
Unfolding the Interactions between Endoplasmic Reticulum Stress and Oxidative Stress
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
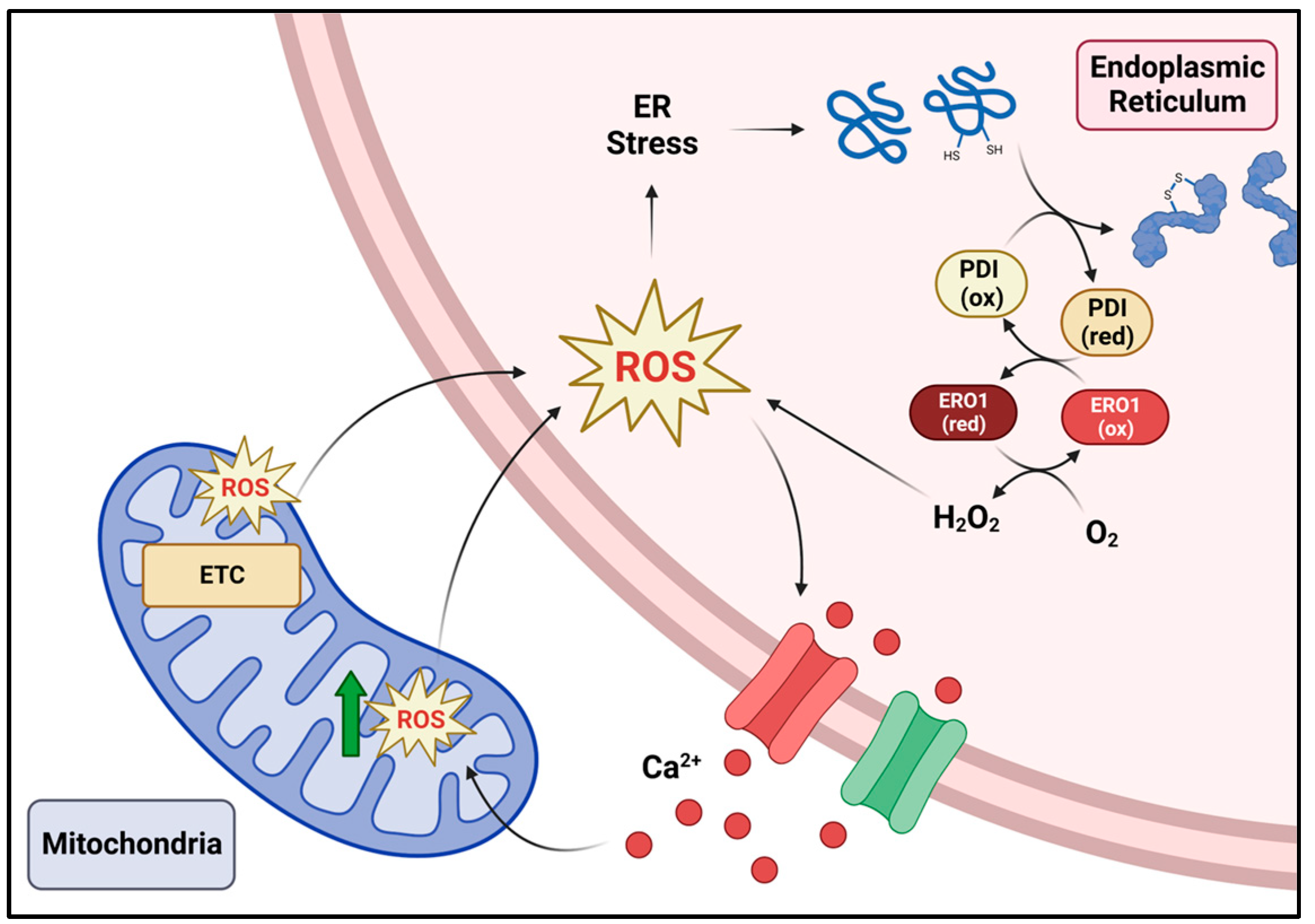
2. The Endoplasmic Reticulum and ROS Production
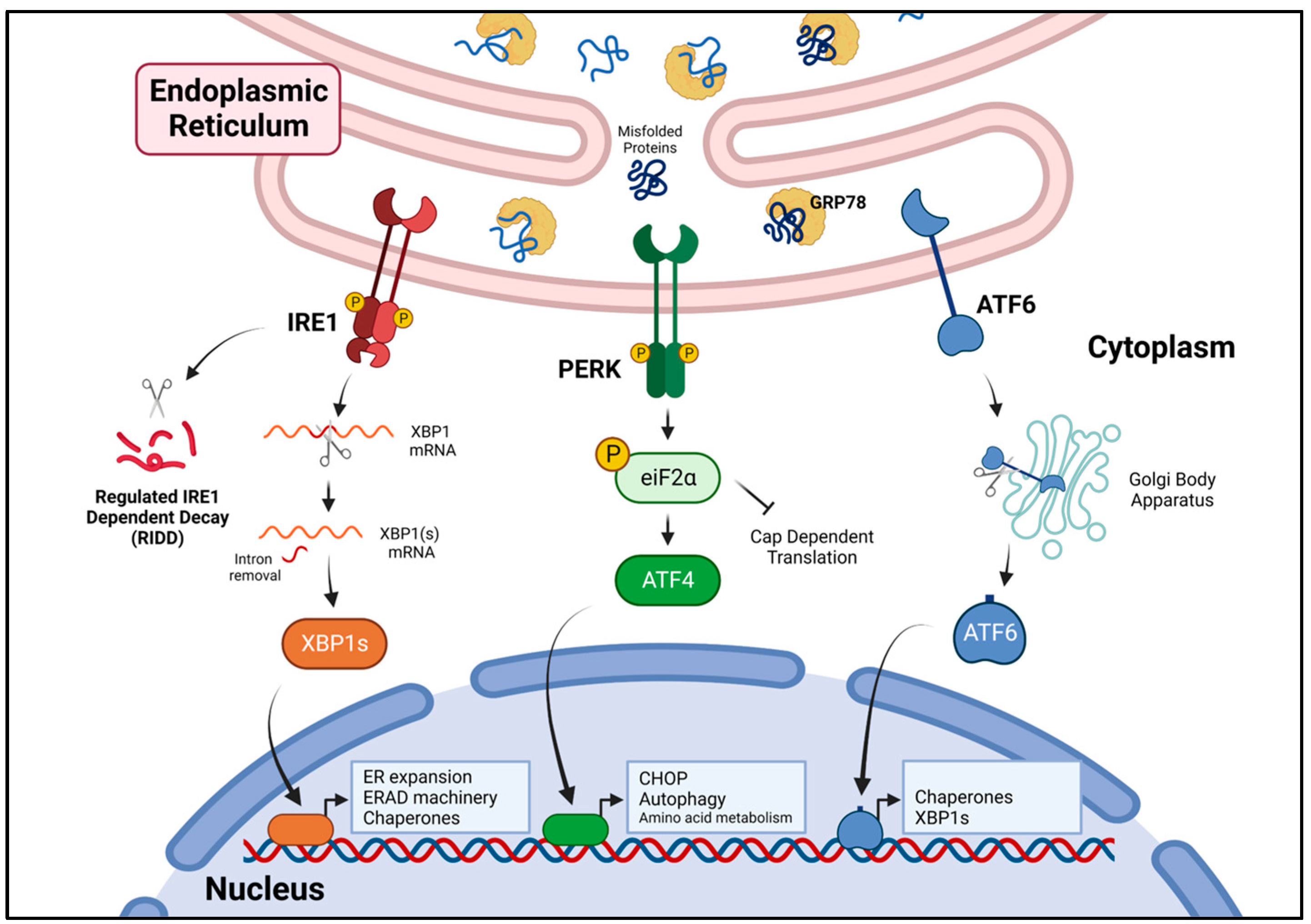
3. The Unfolded Protein Response
3.1. IRE1 Signaling
3.2. PERK Signaling
3.3. ATF6 Signaling
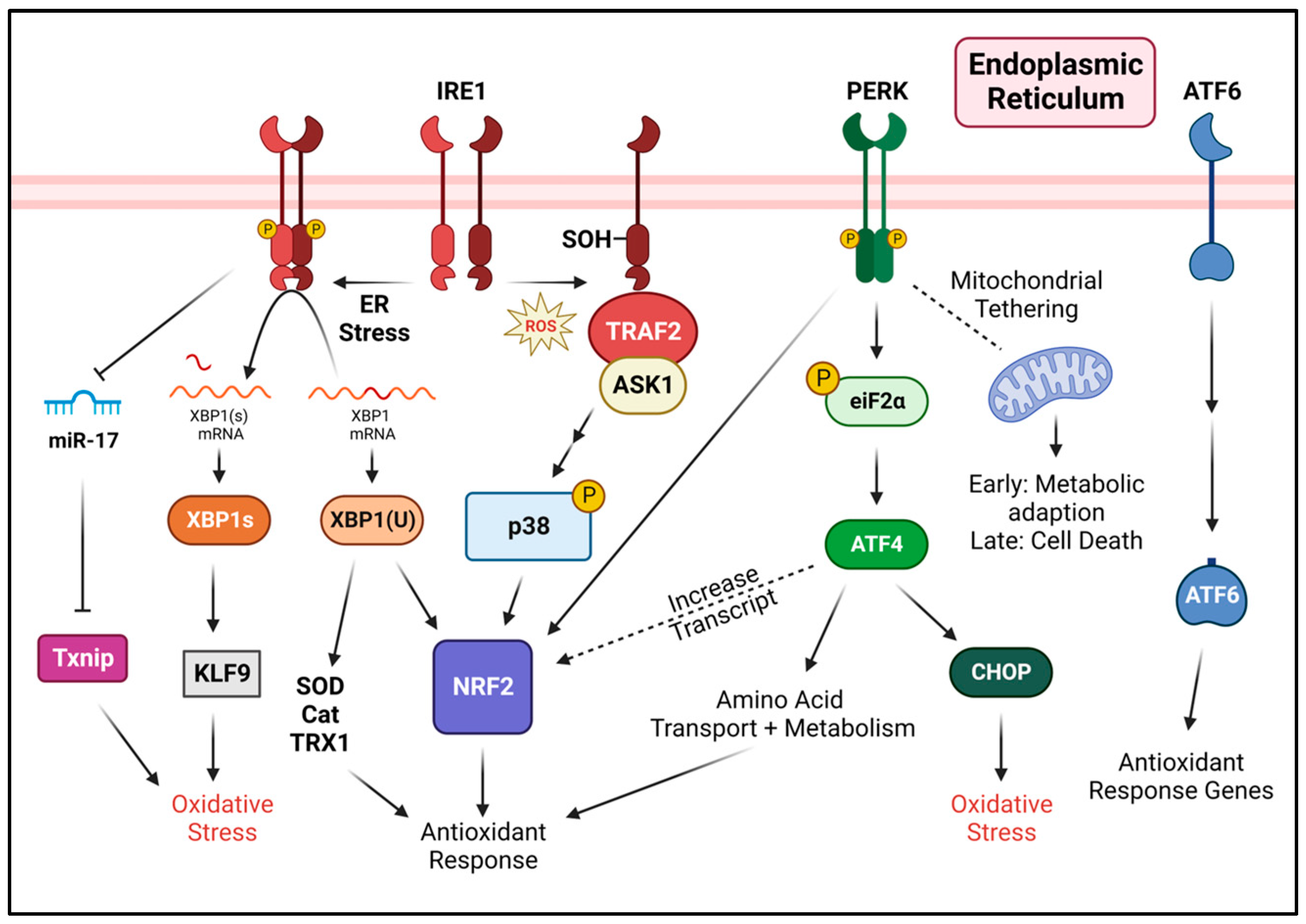
3.4. The Intersection of Oxidative Stress, ER Stress, and the Unfolded Protein Response
4. Regulation of Oxidative Stress by the Unfolded Protein Response
4.1. PERK in Response to Oxidative Stress
4.2. IRE1 in Response to Oxidative Stress
4.3. ATF6 in Response to Oxidative Stress
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial Electron Transport Chain, ROS Generation and Uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.B.; Chandel, N.S. Mitochondrial Reactive Oxygen Species and Cancer. Cancer Metab. 2014, 2, 17. [Google Scholar] [CrossRef] [PubMed]
- Tu, B.P.; Weissman, J.S. Oxidative Protein Folding in Eukaryotes: Mechanisms and Consequences. J. Cell Biol. 2004, 164, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Shibata, Y.; Voeltz, G.K.; Rapoport, T.A. Rough Sheets and Smooth Tubules. Cell 2006, 126, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, D.S.; Blower, M.D. The Endoplasmic Reticulum: Structure, Function and Response to Cellular Signaling. Cell Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef]
- Michalak, M.; Robert Parker, J.M.; Opas, M. Ca2+ Signaling and Calcium Binding Chaperones of the Endoplasmic Reticulum. Cell Calcium 2002, 32, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.; Sinskey, A.J.; Lodish, H.F. Oxidized Redox State of Glutathione in the Endoplasmic Reticulum. Science 1992, 257, 1496–1502. [Google Scholar] [CrossRef]
- Kozlov, G.; Määttänen, P.; Thomas, D.Y.; Gehring, K. A Structural Overview of the PDI Family of Proteins. FEBS J. 2010, 277, 3924–3936. [Google Scholar] [CrossRef] [PubMed]
- Frand, A.R.; Kaiser, C.A. Ero1p Oxidizes Protein Disulfide Isomerase in a Pathway for Disulfide Bond Formation in the Endoplasmic Reticulum. Mol. Cell 1999, 4, 469–477. [Google Scholar] [CrossRef]
- Gross, E.; Sevier, C.S.; Heldman, N.; Vitu, E.; Bentzur, M.; Kaiser, C.A.; Thorpe, C.; Fass, D. Generating Disulfides Enzymatically: Reaction Products and Electron Acceptors of the Endoplasmic Reticulum Thiol Oxidase Ero1p. Proc. Natl. Acad. Sci. USA 2006, 103, 299–304. [Google Scholar] [CrossRef]
- Konno, T.; Pinho Melo, E.; Lopes, C.; Mehmeti, I.; Lenzen, S.; Ron, D.; Avezov, E. ERO1-Independent Production of H2O2 within the Endoplasmic Reticulum Fuels Prdx4-Mediated Oxidative Protein Folding. J. Cell Biol. 2015, 211, 253–259. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, L.; Niu, Y.; Sitia, R.; Wang, C.-C. Glutathione Peroxidase 7 Utilizes Hydrogen Peroxide Generated by Ero1α to Promote Oxidative Protein Folding. Antioxid. Redox Signal. 2014, 20, 545–556. [Google Scholar] [CrossRef]
- Ramming, T.; Hansen, H.G.; Nagata, K.; Ellgaard, L.; Appenzeller-Herzog, C. GPx8 Peroxidase Prevents Leakage of H2O2 from the Endoplasmic Reticulum. Free Radic. Biol. Med. 2014, 70, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Schulman, S.; Wang, B.; Li, W.; Rapoport, T.A. Vitamin K Epoxide Reductase Prefers ER Membrane-Anchored Thioredoxin-like Redox Partners. Proc. Natl. Acad. Sci. USA 2010, 107, 15027–15032. [Google Scholar] [CrossRef] [PubMed]
- Rutkevich, L.A.; Williams, D.B. Vitamin K Epoxide Reductase Contributes to Protein Disulfide Formation and Redox Homeostasis within the Endoplasmic Reticulum. Mol. Biol. Cell 2012, 23, 2017–2027. [Google Scholar] [CrossRef] [PubMed]
- Kodali, V.K.; Thorpe, C. Oxidative Protein Folding and the Quiescin–Sulfhydryl Oxidase Family of Flavoproteins. Antioxid. Redox Signal. 2010, 13, 1217–1230. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.; Kim, S.-M.; Correia, M.A. Cytochrome P450 Endoplasmic Reticulum-Associated Degradation (ERAD): Therapeutic and Pathophysiological Implications. Acta Pharm. Sin. B 2020, 10, 42–60. [Google Scholar] [CrossRef]
- Veith, A.; Moorthy, B. Role of Cytochrome P450S in the Generation and Metabolism of Reactive Oxygen Species. Curr. Opin. Toxicol. 2018, 7, 44–51. [Google Scholar] [CrossRef]
- Yue, Z.; Zhang, X.; Yu, Q.; Liu, L.; Zhou, X. Cytochrome P450-Dependent Reactive Oxygen Species (ROS) Production Contributes to Mn3O4 Nanoparticle-Caused Liver Injury. RSC Adv. 2018, 8, 37307–37314. [Google Scholar] [CrossRef]
- Harjumäki, R.; Pridgeon, C.S.; Ingelman-Sundberg, M. CYP2E1 in Alcoholic and Non-Alcoholic Liver Injury. Roles of ROS, Reactive Intermediates and Lipid Overload. Int. J. Mol. Sci. 2021, 22, 8221. [Google Scholar] [CrossRef]
- Gao, H.; Cao, Y.; Xia, H.; Zhu, X.; Jin, Y. CYP4A11 Is Involved in the Development of Nonalcoholic Fatty Liver Disease via ROS-induced Lipid Peroxidation and Inflammation. Int. J. Mol. Med. 2020, 45, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Tabales, J.; García-Martín, E.; Agúndez, J.A.G.; Gutierrez-Merino, C. Modulation of CYP2C9 Activity and Hydrogen Peroxide Production by Cytochrome B5. Sci. Rep. 2020, 10, 15571. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Nakano, M.; Ihara, H.; Ito, R.; Taniguchi, N.; Fujii, J. Different Consequences of Reactions with Hydrogen Peroxide and T-Butyl Hydroperoxide in the Hyperoxidative Inactivation of Rat Peroxiredoxin-4. J. Biochem. 2011, 149, 443–453. [Google Scholar] [CrossRef]
- Elko, E.A.; Manuel, A.M.; White, S.; Zito, E.; van der Vliet, A.; Anathy, V.; Janssen-Heininger, Y.M.W. Oxidation of Peroxiredoxin-4 Induces Oligomerization and Promotes Interaction with Proteins Governing Protein Folding and Endoplasmic Reticulum Stress. J. Biol. Chem. 2021, 296, 100665. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Cycle or a Double-Edged Sword? Antioxid. Redox Signal. 2007, 9, 2277–2293. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic Interaction of BiP and ER Stress Transducers in the Unfolded-Protein Response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Bertolotti, A.; Wang, X.; Novoa, I.; Jungreis, R.; Schlessinger, K.; Cho, J.H.; West, A.B.; Ron, D. Increased Sensitivity to Dextran Sodium Sulfate Colitis in IRE1β-Deficient Mice. J. Clin. Invest. 2001, 107, 585–593. [Google Scholar] [CrossRef]
- Martino, M.B.; Jones, L.; Brighton, B.; Ehre, C.; Abdulah, L.; Davis, C.W.; Ron, D.; O’Neal, W.K.; Ribeiro, C.M.P. The ER Stress Transducer IRE1β Is Required for Airway Epithelial Mucin Production. Mucosal Immunol. 2013, 6, 639–654. [Google Scholar] [CrossRef]
- Mori, K.; Ma, W.; Gething, M.J.; Sambrook, J. A Transmembrane Protein with a Cdc2+/CDC28-Related Kinase Activity Is Required for Signaling from the ER to the Nucleus. Cell 1993, 74, 743–756. [Google Scholar] [CrossRef]
- Shamu, C.E.; Walter, P. Oligomerization and Phosphorylation of the Ire1p Kinase during Intracellular Signaling from the Endoplasmic Reticulum to the Nucleus. EMBO J. 1996, 15, 3028–3039. [Google Scholar] [CrossRef]
- Sidrauski, C.; Walter, P. The Transmembrane Kinase Ire1p Is a Site-Specific Endonuclease That Initiates MRNA Splicing in the Unfolded Protein Response. Cell 1997, 90, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Tirasophon, W.; Shen, X.; Michalak, M.; Prywes, R.; Okada, T.; Yoshida, H.; Mori, K.; Kaufman, R.J. IRE1-Mediated Unconventional MRNA Splicing and S2P-Mediated ATF6 Cleavage Merge to Regulate XBP1 in Signaling the Unfolded Protein Response. Genes Dev. 2002, 16, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Liang, F.-X.; Wang, X. A Synthetic Biology Approach Identifies the Mammalian UPR RNA Ligase RtcB. Mol. Cell 2014, 55, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Haze, K.; Yanagi, H.; Yura, T.; Mori, K. Identification of the Cis-Acting Endoplasmic Reticulum Stress Response Element Responsible for Transcriptional Induction of Mammalian Glucose-Regulated Proteins. Involvement of Basic Leucine Zipper Transcription Factors. J. Biol. Chem. 1998, 273, 33741–33749. [Google Scholar] [CrossRef]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional Induction of Mammalian ER Quality Control Proteins Is Mediated by Single or Combined Action of ATF6alpha and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Hosokawa, N.; Kaufman, R.J.; Nagata, K.; Mori, K. A Time-Dependent Phase Shift in the Mammalian Unfolded Protein Response. Dev. Cell 2003, 4, 265–271. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 MRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Shaffer, A.L.; Shapiro-Shelef, M.; Iwakoshi, N.N.; Lee, A.-H.; Qian, S.-B.; Zhao, H.; Yu, X.; Yang, L.; Tan, B.K.; Rosenwald, A.; et al. XBP1, Downstream of Blimp-1, Expands the Secretory Apparatus and Other Organelles, and Increases Protein Synthesis in Plasma Cell Differentiation. Immunity 2004, 21, 81–93. [Google Scholar] [CrossRef]
- Hollien, J.; Weissman, J.S. Decay of Endoplasmic Reticulum-Localized MRNAs during the Unfolded Protein Response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-Dependent Decay of Messenger RNAs in Mammalian Cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef]
- Oikawa, D.; Tokuda, M.; Hosoda, A.; Iwawaki, T. Identification of a Consensus Element Recognized and Cleaved by IRE1α. Nucleic Acids Res. 2010, 38, 6265–6273. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.L.; Ferri, E.; Marsters, S.; Harnoss, J.M.; Modrusan, Z.; Li, W.; Rudolph, J.; Wang, W.; Wu, T.D.; Walter, P.; et al. Noncanonical MRNA Decay by the Endoplasmic-Reticulum Stress Sensor IRE1α Promotes Cancer-Cell Survival. bioRxiv 2021, 16. [Google Scholar] [CrossRef]
- Lu, M.; Lawrence, D.A.; Marsters, S.; Acosta-Alvear, D.; Kimmig, P.; Mendez, A.S.; Paton, A.W.; Paton, J.C.; Walter, P.; Ashkenazi, A. Opposing Unfolded-Protein-Response Signals Converge on Death Receptor 5 to Control Apoptosis. Science 2014, 345, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.-K.; Lawrence, D.A.; Lu, M.; Tan, J.; Harnoss, J.M.; Marsters, S.A.; Liu, P.; Sandoval, W.; Martin, S.E.; Ashkenazi, A. Coordination between Two Branches of the Unfolded Protein Response Determines Apoptotic Cell Fate. Mol. Cell 2018, 71, 629–636.e5. [Google Scholar] [CrossRef]
- Coelho, D.S.; Domingos, P.M. Physiological Roles of Regulated Ire1 Dependent Decay. Front. Genet. 2014, 5, 76. [Google Scholar] [CrossRef]
- Eletto, D.; Eletto, D.; Boyle, S.; Argon, Y. PDIA6 Regulates Insulin Secretion by Selectively Inhibiting the RIDD Activity of IRE1. FASEB J. 2016, 30, 653–665. [Google Scholar] [CrossRef]
- Almanza, A.; Mnich, K.; Blomme, A.; Robinson, C.M.; Rodriguez-Blanco, G.; Kierszniowska, S.; McGrath, E.P.; Le Gallo, M.; Pilalis, E.; Swinnen, J.V.; et al. Regulated IRE1α-Dependent Decay (RIDD)-Mediated Reprograming of Lipid Metabolism in Cancer. Nat. Commun. 2022, 13, 2493. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of Stress in the ER to Activation of JNK Protein Kinases by Transmembrane Protein Kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of Endoplasmic Reticulum Stress-Induced Apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef]
- Liu, C.Y.; Schröder, M.; Kaufman, R.J. Ligand-Independent Dimerization Activates the Stress Response Kinases IRE1 and PERK in the Lumen of the Endoplasmic Reticulum. J. Biol. Chem. 2000, 275, 24881–24885. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein Translation and Folding Are Coupled by an Endoplasmic-Reticulum-Resident Kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk Is Essential for Translational Regulation and Cell Survival during the Unfolded Protein Response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Vattem, K.M.; Wek, R.C. Reinitiation Involving Upstream ORFs Regulates ATF4 MRNA Translation in Mammalian Cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269–11274. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An Integrated Stress Response Regulates Amino Acid Metabolism and Resistance to Oxidative Stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- B’chir, W.; Maurin, A.-C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The EIF2α/ATF4 Pathway Is Essential for Stress-Induced Autophagy Gene Expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef]
- Fawcett, T.W.; Martindale, J.L.; Guyton, K.Z.; Hai, T.; Holbrook, N.J. Complexes Containing Activating Transcription Factor (ATF)/CAMP-Responsive-Element-Binding Protein (CREB) Interact with the CCAAT/Enhancer-Binding Protein (C/EBP)-ATF Composite Site to Regulate Gadd153 Expression during the Stress Response. Biochem. J. 1999, 339 Pt 1, 135–141. [Google Scholar] [CrossRef]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.O.; Aw, T.Y.; Holbrook, N.J. Gadd153 Sensitizes Cells to Endoplasmic Reticulum Stress by Down-Regulating Bcl2 and Perturbing the Cellular Redox State. Mol. Cell Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback Inhibition of the Unfolded Protein Response by GADD34-Mediated Dephosphorylation of EIF2α. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.-H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-Stress-Induced Transcriptional Regulation Increases Protein Synthesis Leading to Cell Death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef]
- Nadanaka, S.; Okada, T.; Yoshida, H.; Mori, K. Role of Disulfide Bridges Formed in the Luminal Domain of ATF6 in Sensing Endoplasmic Reticulum Stress. Mol. Cell Biol. 2007, 27, 1027–1043. [Google Scholar] [CrossRef]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER Stress Induces Cleavage of Membrane-Bound ATF6 by the Same Proteases That Process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Oka, O.B.V.; Pierre, A.S.; Pringle, M.A.; Tungkum, W.; Cao, Z.; Fleming, B.; Bulleid, N.J. Activation of the UPR Sensor ATF6α Is Regulated by Its Redox-Dependent Dimerization and ER Retention by ERp18. Proc. Natl. Acad. Sci. USA 2022, 119, e2122657119. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shen, J.; Arenzana, N.; Tirasophon, W.; Kaufman, R.J.; Prywes, R. Activation of ATF6 and an ATF6 DNA Binding Site by the Endoplasmic Reticulum Stress Response. J. Biol. Chem. 2000, 275, 27013–27020. [Google Scholar] [CrossRef]
- Kokame, K.; Kato, H.; Miyata, T. Identification of ERSE-II, a New Cis-Acting Element Responsible for the ATF6-Dependent Mammalian Unfolded Protein Response. J. Biol. Chem. 2001, 276, 9199–9205. [Google Scholar] [CrossRef]
- Adachi, Y.; Yamamoto, K.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. ATF6 Is a Transcription Factor Specializing in the Regulation of Quality Control Proteins in the Endoplasmic Reticulum. Cell Struct. Funct. 2008, 33, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Wufuer, R.; Fan, Z.; Yuan, J.; Zheng, Z.; Hu, S.; Sun, G.; Zhang, Y. Distinct Roles of Nrf1 and Nrf2 in Monitoring the Reductive Stress Response to Dithiothreitol (DTT). Antioxidants 2022, 11, 1535. [Google Scholar] [CrossRef] [PubMed]
- Pibiri, M.; Sulas, P.; Camboni, T.; Leoni, V.P.; Simbula, G. α-Lipoic Acid Induces Endoplasmic Reticulum Stress-Mediated Apoptosis in Hepatoma Cells. Sci. Rep. 2020, 10, 7139. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, T.; Tang, S.; Zhao, D.; Zhang, C.; Zhang, S.; Deng, S.; Zhou, Y.; Xiao, X. Thapsigargin Induces Apoptosis When Autophagy Is Inhibited in HepG2 Cells and Both Processes Are Regulated by ROS-Dependent Pathway. Environ. Toxicol. Pharmacol. 2016, 41, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Yen, J.-H.; Wu, P.-S.; Chen, S.-F.; Wu, M.-J. Fisetin Protects PC12 Cells from Tunicamycin-Mediated Cell Death via Reactive Oxygen Species Scavenging and Modulation of Nrf2-Driven Gene Expression, SIRT1 and MAPK Signaling in PC12 Cells. Int. J. Mol. Sci. 2017, 18, 852. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress in Cell Fate Decision and Human Disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Decuypere, J.-P.; Monaco, G.; Missiaen, L.; De Smedt, H.; Parys, J.B.; Bultynck, G. IP3 Receptors, Mitochondria, and Ca2+ Signaling: Implications for Aging. J. Aging Res. 2011, 2011, 920178. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP Induces Death by Promoting Protein Synthesis and Oxidation in the Stressed Endoplasmic Reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, K.R.; Riaz, T.A.; Kim, H.-R.; Chae, H.-J. The Aftermath of the Interplay between the Endoplasmic Reticulum Stress Response and Redox Signaling. Exp. Mol. Med. 2021, 53, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 Is a Direct PERK Substrate and Effector of PERK-Dependent Cell Survival. Mol. Cell Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [PubMed]
- Alam, J.; Stewart, D.; Touchard, C.; Boinapally, S.; Choi, A.M.; Cook, J.L. Nrf2, a Cap’n’Collar Transcription Factor, Regulates Induction of the Heme Oxygenase-1 Gene. J. Biol. Chem. 1999, 274, 26071–26078. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Villeneuve, N.F.; Lau, A.; Zhang, D.D. Regulation of the Nrf2-Keap1 Antioxidant Response by the Ubiquitin Proteasome System: An Insight into Cullin-Ring Ubiquitin Ligases. Antioxid. Redox Signal. 2010, 13, 1699–1712. [Google Scholar] [CrossRef]
- Suzuki, T.; Muramatsu, A.; Saito, R.; Iso, T.; Shibata, T.; Kuwata, K.; Kawaguchi, S.-I.; Iwawaki, T.; Adachi, S.; Suda, H.; et al. Molecular Mechanism of Cellular Oxidative Stress Sensing by Keap1. Cell Rep. 2019, 28, 746–758.e4. [Google Scholar] [CrossRef]
- Bobrovnikova-Marjon, E.; Grigoriadou, C.; Pytel, D.; Zhang, F.; Ye, J.; Koumenis, C.; Cavener, D.; Diehl, J.A. PERK Promotes Cancer Cell Proliferation and Tumor Growth by Limiting Oxidative DNA Damage. Oncogene 2010, 29, 3881–3895. [Google Scholar] [CrossRef]
- Küper, A.; Baumann, J.; Göpelt, K.; Baumann, M.; Sänger, C.; Metzen, E.; Kranz, P.; Brockmeier, U. Overcoming Hypoxia-Induced Resistance of Pancreatic and Lung Tumor Cells by Disrupting the PERK-NRF2-HIF-Axis. Cell Death Dis. 2021, 12, 82. [Google Scholar] [CrossRef]
- Wang, J.; Lu, L.; Chen, S.; Xie, J.; Lu, S.; Zhou, Y.; Jiang, H. Up-Regulation of PERK/Nrf2/HO-1 Axis Protects Myocardial Tissues of Mice from Damage Triggered by Ischemia-Reperfusion through Ameliorating Endoplasmic Reticulum Stress. Cardiovasc. Diagn. Ther. 2020, 10, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Sarcinelli, C.; Dragic, H.; Piecyk, M.; Barbet, V.; Duret, C.; Barthelaix, A.; Ferraro-Peyret, C.; Fauvre, J.; Renno, T.; Chaveroux, C.; et al. ATF4-Dependent NRF2 Transcriptional Regulation Promotes Antioxidant Protection during Endoplasmic Reticulum Stress. Cancers 2020, 12, 569. [Google Scholar] [CrossRef] [PubMed]
- He, C.H.; Gong, P.; Hu, B.; Stewart, D.; Choi, M.E.; Choi, A.M.; Alam, J. Identification of Activating Transcription Factor 4 (ATF4) as an Nrf2-Interacting Protein. Implication for Heme Oxygenase-1 Gene Regulation. J. Biol. Chem. 2001, 276, 20858–20865. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Mimura, J.; Okada, T.; Sato, H.; Liu, T.; Maruyama, A.; Ohyama, C.; Itoh, K. Nrf2- and ATF4-Dependent Upregulation of XCT Modulates the Sensitivity of T24 Bladder Carcinoma Cells to Proteasome Inhibition. Mol. Cell Biol. 2014, 34, 3421–3434. [Google Scholar] [CrossRef]
- Bai, X.; Ni, J.; Beretov, J.; Wasinger, V.C.; Wang, S.; Zhu, Y.; Graham, P.; Li, Y. Activation of the EIF2α/ATF4 Axis Drives Triple-Negative Breast Cancer Radioresistance by Promoting Glutathione Biosynthesis. Redox Biol. 2021, 43, 101993. [Google Scholar] [CrossRef]
- Song, B.; Scheuner, D.; Ron, D.; Pennathur, S.; Kaufman, R.J. Chop Deletion Reduces Oxidative Stress, Improves Beta Cell Function, and Promotes Cell Survival in Multiple Mouse Models of Diabetes. J. Clin. Investig. 2008, 118, 3378–3389. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.-P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK Is Required at the ER-Mitochondrial Contact Sites to Convey Apoptosis after ROS-Based ER Stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-W.; Zhu, H.-T.; Chen, K.-L.; Dong, X.; Wei, J.; Qiu, C.; Xue, J.-H. Protein Kinase RNA-like Endoplasmic Reticulum Kinase (PERK) Signaling Pathway Plays a Major Role in Reactive Oxygen Species (ROS)-Mediated Endoplasmic Reticulum Stress-Induced Apoptosis in Diabetic Cardiomyopathy. Cardiovasc. Diabetol. 2013, 12, 158. [Google Scholar] [CrossRef] [PubMed]
- Bassot, A.; Chen, J.; Takahashi-Yamashiro, K.; Yap, M.C.; Gibhardt, C.S.; Le, G.N.T.; Hario, S.; Nasu, Y.; Moore, J.; Gutiérrez, T.; et al. The Endoplasmic Reticulum Kinase PERK Interacts with the Oxidoreductase ERO1 to Metabolically Adapt Mitochondria. Cell Rep. 2023, 42, 111899. [Google Scholar] [CrossRef]
- Guerra-Moreno, A.; Ang, J.; Welsch, H.; Jochem, M.; Hanna, J. Regulation of the Unfolded Protein Response in Yeast by Oxidative Stress. FEBS Lett. 2019, 593, 1080–1088. [Google Scholar] [CrossRef]
- Hourihan, J.M.; Moronetti Mazzeo, L.E.; Fernández-Cárdenas, L.P.; Blackwell, T.K. Cysteine Sulfenylation Directs IRE-1 to Activate the SKN-1/Nrf2 Antioxidant Response. Mol. Cell 2016, 63, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Fink, E.E.; Moparthy, S.; Bagati, A.; Bianchi-Smiraglia, A.; Lipchick, B.C.; Wolff, D.W.; Roll, M.V.; Wang, J.; Liu, S.; Bakin, A.V.; et al. XBP1-KLF9 Axis Acts as a Molecular Rheostat to Control the Transition from Adaptive to Cytotoxic Unfolded Protein Response. Cell Rep. 2018, 25, 212–223.e4. [Google Scholar] [CrossRef] [PubMed]
- Zucker, S.N.; Fink, E.E.; Bagati, A.; Mannava, S.; Bianchi-Smiraglia, A.; Bogner, P.N.; Wawrzyniak, J.A.; Foley, C.; Leonova, K.I.; Grimm, M.J.; et al. Nrf2 Amplifies Oxidative Stress via Induction of Klf9. Mol. Cell 2014, 53, 916–928. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Sun, S.; Sha, H.; Liu, Z.; Yang, L.; Xue, Z.; Chen, H.; Qi, L. Emerging Roles for XBP1, a SUPeR Transcription Factor. Gene Expr. 2010, 15, 13–25. [Google Scholar] [CrossRef]
- Liu, Y.; Adachi, M.; Zhao, S.; Hareyama, M.; Koong, A.C.; Luo, D.; Rando, T.A.; Imai, K.; Shinomura, Y. Preventing Oxidative Stress: A New Role for XBP1. Cell Death Differ. 2009, 16, 847–857. [Google Scholar] [CrossRef]
- Martin, D.; Li, Y.; Yang, J.; Wang, G.; Margariti, A.; Jiang, Z.; Yu, H.; Zampetaki, A.; Hu, Y.; Xu, Q.; et al. Unspliced X-Box-Binding Protein 1 (XBP1) Protects Endothelial Cells from Oxidative Stress through Interaction with Histone Deacetylase 3. J. Biol. Chem. 2014, 289, 30625–30634. [Google Scholar] [CrossRef]
- Nishiyama, A.; Matsui, M.; Iwata, S.; Hirota, K.; Masutani, H.; Nakamura, H.; Takagi, Y.; Sono, H.; Gon, Y.; Yodoi, J. Identification of Thioredoxin-Binding Protein-2/Vitamin D(3) up-Regulated Protein 1 as a Negative Regulator of Thioredoxin Function and Expression. J. Biol. Chem. 1999, 274, 21645–21650. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.G.; Upton, J.-P.; Praveen, P.V.K.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1α Induces Thioredoxin-Interacting Protein to Activate the NLRP3 Inflammasome and Promote Programmed Cell Death under Irremediable ER Stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef]
- Dostert, C.; Pétrilli, V.; Van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate Immune Activation through Nalp3 Inflammasome Sensing of Asbestos and Silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-Interacting Protein Links Oxidative Stress to Inflammasome Activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Jin, J.-K.; Blackwood, E.A.; Azizi, K.; Thuerauf, D.J.; Fahem, A.G.; Hofmann, C.; Kaufman, R.J.; Doroudgar, S.; Glembotski, C.C. ATF6 Decreases Myocardial Ischemia/Reperfusion Damage and Links ER Stress and Oxidative Stress Signaling Pathways in the Heart. Circ. Res. 2017, 120, 862–875. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Gong, M.; He, J.; Xie, B.; Zhang, Z.; Meng, L.; Tse, G.; Zhao, Y.; Bao, Q.; Zhang, Y.; et al. IP3R1/GRP75/VDAC1 Complex Mediates Endoplasmic Reticulum Stress-Mitochondrial Oxidative Stress in Diabetic Atrial Remodeling. Redox Biol. 2022, 52, 102289. [Google Scholar] [CrossRef] [PubMed]
- Pallepati, P.; Averill-Bates, D.A. Activation of ER Stress and Apoptosis by Hydrogen Peroxide in HeLa Cells: Protective Role of Mild Heat Preconditioning at 40 °C. Biochim. Biophys. Acta 2011, 1813, 1987–1999. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.X.C.; Tanaka, L.Y.; Wosniak, J.; Laurindo, F.R.M. Mechanisms and Implications of Reactive Oxygen Species Generation during the Unfolded Protein Response: Roles of Endoplasmic Reticulum Oxidoreductases, Mitochondrial Electron Transport, and NADPH Oxidase. Antioxid. Redox Signal. 2009, 11, 2409–2427. [Google Scholar] [CrossRef]
- Yun, H.R.; Jo, Y.H.; Kim, J.; Shin, Y.; Kim, S.S.; Choi, T.G. Roles of Autophagy in Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 3289. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Hernández, A.; Leon-Aparicio, D.; Chavez-Reyes, J.; Olivares-Reyes, J.A.; DeJesus, S. Endoplasmic Reticulum Stress in Insulin Resistance and Diabetes. Cell Calcium 2014, 56, 311–322. [Google Scholar] [CrossRef]
- Reeg, S.; Jung, T.; Castro, J.P.; Davies, K.J.A.; Henze, A.; Grune, T. The Molecular Chaperone Hsp70 Promotes the Proteolytic Removal of Oxidatively Damaged Proteins by the Proteasome. Free Radic. Biol. Med. 2016, 99, 153–166. [Google Scholar] [CrossRef]
- Szyller, J.; Bil-Lula, I. Heat Shock Proteins in Oxidative Stress and Ischemia/Reperfusion Injury and Benefits from Physical Exercises: A Review to the Current Knowledge. Oxid. Med. Cell Longev. 2021, 2021, 6678457. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ong, G.; Logue, S.E. Unfolding the Interactions between Endoplasmic Reticulum Stress and Oxidative Stress. Antioxidants 2023, 12, 981. https://doi.org/10.3390/antiox12050981
Ong G, Logue SE. Unfolding the Interactions between Endoplasmic Reticulum Stress and Oxidative Stress. Antioxidants. 2023; 12(5):981. https://doi.org/10.3390/antiox12050981
Chicago/Turabian StyleOng, Gideon, and Susan E. Logue. 2023. "Unfolding the Interactions between Endoplasmic Reticulum Stress and Oxidative Stress" Antioxidants 12, no. 5: 981. https://doi.org/10.3390/antiox12050981