylation mechanisms in dopamine transporter regulation. J. Chem. Neuroanat. 2017, 83–84, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, G.; Ariga, H.; Nakagawa, Y.; Iwatsubo, T. Roles of distinct cysteine residues in S-nitrosylation and dimerization of DJ-1. Biochem. Biophys. Res. Commun. 2006, 339, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.S.; Nakamura, T.; Cho, S.J.; Han, X.; Holland, E.A.; Qu, J.; Petsko, G.A.; Yates, J.R., 3rd; Liddington, R.C.; Lipton, S.A. Transnitrosylation from DJ-1 to PTEN attenuates neuronal cell death in Parkinson’s disease models. J. Neurosci. 2014, 34, 15123–15131. [Google Scholar] [CrossRef] [Green Version]
- Coetzee, W.A.; Nakamura, T.Y.; Faivre, J.F. Effects of thiol-modifying agents on KATP channels in guinea pig ventricular cells. Am. J. Physiol. 1995, 269, H1625–H1633. [Google Scholar] [CrossRef]
- Jiang, B.; Tang, G.; Cao, K.; Wu, L.; Wang, R. Molecular mechanism for H2S-induced activation of KATP channels. Antioxid. Redox Signal. 2010, 12, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Avshalumov, M.V.; Chen, B.T.; Koós, T.; Tepper, J.M.; Rice, M.E. Endogenous hydrogen peroxide regulates the excitability of midbrain dopamine neurons via ATP-sensitive potassium channels. J. Neurosci. 2005, 25, 4222–4231. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Nakamura, T.; Cho, D.H.; Gu, Z.; Lipton, S.A. S-nitrosylation of peroxiredoxin 2 promotes oxidative stress-induced neuronal cell death in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 18742–18747. [Google Scholar] [CrossRef] [Green Version]
- Mailloux, R.J.; Jin, X.; Willmore, W.G. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol. 2013, 2, 123–139. [Google Scholar] [CrossRef] [Green Version]
- Soeda, Y.; Yoshikawa, M.; Almeida, O.F.; Sumioka, A.; Maeda, S.; Osada, H.; Kondoh, Y.; Saito, A.; Miyasaka, T.; Kimura, T.; et al. Toxic tau oligomer formation blocked by capping of cysteine residues with 1,2-dihydroxybenzene groups. Nat. Commun. 2015, 6, 10216. [Google Scholar] [CrossRef] [Green Version]
- Schweers, O.; Mandelkow, E.; Biernat, J.; Mandelkow, E. Oxidation of cysteine-322 in the repeat domain of microtubule-associated protein T controls the in vitro assembly of paired helical filaments. Proc. Natl. Acad. Sci. USA 1995, 92, 8463–8467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecke, S.H.; Goldberg, A.L.; Mitch, W.E. Protein degradation by the ubiquitin–proteasome pathway in normal and disease states. J. Am. Soc. Nephrol. 2006, 17, 1807–1819. [Google Scholar] [CrossRef] [Green Version]
- Jahngen-Hodge, J.; Obin, M.S.; Gong, X.; Shang, F.; Nowell, T.R., Jr.; Gong, J.; Abasi, H.; Blumberg, J.; Taylor, A. Regulation of ubiquitin-conjugating enzymes by glutathione following oxidative stress. J. Biol. Chem. 1997, 272, 28218–28226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landino, L.M.; Skreslet, T.E.; Alston, J.A. Cysteine oxidation of Tau and microtubule-associated protein-2 by peroxynitrite. J. Biol. Chem. 2004, 279, 35101–35105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouatt-Prigent, A.; Agid, Y.; Hirsch, E.C. Does the calcium binding protein calretinin protect dopaminergic neurons against degeneration in Parkinson’s disease? Brain Res. 1994, 668, 62–70. [Google Scholar] [CrossRef]
- Cohen, T.J.; Friedmann, D.; Hwang, A.W.; Marmorstein, R.; Lee, V.M. The microtubule-associated tau protein has intrinsic acetyltransferase activity. Nat. Struct. Mol. Biol. 2013, 20, 756–762. [Google Scholar] [CrossRef] [Green Version]
- Wegmann, S.; Medalsy, I.D.; Mandelkow, E.; Müller, D.J. The fuzzy coat of pathological human Tau fibrils is a two-layered polyelectric brush. Proc. Natl. Acad. Sci. USA 2012, 109, E313–E321. [Google Scholar]
- Chidambaram, H.; Chinnathambi, S. Role of cysteines in accelerating Tau filament formation. J. Biomol. Struct. Dyn. 2022, 40, 4366–4375. [Google Scholar] [CrossRef]
- Breydo, L.; Wu, J.W.; Uversky, V.N. α-synuclein misfolding and Parkinson’s disease. Biochim. Biophys. Acta 2012, 1822, 261–285. [Google Scholar] [CrossRef] [Green Version]
- Recchia, A.; Debetto, P.; Negro, A.; Guidolin, D.; Skaper, S.D.; Giusti, P. α-Synuclein and Parkinson’s disease. FASEB J. 2004, 18, 617–626. [Google Scholar] [CrossRef] [Green Version]
- Jha, N.; Kumar, M.J.; Boonplueang, R.; Andersen, J.K. Glutathione decreases in dopaminergic PC12 cells interfere with the ubiquitin protein degradation pathway: Relevance for Parkinson’s disease? J. Neurochem. 2002, 80, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Amm, I.; Sommer, T.; Wolf, D.H. Protein quality control and elimination of protein waste: The role of the ubiquitin–proteasome system. Biochim. Biophys. Acta 2014, 1843, 182–196. [Google Scholar] [CrossRef] [Green Version]
- Bellucci, A.; Bubacco, L.; Longhena, F.; Parrella, E.; Faustini, G.; Porrini, V.; Bono, F.; Missale, C.; Pizzi, M. Nuclear Factor-B dysregulation and α-synuclein pathology: Critical interplay in the pathogenesis of Parkinson’s disease. Front. Aging Neurosci. 2020, 12, 68. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Becker, K.; Levine, N.; Zhang, M.; Lieberman, A.P.; Moore, D.J.; Ma, J. Pathogenic alpha-synuclein aggregates preferentially bind to mitochondria and affect cellular respiration. Acta Neuropathol. Commun. 2019, 7, 41. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Conde, L.D.; Ramos-Acevedo, R.; Reyes-Hernández, M.A.; Balbuena-Olvera, A.J.; Morales-Moreno, I.D.; Argüero-Sánchez, R.; Schüle, B.; Guerra-Crespo, M. Alpha-synuclein physiology and pathology: A perspective on cellular structures and organelles. Front. Neurosci. 2020, 13, 1399. [Google Scholar] [CrossRef] [Green Version]
- Deleidi, M.; Cooper, O.; Hargus, G.; Levy, A.; Isacson, O. Oct4-induced reprogramming is required for adult brain neural stem cell differentiation into midbrain dopaminergic neurons. PLoS ONE 2011, 6, e19926. [Google Scholar] [CrossRef] [PubMed]
- Marsboom, G.; Zhang, G.F.; Pohl-Avila, N.; Zhang, Y.; Yuan, Y.; Kang, H.; Hao, B.; Brunengraber, H.; Malik, A.B.; Rehman, J. Glutamine metabolism regulates the pluripotency transcription factor OCT4. Cell Rep. 2016, 16, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Alves da Costa, C.; Checler, F. Apoptosis in Parkinson’s disease: Is p53 the missing link between genetic and sporadic Parkinsonism? Cell Signal. 2011, 23, 963–968. [Google Scholar] [CrossRef]
- Luo, Q.; Sun, W.; Wang, Y.F.; Li, J.; Li, D.W. Association of p53 with neurodegeneration in Parkinson’s disease. Parkinsons Dis. 2022, 2022, 6600944. [Google Scholar] [CrossRef]
- Peng, J.; Andersen, J.K. The role of c-Jun N-terminal kinase (JNK) in Parkinson’s disease. IUBMB Life 2003, 55, 267–271. [Google Scholar] [CrossRef]
- Dolatshahi, M.; Ranjbar Hameghavandi, M.H.; Sabahi, M.; Rostamkhani, S. Nuclear factor-kappa B (NF-κB) in pathophysiology of Parkinson disease: Diverse patterns and mechanisms contributing to neurodegeneration. Eur. J. Neurosci. 2021, 54, 4101–4123. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.R.; Wakasugi, N.; Virelizier, J.L.; Yodoi, J.; Hay, R.T. Thioredoxin regulates the DNA binding activity of NF-kappa B by reduction of a disulphide bond involving cysteine 62. Nucleic Acids Res. 1992, 20, 3821–3830. [Google Scholar] [CrossRef] [PubMed]
- Hui, C.C.; Angers, S. Gli proteins in development and disease. Ann. Rev. Cell Dev. Biol. 2011, 27, 513–537. [Google Scholar] [CrossRef] [Green Version]
- Crapster, J.A.; Hudgins, L.; Chen, J.K.; Gomez-Ospina, N. A novel missense variant in the GLI3 zinc finger domain in a family with digital anomalies. Am. J. Med. Genet. A 2017, 173, 3221–3225. [Google Scholar] [CrossRef] [PubMed]
- Joo, Y.; Xue, Y.; Wang, Y.; McDevitt, R.A.; Sah, N.; Bossi, S.; Su, S.; Lee, S.K.; Peng, W.; Xie, A.; et al. Topoisomerase 3β knockout mice show transcriptional and behavioural impairments associated with neurogenesis and synaptic plasticity. Nat. Commun. 2020, 11, 3143. [Google Scholar] [CrossRef] [PubMed]
- Rahman, F.U.; Kim, Y.R.; Kim, E.K.; Kim, H.R.; Cho, S.M.; Lee, C.S.; Kim, S.J.; Araki, K.; Yamamura, K.I.; Lee, M.N.; et al. Topoisomerase IIIβ Deficiency induces neuro behavioral changes and brain connectivity alterations in mice. Int. J. Mol. Sci. 2021, 22, 12806. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.W.; Liu, Y.; Hasselblatt, K.T.; Mok, S.C.; Berkowitz, R.S. A new human topoisomerase III that interacts with SGS1 protein. Nucleic Acids Res. 1999, 27, 993–1000. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Wang, X.X.; Zhuang, Y.W.; Jiang, Y.; Melcher, K.; Xu, H.E. Structure of the PRC2 complex and application to drug discovery. Acta Pharmacol. Sin. 2017, 38, 963–976. [Google Scholar] [CrossRef] [Green Version]
- Toskas, K.; Yaghmaeian-Salmani, B.; Skiteva, O.; Paslawski, W.; Gillberg, L.; Skara, V.; Antoniou, I.; Södersten, E.; Svenningsson, P.; Chergui, K.; et al. PRC2-mediated repression is essential to maintain identity and function of differentiated dopaminergic and serotonergic neurons. Sci. Adv. 2022, 8, eabo1543. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, T.; Dong, Q.; Nice, E.C.; Huang, C.; Wei, Y. Redox homeostasis: The linchpin in stem cell self-renewal and differentiation. Cell Death Dis. 2013, 4, e537. [Google Scholar] [CrossRef] [Green Version]
- Camins, A.; Pizarro, J.G.; Alvira, D.; Gutierrez-Cuesta, J.; de la Torre, A.V.; Folch, J.; Sureda, F.X.; Verdaguer, E.; Junyent, F.; Jordán, J.; et al. Activation of ataxia telangiectasia muted under experimental models and human Parkinson’s disease. Cell. Mol. Life Sci. 2010, 67, 3865–3882. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.S.; You, J.S.; Kim, T.K.; Ahn, W.J.; Kim, M.J.; Son, K.H.; Ricarte, D.; Ortiz, D.; Lee, S.J.; Lee, H.J. Senescence and impaired DNA damage responses in alpha-synucleinopathy models. Exp. Mol. Med. 2022, 54, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Milanese, C.; Cerri, S.; Ulusoy, A.; Gornati, S.V.; Plat, A.; Gabriels, S.; Blandini, F.; Di Monte, D.A.; Hoeijmakers, J.H.; Mastroberardino, P.G. Activation of the DNA damage response in vivo in synucleinopathy models of Parkinson’s disease. Cell Death Dis. 2018, 9, 818. [Google Scholar] [CrossRef] [Green Version]
- Itoh, F.; Asao, H.; Sugamura, K.; Heldin, C.H.; ten Dijke, P.; Itoh, S. Promoting bone morphogenetic protein signaling through negative regulation of inhibitory Smads. EMBO J. 2001, 20, 4132–4142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitic, Z.; Safory, H.; Jovanovic, V.M.; Sarusi, Y.; Stavsky, A.; Kahn, J.; Kuzmina, A.; Toker, L.; Gitler, D.; Taube, R.; et al. BMP5/7 protect dopaminergic neurons in an α-synuclein mouse model of Parkinson’s disease. Brain 2021, 144, e15. [Google Scholar] [CrossRef]
- Mitu, G.; Hirschberg, R. Bone morphogenetic protein-7 (BMP7) in chronic kidney disease. Front. Biosci. 2008, 13, 4726–4739. [Google Scholar] [CrossRef]
- Forero, D.A.; González-Giraldo, Y.; López-Quintero, C.; Castro-Vega, L.J.; Barreto, G.E.; Perry, G. Telomere length in Parkinson’s disease: A meta-analysis. Exp. Gerontol. 2016, 75, 53–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levstek, T.; Kozjek, E.; DolŽan, V.; Trebušak Podkrajšek, K. Telomere attrition in neurodegenerative disorders. Front. Cell Neurosci. 2020, 14, 219. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H.; Collins, K. Telomerase: An RNP enzyme synthesizes DNA. Cold Spring Harb. Perspect. Biol. 2011, 3, a003558. [Google Scholar] [CrossRef] [Green Version]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial cell senescence in human atherosclerosis: Role of telomere in endothelial dysfunction. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef] [Green Version]
- Colgin, L.M.; Reddel, R.R. Telomere maintenance mechanisms and celular immortalization. Curr. Opin. Genet. Dev. 1999, 9, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H. Telomere states and cell fates. Nature 2000, 408, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Bosoy, D.; Peng, Y.; Mian, I.S.; Lue, N.F. Conserved N-terminal motifs of telomerase reverse transcriptase required for ribonucleoprotein assembly in vivo. J. Biol. Chem. 2003, 278, 3882–3890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minamino, T.; Mitsialis, S.A.; Kourembanas, S. Hypoxia extends the life span of vascular smooth muscle cells through telomerase activation. Mol. Cell. Biol. 2001, 21, 3336–3342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Hodes, R.J.; Weng, N.-P. Telomerase activation in human T lymphocytes does not require increase in telomerase reverse transcriptase (hTERT) protein but is associated with hTERT phosphorylation and nuclear translocation. J. Immunol. 2001, 166, 4826–4830. [Google Scholar] [CrossRef] [Green Version]
- Haendeler, J.; Hoffmann, J.; Brandes, R.P.; Zeiher, A.M.; Dimmeler, S. Hydrogen peroxide triggers nuclear export of telomerase reverse transcriptasevia Src kinase family-dependent phosphorylation of tyrosine 707. Mol. Cell. Biol. 2003, 23, 4598–4610. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Kellermann, G.; Dingli, F.; Masson, V.; Dauzonne, D.; Ségal-Bendirdjian, E.; Teulade-Fichou, M.P.; Loew, D.; Bombard, S. Exploring the mechanism of inhibition of human telomerase by cysteine-reactive compounds. FEBS Lett. 2017, 591, 863–874. [Google Scholar] [CrossRef] [Green Version]
- Cramer, S.L.; Saha, A.; Liu, J.; Tadi, S.; Tiziani, S.; Yan, W.; Triplett, K.; Lamb, C.; Alters, S.E.; Rowlinson, S.; et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat. Med. 2017, 23, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Wang, R. Physiological implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef] [Green Version]
- Lee, Z.W.; Low, Y.L.; Huang, S.; Wang, T.; Deng, L.W. The cystathionine γ-lyase/hydrogen sulfide system maintains cellular glutathione status. Biochem. J. 2014, 460, 425–435. [Google Scholar] [CrossRef]
- Lux, O.; Naidoo, D. Biological variability of superoxide dismutase and glutathione peroxidase in blood. Redox Rep. 1995, 1, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Tosukhowong, P.; Boonla, C.; Dissayabutra, T.; Kaewwilai, L.; Muensri, S.; Chotipanich, C.; Joutsa, J.; Rinne, J.; Bhidayasiri, R. Biochemical and clinical effects of Whey protein supplementation in Parkinson’s disease: A pilot study. J. Neurol. Sci. 2016, 367, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Zavorsky, G.S.; Kubow, S.; Grey, V.; Riverin, V.; Lands, L.C. An open-label dose-response study of lymphocyte glutathione levels in healthy men and women receiving pressurized whey protein isolate supplements. Int. J. Food Sci. Nutr. 2007, 58, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Sechi, G.; Deledda, M.G.; Bua, G.; Satta, W.M.; Deiana, G.A.; Pes, G.M.; Rosati, G. Reduced intravenous glutathione in the treatment of early Parkinson’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 1996, 20, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Meister, A. Glutathione deficiency produced by inhibition of its synthesis, and its reversal; applications in research and therapy. Pharmacol. Ther. 1991, 51, 155–194. [Google Scholar] [CrossRef]
- Anderson, M.E.; Meister, A. Glutathione monoesters. Anal. Biochem. 1989, 183, 16–20. [Google Scholar] [CrossRef]
- Johnston, C.S.; Meyer, C.G.; Srilakshmi, J.C. Vitamin C elevates red blood cell glutathione in healthy adults. Am. J. Clin. Nutr. 1993, 58, 103–105. [Google Scholar] [CrossRef]
- Sharma, A.; Kharb, S.; Chugh, S.N.; Kakkar, R.; Singh, G.P. Evaluation of oxidative stress before and after control of glycemia and after vitamin E supplementation in diabetic patients. Metabolism 2000, 49, 160–162. [Google Scholar] [CrossRef]
- Jain, S.K.; McVie, R.; Smith, T. Vitamin E supplementation restores glutathione and malondialdehyde to normal concentrations in erythrocytes of type 1 diabetic children. Diabetes Care 2000, 23, 1389–1394. [Google Scholar] [CrossRef] [Green Version]
- Ashoori, M.; Saedisomeolia, A. Riboflavin (vitamin B2) and oxidative stress: A review. Br. J. Nutr. 2014, 111, 1985–1991. [Google Scholar] [CrossRef] [Green Version]
- Martins, V.D.; Manfredini, V.; Peralba, M.C.; Benfato, M.S. Alpha-lipoic acid modifies oxidative stress parameters in sickle cell trait subjects and sickle cell patients. Clin. Nutr. 2009, 28, 192–197. [Google Scholar] [CrossRef]
- Khalili, M.; Eghtesadi, S.; Mirshafiey, A.; Eskandari, G.; Sanoobar, M.; Sahraian, M.A.; Motevalian, A.; Norouzi, A.; Moftakhar, S.; Azimi, A. Effect of lipoic acid consumption on oxidative stress among multiple sclerosis patients: A randomized controlled clinical trial. Nutr. Neurosci. 2014, 17, 16–20. [Google Scholar] [CrossRef]
- Minich, D.M.; Brown, B.I. A review of dietary (phyto)nutrients for glutathione support. Nutrients 2019, 11, 2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.; Zhang, B.; Ge, C.; Peng, S.; Fang, J. Xanthohumol, a polyphenol chalcone present in hops, activating Nrf2 enzymes to confer protection against oxidative damage in PC12 cells. J. Agric. Food Chem. 2015, 63, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.S.; Lim, J.; Gal, J.; Kang, J.C.; Kim, H.J.; Kang, B.Y.; Choi, H.J. Antiinflammatory activity of xanthohumol involves heme oxygenase-1 induction via NRF2-ARE signaling in microglial BV2 cells. Neurochem. Int. 2011, 58, 153–160. [Google Scholar] [CrossRef]
- Hearn, B.R.; Fontaine, S.D.; Schneider, E.L.; Kraemer, Y.; Ashley, G.W.; Santi, D.V. Attenuation of the reaction of Michael acceptors with biologically important nucleophiles. Bioconjug. Chem. 2021, 32, 794–800. [Google Scholar] [CrossRef]
- Liang, S.T.; Chen, C.; Chen, R.X.; Li, R.; Chen, W.L.; Jiang, G.H.; Du, L.L. Michael acceptor molecules in natural products and their mechanism of action. Front. Pharmacol. 2022, 13, 1033003. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Wang, J.; Wang, C. Zerumbone attenuates MPP+-induced cytotoxicity in human neuroblasto-ma SH-SY5Y cells by inhibition of oxidative stress. Chin. J. Pathophysiol. 2018, 34, 1061–1066. [Google Scholar]
- Peng, S.; Hou, Y.; Yao, J.; Fang, J. Activation of Nrf2-driven antioxidant enzymes by cardamonin confers neuroprotection of PC12 cells against oxidative damage. Food Funct. 2017, 8, 997–1007. [Google Scholar] [CrossRef]
- Ji, L.; Yuan, Y.; Luo, L.; Chen, Z.; Ma, X.; Ma, Z.; Cheng, L. Physalins with antiinflammatory activity are present in Physalis alkekengi var. franchetii and can function as Michael reaction acceptors. Steroids 2012, 77, 441–447. [Google Scholar] [CrossRef]
- Xu, Z.; Wu, G.; Li, K.; Qian, G.; Wang, X.; Chen, W. Role of PKC/MAPK/NF-kappa B signal cascade in expression of IL-1β in rat monocytes exposed to hypoxia. Chin. J. Crit. Care Med. 2008, 9, 803–807. [Google Scholar]
- Widen, J.C.; Kempema, A.M.; Baur, J.W.; Skopec, H.M.; Edwards, J.T.; Brown, T.J.; Brown, D.A.; Meece, F.A.; Harki, D.A. Helenalin analogues targeting NF-κB p65: Thiol reactivity and cellular potency studies of varied electrophiles. ChemMedChem 2018, 13, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Almannai, M.; El-Hattab, A.W.; Ali, M.; Soler-Alfonso, C.; Scaglia, F. Clinical trials in mitochondrial disorders, an update. Mol. Genet. Metab. 2020, 131, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Enns, G.M.; Kinsman, S.L.; Perlman, S.L.; Spicer, K.M.; Abdenur, J.E.; Cohen, B.H.; Amagata, A.; Barnes, A.; Kheifets, V.; Shrader, W.D.; et al. Initial experience in the treatment of inherited mitochondrial disease with EPI-743. Mol. Genet. Metab. 2012, 105, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Zesiewicz, T.; Salemi, J.L.; Perlman, S.; Sullivan, K.L.; Shaw, J.D.; Huang, Y.; Isaacs, C.; Gooch, C.; Lynch, D.R.; Klein, M.B. Double-blind, randomized and controlled trial of EPI-743 in Friedreich’s ataxia. Neurodegener. Dis. Manag. 2018, 8, 233–242. [Google Scholar] [CrossRef]
- Pastore, A.; Petrillo, S.; Tozzi, G.; Carrozzo, R.; Martinelli, D.; Dionisi-Vici, C.; Di Giovamberardino, G.; Ceravolo, F.; Klein, M.B.; Miller, G.; et al. Glutathione: A redox signature in monitoring EPI-743 therapy in children with mitochondrial encephalomyopathies. Mol. Genet. Metab. 2013, 109, 208–214. [Google Scholar] [CrossRef]
- Martinelli, D.; Catteruccia, M.; Piemonte, F.; Pastore, A.; Tozzi, G.; Dionisi-Vici, C.; Pontrelli, G.; Corsetti, T.; Livadiotti, S.; Kheifets, V.; et al. EPI-743 reverses the progression of the pediatric mitochondrial disease–genetically defined Leigh Syndrome. Mol. Genet. Metab. 2012, 107, 383–388. [Google Scholar] [CrossRef]
- Zesiewicz, T.; Allison, K.; Jahan, I.; Shaw, J.; Murtagh, F.; Jones, T.; Gooch, C.; Salemi, J.; Klein, M.; Miller, G.; et al. EPI-743 improves motor function and CNS biomarkers in PD: Results from a phase 2A pilot trial (S40.004). Neurology 2016, 86 (Suppl. S16), I1.012. [Google Scholar]
- Martinez-Banaclocha, M. N-acetyl-cysteine in schizophrenia: Potential role on the sensitive cysteine proteome. Curr. Med. Chem. 2020, 27, 6424–6439. [Google Scholar] [CrossRef]
- Martinez-Banaclocha, M. N-acetylcysteine in psychiatric disorders: Possible role of cysteinet deregulation. Inter. Neuropsy. Dis. J. 2018, 12, 1–6. [Google Scholar] [CrossRef]
- Medina, S.; Martinez-Banaclocha, M.; Hernanz, A. Antioxidants inhibit the human cortical neuron apoptosis induced by hydrogen peroxide, tumor necrosis factor alpha, dopamine and beta-amyloid peptide 1–42. Free Radic. Res. 2002, 36, 1179–1184. [Google Scholar] [CrossRef]
- Martinez-Banaclocha, M. N-acetylcysteine elicited increase in complex I activity in synaptic mitochondria from aged mice: Implications for treatment of Parkinson’s disease. Brain Res. 2000, 859, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Banaclocha, M. Therapeutic potential of N-acetylcysteine in age-related mitochondrial neurodegenerative diseases. Med. Hypotheses 2001, 56, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Banaclocha, M. N-acetyl-cysteine in the treatment of Parkinson’s disease. What are we waiting for? Med. Hypotheses 2012, 79, 8–12. [Google Scholar] [CrossRef]
- Martinez-Banaclocha, M.; Martínez, N. N-acetylcysteine elicited increase in cytochrome c oxidase activity in mice synaptic mitochondria. Brain Res. 1999, 842, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Banaclocha, M.; Martinez, N.; Hernandez, A.I.; Ferrandiz, M.L. Hypothesis: Can N-acetylcysteine be beneficial in Parkinson’s disease? Life Sci. 1999, 64, 1253–1257. [Google Scholar] [CrossRef]
- Martinez-Banaclocha, M. Cysteinet dysregulation in muscular dystrophies: A pathogenic network susceptible to therapy. Curr. Med. Chem. 2017, 24, 312–330. [Google Scholar] [CrossRef]
- Martinez-Banaclocha, M.; Hernandez, A.I.; Martinez, N. N-Acetylcysteine delays age-associated memory impairment in mice: Role in synaptic mitochondria. Brain Res. 2000, 855, 100–106. [Google Scholar] [CrossRef]
- Martinez-Banaclocha, M.; Hernandez, A.I.; Martínez, N.; Ferrándiz, M.L. N-acetylcysteine protects against age-related increase in oxidized proteins in mouse synaptic mitochondria. Brain Res. 1997, 762, 256–258. [Google Scholar] [CrossRef]
- Miquel, J.; Martinez-Banaclocha, M.; Díez, A.; De Juan, E.; Soler, A.; Ramirez, A.; Laborda, J.; Carrión, M. Effects of turmeric on blood and liver lipoperoxide levels of mice: Lack of toxicity. Age 1995, 18, 171–174. [Google Scholar] [CrossRef]
- Martinez-Banaclocha, M. Interfering with the reactive cysteine proteome in COVID-19. Curr. Med. Chem. 2022, 29, 1657–1663. [Google Scholar] [CrossRef]
- Sekhar, R.V.; Patel, S.G.; Guthikonda, A.P.; Reid, M.; Balasubramanyam, A.; Taffet, G.E.; Jahoor, F. Deficient synthesis of glutathione underlies oxidative stress in aging and can be corrected by dietary cysteine and glycine supplementation. Am. J. Clin. Nutr. 2011, 94, 847–853. [Google Scholar] [CrossRef] [Green Version]
- Holmay, M.J.; Terpstra, M.; Coles, L.D.; Mishra, U.; Ahlskog, M.; Oz, G.; Cloyd, J.C.; Tuite, P.J. N-Acetylcysteine boosts brain and blood glutathione in Gaucher and Parkinson diseases. Clin. Neuropharmacol. 2013, 36, 103–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caro, L.; Ghizzi, A.; Costa, R.; Longo, A.; Ventresca, G.P.; Lodola, E. Pharmacokinetics and bioavailability of oral acetylcysteine in healthy volunteers. Arzneimittelforschung 1989, 39, 382–386. [Google Scholar] [PubMed]
- Van Zandwijk, N. N-Acetylcysteine for lung cancer prevention. Chest 1995, 107, 1437–1441. [Google Scholar] [CrossRef] [Green Version]
- De Flora, S.; Astengo, M.; Serra, D.; Bennicelli, C. Inhibition of urethan-induced lung tumors in mice by dietary N-acetylcysteine. Cancer Lett. 1986, 32, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Samuni, Y.; Goldstein, S.; Dean, O.M.; Berk, M. The chemistry and biological activities of N-acetylcysteine. Biochim. Biophys. Acta 2013, 1830, 4117–4129. [Google Scholar] [CrossRef]
- Schwalfenberg, G.K. N-Acetylcysteine: A review of clinical usefulness (an old drug with new tricks). J. Nutr. Metab. 2021, 2021, 9949453. [Google Scholar] [CrossRef] [PubMed]
- De Flora, S.; Izzotti, A.; D’Agostini, F.; Balansky, R.M. Mechanisms of N-acetylcysteine in the prevention of DNA damage and cancer, with special reference to smoking-related end-points. Carcinogenesis 2001, 22, 999–1013. [Google Scholar] [CrossRef] [Green Version]
- Dekhuijzen, P.N.; van Beurden, W.J. The role for N-acetylcysteine in the management of COPD. Int. J. Chron. Obstruct. Pulmon Dis. 2006, 1, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Banaclocha, M.; Hernandez, A.I.; Martinez, N.; Ferrandiz, M.L. Age-related increase in oxidized proteins in mouse synaptic mitochondria. Brain Res. 1996, 731, 246–248. [Google Scholar] [CrossRef]
- Martinez-Banaclocha, M.; Ferrandiz, M.L.; Díez, A.; Miquel, J. Depletion of cytosolic GSH decreases the ATP levels and viability of synaptosomes from aged mice but not from young mice. Mech. Ageing Dev. 1995, 84, 77–81. [Google Scholar] [CrossRef]
- Ferrandiz, M.L.; Martinez-Banaclocha, M.; De Juan, E.; Díez, A.; Bustos, G.; Miquel, J. Impairment of mitochondrial oxidative phosphorylation in the brain of aged mice. Brain Res. 1994, 644, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Banaclocha, M.; Ferrandiz, M.L.; De Juan, E.; Miquel, J. Age-related changes in glutathione and lipid peroxide content in mouse synaptic mitochondria: Relationship to cytochrome c oxidase decline. Neurosci. Lett. 1994, 170, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.Y.; Greene, L.A. Prevention of PC12 cell death by NAcetylcysteine requires activation of the Ras pathway. J. Neurosci. 1998, 18, 4042–4049. [Google Scholar] [CrossRef] [Green Version]
- Hobbs, G.A.; Gunawardena, H.P.; Campbell, S.L. Biophysical and proteomic characterization strategies for cysteine modifications in Ras GTPases. Meth. Mol. Biol. 2014, 1120, 75–96. [Google Scholar]
- Tenório, M.C.D.S.; Graciliano, N.G.; Moura, F.A.; Oliveira, A.C.M.; Goulart, M.O.F. N-Acetylcysteine (NAC): Impacts on human health. Antioxidants 2021, 10, 967. [Google Scholar] [CrossRef] [PubMed]
- Studer, R.; Baysang, G.; Brack, C. N-acetyl-L-cystein downregulates beta-amyloid precursor protein gene transcription in human neuroblastoma cells. Biogerontology 2001, 2, 55–60. [Google Scholar] [CrossRef]
- García-Piñeres, A.J.; Castro, V.; Mora, G.; Schmidt, T.J.; Strunck, E.; Pahl, H.L.; Merfort, I. Cysteine 38 in p65/NF-κB plays a crucial role in DNA binding inhibition by sesquiterpene lactones. J. Biol. Chem. 2001, 276, 39713–39720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilmore, T.D.; Herscovitch, M. Inhibitors of NF-B signaling: 785 and counting. Oncogene 2006, 25, 6887–6899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Z.; Machius, M.; Nestler, E.J.; Rudenko, G. Activator protein-1: Redox switch controlling structure and DNA-binding. Nucleic Acids Res. 2017, 45, 11425–11436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Sorbo, L.; Zhang, H. Is there a place for N-acetylcysteine in the treatment of septic shock? Crit. Care 2004, 8, 93–95. [Google Scholar] [CrossRef] [Green Version]
- Mokhtari, V.; Afsharian, P.; Shahhoseini, M.; Kalantar, S.M.; Moini, A. A review on various uses of N-acetyl cysteine. Cell J. 2017, 19, 11–17. [Google Scholar]
- Tardiolo, G.; Bramanti, P.; Mazzon, E. Overview on the effects of N-acetylcysteine in neurodegenerative diseases. Molecules 2018, 23, 3305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynaud, E. Protein misfolding and degenerative diseases. Nat. Educ. 2010, 3, 28. [Google Scholar]
- Takai, E.; Uda, K.; Yoshida, T.; Zako, T.; Maeda, M.; Shiraki, K. Cysteine inhibits the fibrillisation and cytotoxicity of amyloid-β 40 and 42: Implications for the contribution of the thiophilic interaction. Phys. Chem. Chem. Phys. 2014, 16, 3566–3572. [Google Scholar] [CrossRef]
- Wright, D.J.; Renoir, T.; Smith, Z.M.; Frazier, A.E.; Francis, P.S.; Thorburn, D.R.; McGee, S.L.; Hannan, A.J.; Gray, L.J. N-acetylcysteine improves mitochondrial function and ameliorates behavioral deficits in the R6/1 mouse model of Huntington’s disease. Transl. Psychiatry 2015, 5, e492. [Google Scholar] [CrossRef] [Green Version]
- Mas-Bargues, C.; Sanz-Ros, J.; Romero-García, N.; Huete-Acevedo, J.; Dromant, M.; Borrás, C. Small extracellular vesicles from senescent stem cells trigger adaptive mechanisms in young stem cells by increasing antioxidant enzyme expression. Redox Biol. 2023, 62, 102668. [Google Scholar] [CrossRef]
- Borras, C.; Mas-Bargues, C.; Sanz-Ros, J.; Román-Domínguez, A.; Gimeno-Mallench, L.; Inglés, M.; Gambini, J.; Viña, J. Extracellular vesicles and redox modulation in aging. Free Radic. Biol. Med. 2020, 149, 44–50. [Google Scholar] [CrossRef]
- Burgoyne, R.D.; Morgan, A. Cysteine string protein (CSP) and its role in preventing neurodegeneration. Semin. Cell Dev. Biol. 2015, 40, 153–159. [Google Scholar]
- Stefanis, L. α-synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Südhof, T.C. Synaptotagmins: Why so many? J. Biol. Chem. 2002, 277, 7629–7632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoyama, K.; Suh, S.W.; Hamby, A.M.; Liu, J.; Chan, W.Y.; Chen, Y.; Swanson, R.A. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci. 2006, 9, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Tsai, L.H.; Delalle, I.; Caviness, V.S., Jr.; Chae, T.; Harlow, E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature 1994, 371, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Lew, J.; Huang, Q.Q.; Qi, Z.; Winkfein, R.J.; Aebersold, R.; Hunt, T.; Wang, J.H. A brain-specific activator of cyclin-dependent kinase 5. Nature 1994, 371, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, T.; Ward, J.M.; Huh, C.G.; Longenecker, G.; Pant, H.C.; Brady, R.O.; Martin, L.J.; Kulkarni, A.B. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc. Natl. Acad. Sci. USA 1996, 93, 11173–11178. [Google Scholar] [CrossRef] [Green Version]
- Qu, J.; Nakamura, T.; Cao, G.; Holland, E.A.; McKercher, S.R.; Lipton, S.A. S-Nitrosylation activates Cdk5 and contributes to synaptic spine loss induced by beta-amyloid peptide. Proc. Natl. Acad. Sci. USA 2011, 108, 14330–14335. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Yu, P.C.; Tsang, A.H.; Chen, Y.; Fu, A.K.; Fu, W.Y.; Chung, K.K.; Ip, N.Y. S-nitrosylation of cyclin-dependent kinase 5 (cdk5) regulates its kinase activity and dendrite growth during neuronal development. J. Neurosci. 2010, 30, 14366–14370. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.Q.; Ye, J.S.; Zong, Y.Y.; Sun, C.C.; Liu, D.H.; Wu, Y.P.; Song, T.; Zhang, G.Y. S-nitrosylation of mixed lineage kinase 3 contributes to its activation after cerebral ischemia. J. Biol. Chem. 2011, 287, 2364–2377. [Google Scholar] [CrossRef] [Green Version]
- Tian, H.; Zhang, Q.; Li, H.; Zhang, G. Antioxidant N-acetylcysteine and AMPA/KA receptor antagonist DNQX inhibited mixed lineage kinase-3 activation following cerebral ischemia in rat hippocampus. Neurosci. Res. 2003, 47, 47–53. [Google Scholar] [CrossRef]
- Chen, C.H.; Li, W.; Sultana, R.; You, M.H.; Kondo, A.; Shahpasand, K.; Kim, B.M.; Luo, M.L.; Nechama, M.; Lin, Y.M.; et al. Pin1 cysteine-113 oxidation inhibits its catalytic activity and cellular function in Alzheimer’s disease. Neurobiol. Dis. 2015, 76, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Rangasamy, V.; Mishra, R.; Sondarva, G.; Das, S.; Lee, T.H.; Bakowska, J.C.; Tzivion, G.; Malter, J.S.; Rana, B.; Lu, K.P.; et al. Mixed-lineage kinase 3 phosphorylates prolyl-isomerase Pin1 to regulate its nuclear translocation and cellular function. Proc. Natl. Acad. Sci. USA 2012, 109, 8149–8154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haendeler, J.; Hoffmann, J.; Diehl, J.F.; Vasa, M.; Spyridopoulos, I.; Zeiher, A.M.; Dimmeler, S. Antioxidants inhibit nuclear export of telomerase reverse transcriptase and delay replicative senescence of endothelial cells. Circ. Res. 2004, 94, 768–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.P. Studies of the molecular mechanisms in the regulation of telomerase activity. FASEB J. 1999, 13, 2091–2104. [Google Scholar] [CrossRef] [PubMed]
- Voghel, G.; Thorin-Trescases, N.; Farhat, N.; Mamarbachi, A.M.; Villeneuve, L.; Fortier, A.; Perrault, L.P.; Carrier, M.; Thorin, E. Chronic treatment with N-acetyl-cystein delays cellular senescence in endothelial cells isolated from a subgroup of atherosclerotic patients. Mech. Ageing Dev. 2008, 129, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Gaucher, C.; Boudier, A.; Bonetti, J.; Clarot, I.; Leroy, P.; Parent, M. Glutathione: Antioxidant properties dedicated to nanotechnologies. Antioxidants 2018, 7, 62. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Won, S.J.; Wang, J.; Fong, R.; Butler, N.J.M.; Moss, A.; Wong, C.; Pan, J.; Sanchez, J.; Huynh, A.; et al. α-synuclein aggregates induce c-Abl activation and dopaminergic neuronal loss by a feed-forward redox stress mechanism. Prog. Neurobiol. 2021, 202, 102070. [Google Scholar] [CrossRef]
- Lee, Y.M.; Park, S.H.; Shin, D.I.; Hwang, J.Y.; Park, B.; Park, Y.J.; Lee, T.H.; Chae, H.Z.; Jin, B.K.; Oh, T.H.; et al. Oxidative modification of peroxiredoxin is associated with drug-induced apoptotic signaling in experimental models of Parkinson disease. J. Biol. Chem. 2008, 283, 9986–9998. [Google Scholar] [CrossRef] [Green Version]
- Monti, D.A.; Zabrecky, G.; Kremens, D.; Liang, T.W.; Wintering, N.A.; Cai, J.; Wei, X.; Bazzan, A.J.; Zhong, L.; Bowen, B.; et al. N-Acetyl-cysteine may support dopamine neurons in Parkinson’s disease: Preliminary clinical and cell line data. PLoS ONE 2016, 11, e0157602. [Google Scholar] [CrossRef] [Green Version]
- Monti, D.A.; Zabrecky, G.; Kremens, D.; Liang, T.W.; Wintering, N.A.; Bazzan, A.J.; Zhong, L.; Bowens, B.K.; Chervoneva, I.; Intenzo, C.; et al. N-acetyl-cysteine is associated with dopaminergic improvement in Parkinson’s disease. Clin. Pharmacol. Ther. 2019, 106, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Coles, L.D.; Tuite, P.J.; Öz, G.; Mishra, U.R.; Kartha, R.V.; Sullivan, K.M.; Cloyd, J.C.; Terpstra, M. Repeated-dose oral N-Acetylcysteine in Parkinson’s disease: Pharmacokinetics and effect on brain glutathione and oxidative stress. J. Clin. Pharmacol. 2018, 58, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Berman, A.E.; Chan, W.Y.; Brennan, A.M.; Reyes, R.C.; Adler, B.L.; Reyes, R.C.; Adler, B.L.; Suh, S.W.; Kauppinen, T.M.; Edling, Y.; et al. N-acetylcysteine prevents loss of dopaminergic neurons in the EAAC1–/– mouse. Ann. Neurol. 2011, 69, 509–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Zerangue, N.; Kavanaugh, M.P. Interaction of L-cysteine with a human excitatory amino acid transporter. J. Physiol. 1996, 493, 419–423. [Google Scholar] [CrossRef]
- Watts, S.D.; Torres-Salazar, D.; Divito, C.B.; Amara, S.G. Cysteine transport through excitatory amino acid transporter 3 (EAAT3). PLoS ONE 2014, 9, e109245. [Google Scholar] [CrossRef] [Green Version]
- Taghizadeh, M.; Tamtaji, O.R.; Dadgostar, E.; Daneshvar Kakhaki, R.; Bahmani, F.; Abolhassani, J.; Aarabi, M.H.; Kouchaki, E.; Memarzadeh, M.R.; Asemi, Z. The effects of omega-3 fatty acids and vitamin E co-supplementation on clinical and metabolic status in patients with Parkinson’s disease: A randomized, double-blind, placebo-controlled trial. Neurochem. Int. 2017, 108, 183–189. [Google Scholar] [CrossRef]
- Perry, T.L.; Godin, D.V.; Hansen, S. Parkinson’s disease: A disorder due to nigral glutathione deficiency? Neurosci. Lett. 1983, 33, 305–310. [Google Scholar] [CrossRef]
- Schapira, A.H.; Mann, V.M.; Cooper, J.M.; Dexter, D.; Daniel, S.E.; Jenner, P.; Clark, J.B.; Marsden, C.D. Anatomic and disease specificity of NADH CoQ1 reductase (complex I) deficiency in Parkinson’s disease. J. Neurochem. 1990, 55, 2142–2145. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Parks, J.K.; Miller, S.W.; Tuttle, J.B.; Trimmer, P.A.; Sheehan, J.P.; Bennett, J.P., Jr.; Davis, R.E.; Parker, W.D., Jr. Origin and functional consequences of the complex I defect in Parkinson’s disease. Ann. Neurol. 1996, 40, 663–671. [Google Scholar] [CrossRef]
- Pérez-Sala, D.; Pajares, M.A. Appraising the role of astrocytes as suppliers of neuronal glutathione precursors. Int. J. Mol. Sci. 2023, 24, 8059. [Google Scholar] [CrossRef]
- Nagumo, K.; Tanaka, M.; Chuang, V.T.; Setoyama, H.; Watanabe, H.; Yamada, N.; Kubota, K.; Tanaka, M.; Matsushita, K.; Yoshida, A.; et al. Cys34-cysteinylated human serum albumin is a sensitive plasma marker in oxidative stress-related chronic diseases. PLoS ONE 2014, 9, e85216. [Google Scholar] [CrossRef] [PubMed]
- Paramasivan, S.; Adav, S.S.; Ngan, S.C.; Dalan, R.; Leow, M.K.; Ho, H.H.; Sze, S.K. Serum albumin cysteine trioxidation is a potential oxidative stress biomarker of type 2 diabetes mellitus. Sci. Rep. 2020, 10, 6475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bocedi, A.; Cattani, G.; Stella, L.; Massoud, R.; Ricci, G. Thiol disulfide exchange reactions in human serum albumin: The apparent paradox of the redox transitions of Cys34. FEBS J. 2018, 285, 3225–3237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Chiku, T.; Oka, M.; Wada-Kakuda, S.; Nobuhara, M.; Oba, T.; Shinno, K.; Abe, S.; Asada, A.; Sumioka, A.; et al. Disulfide bond formation in microtubule-associated tau protein promotes tau accumulation and toxicity in vivo. Hum. Mol. Genet. 2021, 30, 1955–1967. [Google Scholar] [CrossRef]
- Tunold, J.A.; Geut, H.; Rozemuller, J.M.A.; Henriksen, S.P.; Toft, M.; van de Berg, W.D.J.; Pihlstrøm, L. APOE and MAPT are associated with dementia in neuropathologically confirmed Parkinson’s disease. Front. Neurol. 2021, 12, 631145. [Google Scholar] [CrossRef] [PubMed]
- Toshikawa, H.; Ikenaka, A.; Li, L.; Nishinaka-Arai, Y.; Niwa, A.; Ashida, A.; Kazuki, Y.; Nakahata, T.; Tamai, H.; Russell, D.W.; et al. N-acetylcysteine prevents amyloid-β secretion in neurons derived from human pluripotent stem cells with trisomy 21. Sci. Rep. 2021, 11, 17377. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez-Banaclocha, M.A. Targeting the Cysteine Redox Proteome in Parkinson’s Disease: The Role of Glutathione Precursors and Beyond. Antioxidants 2023, 12, 1373. https://doi.org/10.3390/antiox12071373
Martinez-Banaclocha MA. Targeting the Cysteine Redox Proteome in Parkinson’s Disease: The Role of Glutathione Precursors and Beyond. Antioxidants. 2023; 12(7):1373. https://doi.org/10.3390/antiox12071373
Chicago/Turabian StyleMartinez-Banaclocha, Marcos A. 2023. "Targeting the Cysteine Redox Proteome in Parkinson’s Disease: The Role of Glutathione Precursors and Beyond" Antioxidants 12, no. 7: 1373. https://doi.org/10.3390/antiox12071373