
CORM-A1 Alleviates Pro-Atherogenic Manifestations via miR-34a-5p Downregulation and an Improved Mitochondrial Function
 ,
,  , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Material and Methods
2.1. Chemicals and Reagents
2.2. Animal Studies and Experimental Protocol
2.3. Serum Lipid Profile
2.4. Histopathological Study
2.5. Immunohistochemical Analysis
2.6. Cell Culture and Treatment
2.7. Cell Viability Assay
2.8. DNA Methylation Assay
2.9. Cellular Oxidative Stress, Mitochondrial Mass, and Mitochondrial Membrane Potential (MMP) Assessment
2.10. Mitochondrial DNA Copy Number
2.11. ATP Assay
2.12. q-PCR Analysis
2.13. Immunoblot Analysis
2.14. Mitochondrial Bioenergetics XF Assay
2.15. Statistical Analysis
3. Results
3.1. CORM-A1 Ameliorates Pro-Atherogenic Manifestations in Ath Diet-Fed Rats
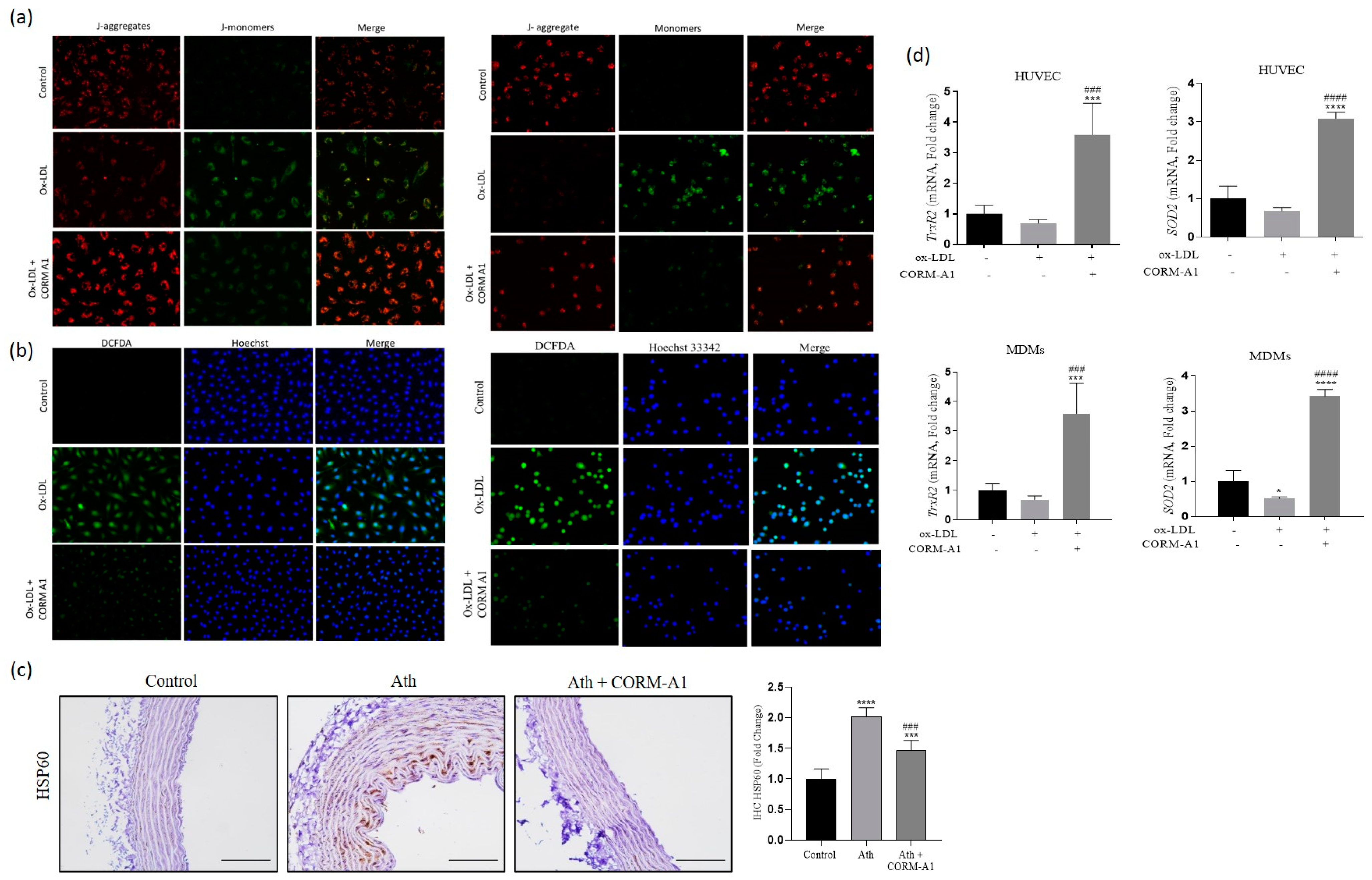
3.2. CORM-A1 Ameliorates Atherogenic Changes in ox-LDL Treated HUVEC and MDMs
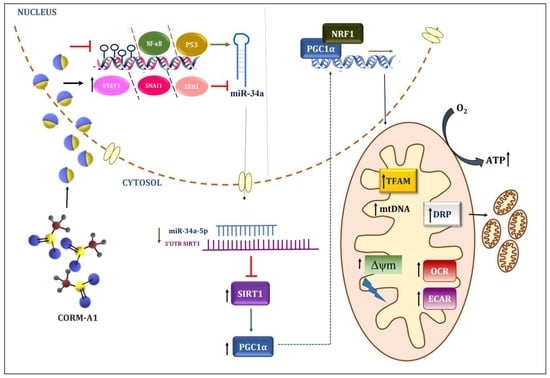
3.3. CORM-A1 Alleviates miR-34a-5p Expression by Altering Its Transcription Factors and Methylation Pattern
3.4. CORM-A1 Lowered miR-34a-5p Expression Correlates with Rescue of SIRT-1 Expression and Improved Mitochondrial Biogenesis
3.5. CORM-A1 Abrogates ox-LDL-Mediated Mitochondrial Stress and Improves Cellular Redox Status
3.6. CORM-A1-Mediated Lowering of miR-34a-5p Improves Mitochondrial Respiration and Function
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davignon, J.; Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004, 109, III-27–III-32. [Google Scholar] [CrossRef] [PubMed]
- Poznyak, A.V.; Wu, W.-K.; Melnichenko, A.A.; Wetzker, R.; Sukhorukov, V.; Markin, A.M.; Khotina, V.A.; Orekhov, A.N. Signaling pathways and key genes involved in regulation of foam cell formation in atherosclerosis. Cells 2020, 9, 584. [Google Scholar] [CrossRef]
- Petrie, J.R.; Guzik, T.J.; Touyz, R.M. Diabetes, hypertension, and cardiovascular disease: Clinical insights and vascular mechanisms. Can. J. Cardiol. 2018, 34, 575–584. [Google Scholar] [CrossRef]
- Coly, P.M.; Boulanger, C.M. Role of extracellular vesicles in atherosclerosis: An update. J. Leukoc. Biol. 2022, 111, 51–62. [Google Scholar] [CrossRef]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive oxygen species in metabolic and inflammatory signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef] [PubMed]
- Sandhir, R.; Halder, A.; Sunkaria, A. Mitochondria as a centrally positioned hub in the innate immune response. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2017, 1863, 1090–1097. [Google Scholar] [CrossRef] [PubMed]
- Kunsch, C.; Medford, R.M. Oxidative stress as a regulator of gene expression in the vasculature. Circ. Res. 1999, 85, 753–766. [Google Scholar] [CrossRef]
- Kietzmann, T.; Petry, A.; Shvetsova, A.; Gerhold, J.M.; Görlach, A. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. Br. J. Pharmacol. 2017, 174, 1533–1554. [Google Scholar] [CrossRef]
- Kitada, M.; Ogura, Y.; Koya, D. The protective role of Sirt1 in vascular tissue: Its relationship to vascular aging and atherosclerosis. Aging 2016, 8, 2290. [Google Scholar] [CrossRef]
- Klotz, L.-O.; Sánchez-Ramos, C.; Prieto-Arroyo, I.; Urbánek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, W.; Pan, H.; Feldser, H.G.; Lainez, E.; Miller, C.; Leung, S.; Zhong, Z.; Zhao, H.; Sweitzer, S. SIRT1 activators suppress inflammatory responses through promotion of p65 deacetylation and inhibition of NF-κB activity. PLoS ONE 2012, 7, e46364. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, S.; Li, Y.; Yu, S.; Zhao, Y. SIRT1/PGC-1α signaling promotes mitochondrial functional recovery and reduces apoptosis after intracerebral hemorrhage in rats. Front. Mol. Neurosci. 2018, 10, 443. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Crosstalk between oxidative stress and SIRT1: Impact on the aging process. Int. J. Mol. Sci. 2013, 14, 3834–3859. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W.; Sontheimer, E.J. Origins and mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef]
- Zhu, J.-N.; Fu, Y.-H.; Hu, Z.-Q.; Li, W.-Y.; Tang, C.-M.; Fei, H.-W.; Yang, H.; Lin, Q.-X.; Gou, D.-M.; Wu, S.-L. Activation of miR-34a-5p/Sirt1/p66shc pathway contributes to doxorubicin-induced cardiotoxicity. Sci. Rep. 2017, 7, 11879. [Google Scholar] [CrossRef]
- Chen, Q.; Li, L.; Tu, Y.; Zheng, L.L.; Liu, W.; Zuo, X.Y.; He, Y.M.; Zhang, S.Y.; Zhu, W.; Cao, J.P. MiR-34a regulates apoptosis in liver cells by targeting the KLF4 gene. Cell. Mol. Biol. Lett. 2014, 19, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Shanesazzade, Z.; Peymani, M.; Ghaedi, K.; Nasr Esfahani, M.H. miR-34a/BCL-2 signaling axis contributes to apoptosis in MPP+-induced SH-SY5Y cells. Mol. Genet. Genom. Med. 2018, 6, 975–981. [Google Scholar] [CrossRef]
- Li, Z.; Chen, H. miR-34a inhibits proliferation, migration and invasion of paediatric neuroblastoma cells via targeting HNF4α. Artif. Cells Nanomed. Biotechnol. 2019, 47, 3072–3078. [Google Scholar] [CrossRef] [PubMed]
- Thounaojam, M.C.; Jadeja, R.N.; Warren, M.; Powell, F.L.; Raju, R.; Gutsaeva, D.; Khurana, S.; Martin, P.M.; Bartoli, M. MicroRNA-34a (miR-34a) mediates retinal endothelial cell premature senescence through mitochondrial dysfunction and loss of antioxidant activities. Antioxidants 2019, 8, 328. [Google Scholar] [CrossRef]
- Xu, Y.; Xu, Y.; Zhu, Y.; Sun, H.; Juguilon, C.; Li, F.; Fan, D.; Yin, L.; Zhang, Y. Macrophage miR-34a is a key regulator of cholesterol efflux and atherosclerosis. Mol. Ther. 2020, 28, 202–216. [Google Scholar] [CrossRef]
- Choi, Y.K.; Elaine, D.; Kwon, Y.-G.; Kim, Y.-M. Regulation of ROS production and vascular function by carbon monoxide. Oxidative Med. Cell. Longev. 2012, 2012, 794237. [Google Scholar] [CrossRef]
- Almeida, A.S.; Figueiredo-Pereira, C.; Vieira, H.L. Carbon monoxide and mitochondria—Modulation of cell metabolism, redox response and cell death. Front. Physiol. 2015, 6, 33. [Google Scholar] [CrossRef]
- Coceani, F. Carbon monoxide in vasoregulation: The promise and the challenge. Circ. Res. 2000, 86, 1184–1186. [Google Scholar] [CrossRef]
- Logan, S.M.; Gupta, A.; Wang, A.; Levy, R.J.; Storey, K.B. Isoflurane and low-level carbon monoxide exposures increase expression of pro-survival miRNA in neonatal mouse heart. Cell Stress Chaperones 2021, 26, 541–548. [Google Scholar] [CrossRef]
- Motterlini, R.; Sawle, P.; Bains, S.; Hammad, J.; Alberto, R.; Foresti, R.; Green, C.J. CORM-A1: A new pharmacologically active carbon monoxide-releasing molecule. FASEB J. 2005, 19, 284–286. [Google Scholar] [CrossRef]
- Nikolic, I.; Saksida, T.; Vujicic, M.; Stojanovic, I.; Stosic-Grujicic, S. Anti-diabetic actions of carbon monoxide-releasing molecule (CORM)-A1: Immunomodulation and regeneration of islet beta cells. Immunol. Lett. 2015, 165, 39–46. [Google Scholar] [CrossRef]
- Varadi, J.; Lekli, I.; Juhasz, B.; Bacskay, I.; Szabo, G.; Gesztelyi, R.; Szendrei, L.; Varga, E.; Bak, I.; Foresti, R. Beneficial effects of carbon monoxide-releasing molecules on post-ischemic myocardial recovery. Life Sci. 2007, 80, 1619–1626. [Google Scholar] [CrossRef]
- Fagone, P.; Mangano, K.; Mammana, S.; Cavalli, E.; Di Marco, R.; Barcellona, M.L.; Salvatorelli, L.; Magro, G.; Nicoletti, F. Carbon monoxide-releasing molecule-A1 (CORM-A1) improves clinical signs of experimental autoimmune uveoretinitis (EAU) in rats. Clin. Immunol. 2015, 157, 198–204. [Google Scholar] [CrossRef]
- Jadeja, R.N.; Thounaojam, M.C.; Jain, M.; Devkar, R.V.; Ramachandran, A. Clerodendron glandulosum. Coleb leaf extract attenuates in vitro macrophage differentiation and expression of VCAM-1 and P-selectin in thoracic aorta of atherogenic diet fed rats. Immunopharmacol. Immunotoxicol. 2012, 34, 443–453. [Google Scholar] [CrossRef]
- Jiang, F.; Qian, J.; Chen, S.; Zhang, W.; Liu, C. Ligustrazine improves atherosclerosis in rat via attenuation of oxidative stress. Pharm. Biol. 2011, 49, 856–863. [Google Scholar] [CrossRef]
- Wu, Y.; Li, J.; Wang, J.; Si, Q.; Zhang, J.; Jiang, Y.; Chu, L. Anti-atherogenic effects of centipede acidic protein in rats fed an atherogenic diet. J. Ethnopharmacol. 2009, 122, 509–516. [Google Scholar] [CrossRef]
- Zou, D.; Yang, P.; Liu, J.; Dai, F.; Xiao, Y.; Zhao, A.; Huang, N. Constructing Mal-Efferocytic Macrophage Model and Its Atherosclerotic Spheroids and Rat Model for Therapeutic Evaluation. Adv. Biol. 2023, 2200277. [Google Scholar] [CrossRef]
- Xu, Y.Y.; Yang, J.; Shen, T.; Zhou, F.; Xia, Y.; Fu, J.Y.; Meng, J.; Zhang, J.; Zheng, Y.F.; Yang, J. Intravenous administration of multi-walled carbon nanotubes affects the formation of atherosclerosis in Sprague-Dawley rats. J. Occup. Health 2012, 54, 361–369. [Google Scholar] [CrossRef]
- Pang, J.; Xu, Q.; Xu, X.; Yin, H.; Xu, R.; Guo, S.; Hao, W.; Wang, L.; Chen, C.; Cao, J.-M. Hexarelin suppresses high lipid diet and vitamin D3-induced atherosclerosis in the rat. Peptides 2010, 31, 630–638. [Google Scholar] [CrossRef]
- Jadeja, R.N.; Thouaojam, M.C.; Sankhari, J.M.; Jain, M.; Devkar, R.V.; Ramachandran, A. Standardized flavonoid-rich Eugenia jambolana seed extract retards in vitro and in vivo LDL oxidation and expression of VCAM-1 and P-selectin in atherogenic rats. Cardiovasc. Toxicol. 2012, 12, 73–82. [Google Scholar] [CrossRef]
- Cai, G.-J.; Miao, C.-Y.; Xie, H.-H.; Lu, L.-H.; Su, D.-F. Arterial baroreflex dysfunction promotes atherosclerosis in rats. Atherosclerosis 2005, 183, 41–47. [Google Scholar] [CrossRef]
- Upadhyay, K.K.; Jadeja, R.N.; Vyas, H.S.; Pandya, B.; Joshi, A.; Vohra, A.; Thounaojam, M.C.; Martin, P.M.; Bartoli, M.; Devkar, R.V. Carbon monoxide releasing molecule-A1 improves nonalcoholic steatohepatitis via Nrf2 activation mediated improvement in oxidative stress and mitochondrial function. Redox Biol. 2020, 28, 101314. [Google Scholar] [CrossRef]
- Meng, F.; Glaser, S.S.; Francis, H.; Yang, F.; Han, Y.; Stokes, A.; Staloch, D.; McCarra, J.; Liu, J.; Venter, J. Epigenetic regulation of miR-34a expression in alcoholic liver injury. Am. J. Pathol. 2012, 181, 804–817. [Google Scholar] [CrossRef]
- Fagone, P.; Mangano, K.; Quattrocchi, C.; Motterlini, R.; Di Marco, R.; Magro, G.; Penacho, N.; Romao, C.; Nicoletti, F. Prevention of clinical and histological signs of proteolipid protein (PLP)-induced experimental allergic encephalomyelitis (EAE) in mice by the water-soluble carbon monoxide-releasing molecule (CORM)-A1. Clin. Exp. Immunol. 2011, 163, 368–374. [Google Scholar] [CrossRef]
- Foresti, R.; Bani-Hani, M.G.; Motterlini, R. Use of carbon monoxide as a therapeutic agent: Promises and challenges. Intensive Care Med. 2008, 34, 649–658. [Google Scholar] [CrossRef]
- Raitoharju, E.; Lyytikäinen, L.-P.; Levula, M.; Oksala, N.; Mennander, A.; Tarkka, M.; Klopp, N.; Illig, T.; Kähönen, M.; Karhunen, P.J. miR-21, miR-210, miR-34a, and miR-146a/b are up-regulated in human atherosclerotic plaques in the Tampere Vascular Study. Atherosclerosis 2011, 219, 211–217. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, Z.; Pang, X.; Yang, J.; Yu, H.; Zhang, Y.; Zhou, H.; Zhao, J. MiR-34a/sirtuin-1/foxo3a is involved in genistein protecting against ox-LDL-induced oxidative damage in HUVECs. Toxicol. Lett. 2017, 277, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Li, P.; Li, J.; He, R.; Cheng, G.; Li, Y. Downregulation of microRNA-34a inhibits oxidized low-density lipoprotein-induced apoptosis and oxidative stress in human umbilical vein endothelial cells. Int. J. Mol. Med. 2018, 42, 1134–1144. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Liu, T.; Yang, Y.; Xiao, W.; Shi, J.; Mei, X.; Tian, S.; Liu, X.; Huang, H.; Bai, Y. Metformin promotes proliferation and suppresses apoptosis in Ox-LDL stimulated macrophages by regulating the miR-34a/Bcl2 axis. RSC Adv. 2019, 9, 14670–14676. [Google Scholar] [CrossRef]
- Hai, Z.; Zuo, W. Aberrant DNA methylation in the pathogenesis of atherosclerosis. Clin. Chim. Acta 2016, 456, 69–74. [Google Scholar] [CrossRef]
- Cheng, Z.; Ristow, M. Mitochondria and Metabolic Homeostasis; Mary Ann Liebert, Inc.: New Rochelle, NY, USA, 2013; pp. 240–242. [Google Scholar]
- Hu, Y.; Xia, W.; Hou, M. Macrophage migration inhibitory factor serves a pivotal role in the regulation of radiation-induced cardiac senescencethrough rebalancing the microRNA-34a/sirtuin 1 signaling pathway. Int. J. Mol. Med. 2018, 42, 2849–2858. [Google Scholar] [CrossRef] [PubMed]
- Khwaja, B.; Thankam, F.G.; Agrawal, D.K. Mitochondrial DAMPs and altered mitochondrial dynamics in OxLDL burden in atherosclerosis. Mol. Cell. Biochem. 2021, 476, 1915–1928. [Google Scholar] [CrossRef]
- Bukeirat, M.; Sarkar, S.N.; Hu, H.; Quintana, D.D.; Simpkins, J.W.; Ren, X. MiR-34a regulates blood–brain barrier permeability and mitochondrial function by targeting cytochrome c. J. Cereb. Blood Flow Metab. 2016, 36, 387–392. [Google Scholar] [CrossRef]
- Hu, H.; Hone, E.A.; Provencher, E.A.; Sprowls, S.A.; Farooqi, I.; Corbin, D.R.; Sarkar, S.N.; Hollander, J.M.; Lockman, P.R.; Simpkins, J.W. MiR-34a interacts with cytochrome c and shapes stroke outcomes. Sci. Rep. 2020, 10, 3233. [Google Scholar] [CrossRef]
- Harrison, D.; Griendling, K.K.; Landmesser, U.; Hornig, B.; Drexler, H. Role of oxidative stress in atherosclerosis. Am. J. Cardiol. 2003, 91, 7–11. [Google Scholar] [CrossRef]
- Tavakoli, S.; Asmis, R. Reactive oxygen species and thiol redox signaling in the macrophage biology of atherosclerosis. Antioxid. Redox Signal. 2012, 17, 1785–1795. [Google Scholar] [CrossRef] [PubMed]
- Motterlini, R.; Otterbein, L.E. The therapeutic potential of carbon monoxide. Nat. Rev. Drug Discov. 2010, 9, 728–743. [Google Scholar] [CrossRef]
- Lee, F.-Y.; Chen, W.-K.; Lin, C.-L.; Kao, C.-H. Carbon monoxide poisoning and subsequent cardiovascular disease risk: A nationwide population-based cohort study. Medicine 2015, 94, e624. [Google Scholar] [CrossRef]
- Wang, P.; Huang, J.; Li, Y.; Chang, R.; Wu, H.; Lin, J.; Huang, Z. Exogenous carbon monoxide decreases sepsis-induced acute kidney injury and inhibits NLRP3 inflammasome activation in rats. Int. J. Mol. Sci. 2015, 16, 20595–20608. [Google Scholar] [CrossRef]
- Nakao, A.; Choi, A.M.; Murase, N. Protective effect of carbon monoxide in transplantation. J. Cell. Mol. Med. 2006, 10, 650–671. [Google Scholar] [CrossRef] [PubMed]
- Bauer, I.; Pannen, B.H. Bench-to-bedside review: Carbon monoxide—From mitochondrial poisoning to therapeutic use. Crit. Care 2009, 13, 220. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, K.K.; Jadeja, R.N.; Thadani, J.M.; Joshi, A.; Vohra, A.; Mevada, V.; Patel, R.; Khurana, S.; Devkar, R.V. Carbon monoxide releasing molecule A-1 attenuates acetaminophen-mediated hepatotoxicity and improves survival of mice by induction of Nrf2 and related genes. Toxicol. Appl. Pharmacol. 2018, 360, 99–108. [Google Scholar] [CrossRef]
- Nizamutdinova, I.T.; Kim, Y.M.; Kim, H.J.; Seo, H.G.; Lee, J.H.; Chang, K.C. Carbon monoxide (from CORM-2) inhibits high glucose-induced ICAM-1 expression via AMP-activated protein kinase and PPAR-γ activations in endothelial cells. Atherosclerosis 2009, 207, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.-J.; Xu, D.-Y.; Sun, Y.-X.; Xue, T.; Zhang, C.-X.; Zhang, Z.-X.; Lin, W.; Li, K.-X. CO-releasing molecules-2 attenuates ox-LDL-induced injury in HUVECs by ameliorating mitochondrial function and inhibiting Wnt/β-catenin pathway. Biochem. Biophys. Res. Commun. 2017, 490, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Qiu, G.; Yu, K.; Yu, C.; Li, W.; Lv, J.; Guo, Y.; Bian, Z.; Yang, L.; Chen, Y.; Chen, Z. Association of exhaled carbon monoxide with risk of cardio-cerebral-vascular disease in the China Kadoorie Biobank cohort study. Sci. Rep. 2020, 10, 19507. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, G.; Gao, X. Attenuation of miR-34a protects cardiomyocytes against hypoxic stress through maintenance of glycolysis. Biosci. Rep. 2017, 37, BSR20170925. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Liang, L.-R.; Wang, L.-F.; Zhong, J.-C. Targeting the microRNA-34a as a Novel Therapeutic Strategy for Cardiovascular Diseases. Front. Cardiovasc. Med. 2022, 8, 2243. [Google Scholar]
- Li, J.; Wang, K.; Chen, X.; Meng, H.; Song, M.; Wang, Y.; Xu, X.; Bai, Y. Transcriptional activation of microRNA-34a by NF-kappa B in human esophageal cancer cells. BMC Mol. Biol. 2012, 13, 4. [Google Scholar] [CrossRef]
- Okada, N.; Lin, C.-P.; Ribeiro, M.C.; Biton, A.; Lai, G.; He, X.; Bu, P.; Vogel, H.; Jablons, D.M.; Keller, A.C. A positive feedback between p53 and miR-34 miRNAs mediates tumor suppression. Genes Dev. 2014, 28, 438–450. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, C.; Liu, X.; Zhang, B.; Wang, P.; Yang, Z.; Niu, N. The regulatory role of aberrant methylation of microRNA-34a promoter CpGs in osteosarcoma. Transl. Cancer Res. 2019, 8, 2328. [Google Scholar] [CrossRef] [PubMed]
- Vogt, M.; Munding, J.; Grüner, M.; Liffers, S.-T.; Verdoodt, B.; Hauk, J.; Steinstraesser, L.; Tannapfel, A.; Hermeking, H. Frequent concomitant inactivation of miR-34a and miR-34b/c by CpG methylation in colorectal, pancreatic, mammary, ovarian, urothelial, and renal cell carcinomas and soft tissue sarcomas. Virchows Arch. 2011, 458, 313–322. [Google Scholar] [CrossRef]
- Rezaei, M.; Eskandari, F.; Mohammadpour-Gharehbagh, A.; Harati-Sadegh, M.; Teimoori, B.; Salimi, S. Hypomethylation of the miRNA-34a gene promoter is associated with Severe Preeclampsia. Clin. Exp. Hypertens. 2019, 41, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.; Matter, C.M. Protective roles of SIRT1 in atherosclerosis. Cell Cycle 2011, 10, 640–647. [Google Scholar] [CrossRef]
- Kim, H.J.; Joe, Y.; Yu, J.K.; Chen, Y.; Jeong, S.O.; Mani, N.; Cho, G.J.; Pae, H.-O.; Ryter, S.W.; Chung, H.T. Carbon monoxide protects against hepatic ischemia/reperfusion injury by modulating the miR-34a/SIRT1 pathway. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2015, 1852, 1550–1559. [Google Scholar] [CrossRef]
- Yu, E.P.; Reinhold, J.; Yu, H.; Starks, L.; Uryga, A.K.; Foote, K.; Finigan, A.; Figg, N.; Pung, Y.-F.; Logan, A. Mitochondrial respiration is reduced in atherosclerosis, promoting necrotic core formation and reducing relative fibrous cap thickness. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2322–2332. [Google Scholar] [CrossRef]
- Peng, W.; Cai, G.; Xia, Y.; Chen, J.; Wu, P.; Wang, Z.; Li, G.; Wei, D. Mitochondrial dysfunction in atherosclerosis. DNA Cell Biol. 2019, 38, 597–606. [Google Scholar] [CrossRef]
- Madamanchi, N.R.; Runge, M.S. Mitochondrial dysfunction in atherosclerosis. Circ. Res. 2007, 100, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Gao, Y.; Zhang, Q.; Wei, S.; Chen, Z.; Dai, X.; Zeng, Z.; Zhao, K.-S. SIRT1/3 activation by resveratrol attenuates acute kidney injury in a septic rat model. Oxidative Med. Cell. Longev. 2016, 2016, 7296092. [Google Scholar] [CrossRef]
- Lu, Z.; Xu, X.; Hu, X.; Fassett, J.; Zhu, G.; Tao, Y.; Li, J.; Huang, Y.; Zhang, P.; Zhao, B. PGC-1α regulates expression of myocardial mitochondrial antioxidants and myocardial oxidative stress after chronic systolic overload. Antioxid. Redox Signal. 2010, 13, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Siasos, G.; Tsigkou, V.; Kosmopoulos, M.; Theodosiadis, D.; Simantiris, S.; Tagkou, N.M.; Tsimpiktsioglou, A.; Stampouloglou, P.K.; Oikonomou, E.; Mourouzis, K. Mitochondria and cardiovascular diseases—From pathophysiology to treatment. Ann. Transl. Med. 2018, 6, 256. [Google Scholar] [CrossRef]
- Babu, D.; Leclercq, G.; Goossens, V.; Remijsen, Q.; Vandenabeele, P.; Motterlini, R.; Lefebvre, R.A. Antioxidant potential of CORM-A1 and resveratrol during TNF-α/cycloheximide-induced oxidative stress and apoptosis in murine intestinal epithelial MODE-K cells. Toxicol. Appl. Pharmacol. 2015, 288, 161–178. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vyas, H.S.; Jadeja, R.N.; Vohra, A.; Upadhyay, K.K.; Thounaojam, M.C.; Bartoli, M.; Devkar, R.V. CORM-A1 Alleviates Pro-Atherogenic Manifestations via miR-34a-5p Downregulation and an Improved Mitochondrial Function. Antioxidants 2023, 12, 997. https://doi.org/10.3390/antiox12050997
Vyas HS, Jadeja RN, Vohra A, Upadhyay KK, Thounaojam MC, Bartoli M, Devkar RV. CORM-A1 Alleviates Pro-Atherogenic Manifestations via miR-34a-5p Downregulation and an Improved Mitochondrial Function. Antioxidants. 2023; 12(5):997. https://doi.org/10.3390/antiox12050997
Chicago/Turabian StyleVyas, Hitarthi S., Ravirajsinh N. Jadeja, Aliasgar Vohra, Kapil K. Upadhyay, Menaka C. Thounaojam, Manuela Bartoli, and Ranjitsinh V. Devkar. 2023. "CORM-A1 Alleviates Pro-Atherogenic Manifestations via miR-34a-5p Downregulation and an Improved Mitochondrial Function" Antioxidants 12, no. 5: 997. https://doi.org/10.3390/antiox12050997