
Mitochondrial Oxidative Stress Mediates Bradyarrhythmia in Leigh Syndrome Mitochondrial Disease Mice

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
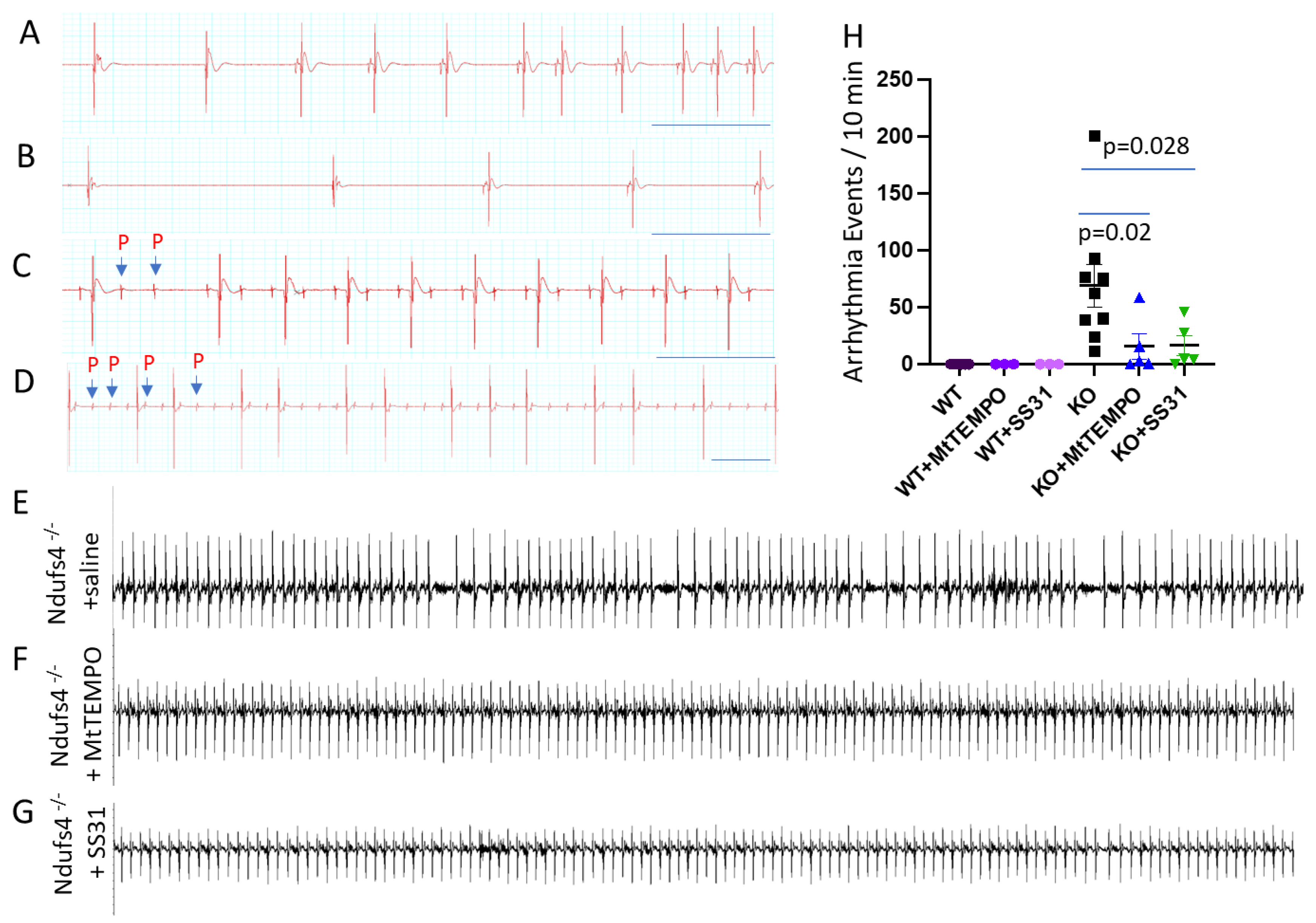
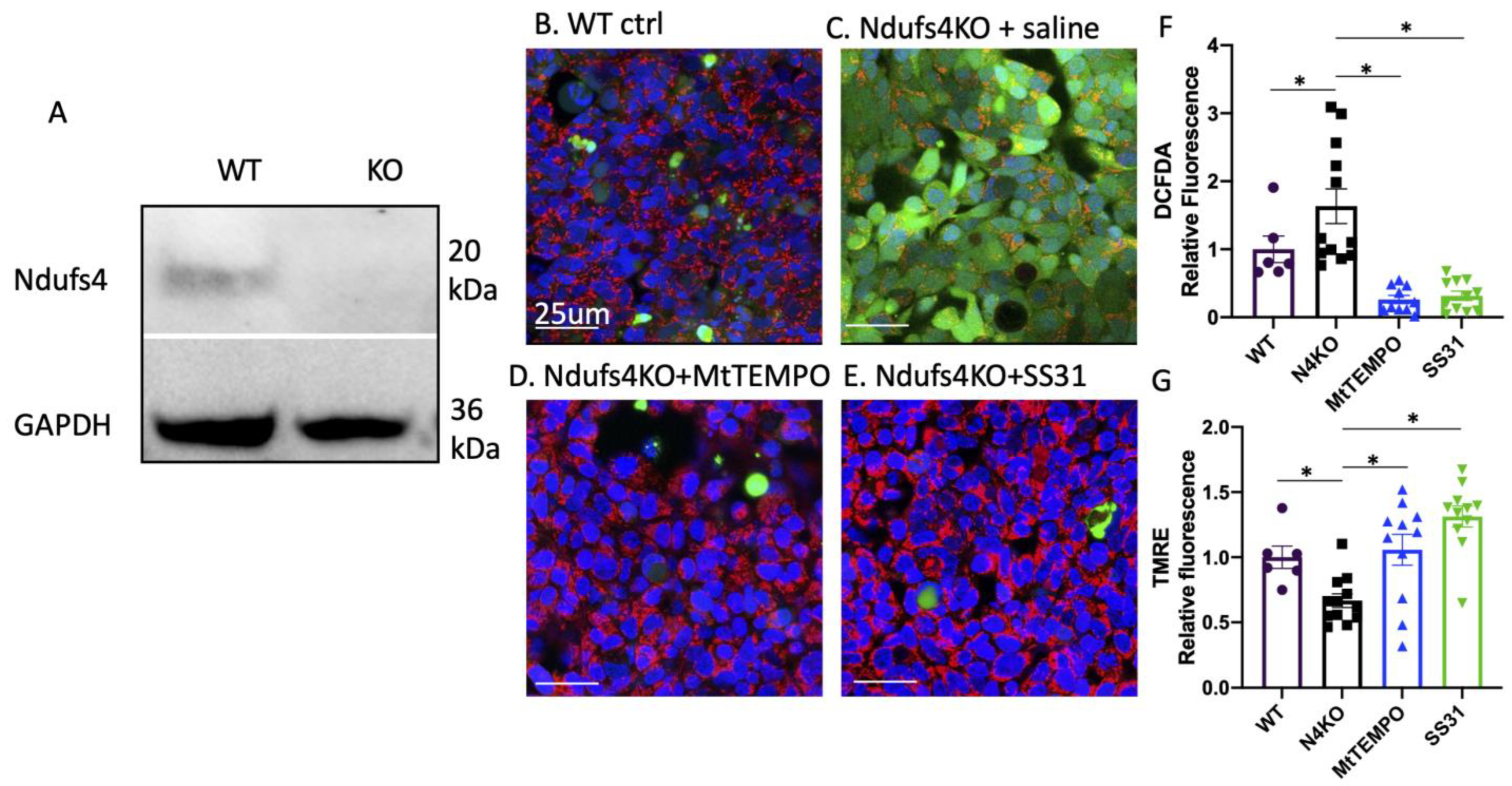
Leigh Syndrome Mice Develop Bradyarrhythmia
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dhalla, N.S.; Temsah, R.M.; Netticadan, T. Role of oxidative stress in cardiovascular diseases. J. Hypertens. 2000, 18, 655–673. [Google Scholar] [CrossRef] [PubMed]
- Senoner, T.; Dichtl, W. Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients 2019, 11, 2090. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Chen, T.; Johnson, S.C.; Szeto, H.; Rabinovitch, P.S. Cardiac aging: From molecular mechanisms to significance in human health and disease. Antioxid. Redox Signal. 2012, 16, 1492–1526. [Google Scholar] [CrossRef]
- Dai, D.F.; Santana, L.F.; Vermulst, M.; Tomazela, D.M.; Emond, M.J.; MacCoss, M.J.; Gollahon, K.; Martin, G.M.; Loeb, L.A.; Ladiges, W.C.; et al. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation 2009, 119, 2789–2797. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Hsieh, E.J.; Chen, T.; Menendez, L.G.; Basisty, N.B.; Tsai, L.; Beyer, R.P.; Crispin, D.A.; Shulman, N.J.; Szeto, H.H.; et al. Global proteomics and pathway analysis of pressure-overload-induced heart failure and its attenuation by mitochondrial-targeted peptides. Circ. Heart Fail. 2013, 6, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Hsieh, E.J.; Liu, Y.; Chen, T.; Beyer, R.P.; Chin, M.T.; MacCoss, M.J.; Rabinovitch, P.S. Mitochondrial proteome remodelling in pressure overload-induced heart failure: The role of mitochondrial oxidative stress. Cardiovasc. Res. 2012, 93, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Johnson, S.C.; Villarin, J.J.; Chin, M.T.; Nieves-Cintron, M.; Chen, T.; Marcinek, D.J.; Dorn, G.W., 2nd; Kang, Y.J.; Prolla, T.A.; et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ. Res. 2011, 108, 837–846. [Google Scholar] [CrossRef]
- Yang, K.C.; Bonini, M.G.; Dudley, S.C., Jr. Mitochondria and arrhythmias. Free Radic. Biol. Med. 2014, 71, 351–361. [Google Scholar] [CrossRef]
- Yang, K.C.; Kyle, J.W.; Makielski, J.C.; Dudley, S.C., Jr. Mechanisms of sudden cardiac death: Oxidants and metabolism. Circ. Res. 2015, 116, 1937–1955. [Google Scholar] [CrossRef]
- Quintana, A.; Kruse, S.E.; Kapur, R.P.; Sanz, E.; Palmiter, R.D. Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 10996–11001. [Google Scholar] [CrossRef]
- Sofou, K.; de Coo, I.F.M.; Ostergaard, E.; Isohanni, P.; Naess, K.; De Meirleir, L.; Tzoulis, C.; Uusimaa, J.; Lonnqvist, T.; Bindoff, L.A.; et al. Phenotype-genotype correlations in Leigh syndrome: New insights from a multicentre study of 96 patients. J. Med. Genet. 2018, 55, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhang, C.; Guo, A.; Song, L.S. In situ single photon confocal imaging of cardiomyocyte T-tubule system from Langendorff-perfused hearts. Front. Physiol. 2015, 6, 134. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.P.; Wu, Y.; Joiner, M.L.; Koval, O.M.; Wilson, N.R.; Luczak, E.D.; Wang, Q.; Chen, B.; Gao, Z.; Zhu, Z.; et al. Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc. Natl. Acad. Sci. USA 2015, 112, 9129–9134. [Google Scholar] [CrossRef]
- Sofou, K.; De Coo, I.F.; Isohanni, P.; Ostergaard, E.; Naess, K.; De Meirleir, L.; Tzoulis, C.; Uusimaa, J.; De Angst, I.B.; Lonnqvist, T.; et al. A multicenter study on Leigh syndrome: Disease course and predictors of survival. Orphanet J. Rare Dis. 2014, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Liu, H.; Dudley, S.C., Jr. Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ. Res. 2010, 107, 967–974. [Google Scholar] [CrossRef]
- Lee, H.F.; Tsai, C.R.; Chi, C.S.; Lee, H.J.; Chen, C.C. Leigh syndrome: Clinical and neuroimaging follow-up. Pediatr. Neurol. 2009, 40, 88–93. [Google Scholar] [CrossRef]
- Savvatis, K.; Vissing, C.R.; Klouvi, L.; Florian, A.; Rahman, M.; Béhin, A.; Fayssoil, A.; Masingue, M.; Stojkovic, T.; Bécane, H.M.; et al. Cardiac Outcomes in Adults with Mitochondrial Diseases. J. Am. Coll. Cardiol. 2022, 80, 1421–1430. [Google Scholar] [CrossRef]
- Quintana, A.; Zanella, S.; Koch, H.; Kruse, S.E.; Lee, D.; Ramirez, J.M.; Palmiter, R.D. Fatal breathing dysfunction in a mouse model of Leigh syndrome. J. Clin. Investig. 2012, 122, 2359–2368. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Methner, C.; Buonincontri, G.; Hu, C.H.; Logan, A.; Sawiak, S.J.; Murphy, M.P.; Krieg, T. Complex I deficiency due to selective loss of Ndufs4 in the mouse heart results in severe hypertrophic cardiomyopathy. PLoS ONE 2014, 9, e94157. [Google Scholar] [CrossRef] [PubMed]
- Karamanlidis, G.; Lee, C.F.; Garcia-Menendez, L.; Kolwicz, S.C., Jr.; Suthammarak, W.; Gong, G.; Sedensky, M.M.; Morgan, P.G.; Wang, W.; Tian, R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 2013, 18, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Zhao, M.; Zhou, B.; Yoshii, A.; Bugg, D.; Villet, O.; Sahu, A.; Olson, G.S.; Davis, J.; Tian, R. Mitochondrial dysfunction in macrophages promotes inflammation and suppresses repair after myocardial infarction. J. Clin. Investig. 2022, 133, e159498. [Google Scholar] [CrossRef]
- Grune, J.; Lewis, A.J.M.; Yamazoe, M.; Hulsmans, M.; Rohde, D.; Xiao, L.; Zhang, S.; Ott, C.; Calcagno, D.M.; Zhou, Y.; et al. Neutrophils incite and macrophages avert electrical storm after myocardial infarction. Nat. Cardiovasc. Res. 2022, 1, 649–664. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.Y.; Daneshgar, N.; Chu, Y.; Chen, B.; Hefti, M.; Vikram, A.; Irani, K.; Song, L.S.; Brenner, C.; Abel, E.D.; et al. Metabolic rescue ameliorates mitochondrial encephalo-cardiomyopathy in murine and human iPSC models of Leigh syndrome. Clin. Transl. Med. 2022, 12, e954. [Google Scholar] [CrossRef]
- Zhang, H.; Gong, G.; Wang, P.; Zhang, Z.; Kolwicz, S.C.; Rabinovitch, P.S.; Tian, R.; Wang, W. Heart specific knockout of Ndufs4 ameliorates ischemia reperfusion injury. J. Mol. Cell Cardiol. 2018, 123, 38–45. [Google Scholar] [CrossRef]
- Birk, A.V.; Chao, W.M.; Bracken, C.; Warren, J.D.; Szeto, H.H. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br. J. Pharmacol. 2014, 171, 2017–2028. [Google Scholar] [CrossRef] [PubMed]
- Daneshgar, N.; Liang, P.I.; Lan, R.S.; Horstmann, M.M.; Pack, L.; Bhardwaj, G.; Penniman, C.M.; O’Neill, B.T.; Dai, D.F. Elamipretide treatment during pregnancy ameliorates the progression of polycystic kidney disease in maternal and neonatal mice with PKD1 mutations. Kidney Int. 2022, 101, 906–911. [Google Scholar] [CrossRef]
- Szeto, H.H. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol. 2014, 171, 2029–2050. [Google Scholar] [CrossRef]
- Mitchell, W.; Ng, E.A.; Tamucci, J.D.; Boyd, K.J.; Sathappa, M.; Coscia, A.; Pan, M.; Han, X.; Eddy, N.A.; May, E.R.; et al. The mitochondria-targeted peptide SS-31 binds lipid bilayers and modulates surface electrostatics as a key component of its mechanism of action. J. Biol. Chem. 2020, 295, 7452–7469. [Google Scholar] [CrossRef]
- Chavez, J.D.; Tang, X.; Campbell, M.D.; Reyes, G.; Kramer, P.A.; Stuppard, R.; Keller, A.; Zhang, H.; Rabinovitch, P.S.; Marcinek, D.J.; et al. Mitochondrial protein interaction landscape of SS-31. Proc. Natl. Acad. Sci. USA 2020, 117, 15363–15373. [Google Scholar] [CrossRef]
- Johnson, S.C.; Yanos, M.E.; Kayser, E.B.; Quintana, A.; Sangesland, M.; Castanza, A.; Uhde, L.; Hui, J.; Wall, V.Z.; Gagnidze, A.; et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 2013, 342, 1524–1528. [Google Scholar] [CrossRef] [PubMed]
- Jain, I.H.; Zazzeron, L.; Goli, R.; Alexa, K.; Schatzman-Bone, S.; Dhillon, H.; Goldberger, O.; Peng, J.; Shalem, O.; Sanjana, N.E.; et al. Hypoxia as a therapy for mitochondrial disease. Science 2016, 352, 54–61. [Google Scholar] [CrossRef]
- Daneshgar, N.; Leidinger, M.R.; Le, S.; Hefti, M.; Prigione, A.; Dai, D.F. Activated microglia and neuroinflammation as a pathogenic mechanism in Leigh syndrome. Front. Neurosci. 2022, 16, 1068498. [Google Scholar] [CrossRef] [PubMed]
- Grange, R.M.H.; Sharma, R.; Shah, H.; Reinstadler, B.; Goldberger, O.; Cooper, M.K.; Nakagawa, A.; Miyazaki, Y.; Hindle, A.G.; Batten, A.J.; et al. Hypoxia ameliorates brain hyperoxia and NAD(+) deficiency in a murine model of Leigh syndrome. Mol. Genet. Metab. 2021, 133, 83–93. [Google Scholar] [CrossRef]
- Jain, I.H.; Zazzeron, L.; Goldberger, O.; Marutani, E.; Wojtkiewicz, G.R.; Ast, T.; Wang, H.; Schleifer, G.; Stepanova, A.; Brepoels, K.; et al. Leigh Syndrome Mouse Model Can Be Rescued by Interventions that Normalize Brain Hyperoxia, but Not HIF Activation. Cell Metab. 2019, 30, 824–832.e3. [Google Scholar] [CrossRef]
- Lee, C.F.; Caudal, A.; Abell, L.; Nagana Gowda, G.A.; Tian, R. Targeting NAD(+) Metabolism as Interventions for Mitochondrial Disease. Sci. Rep. 2019, 9, 3073. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, K.; Comes, G.; Canal, C.; Quintana, A.; Sanz, E.; Hidalgo, J. Microglial response promotes neurodegeneration in the Ndufs4 KO mouse model of Leigh syndrome. Glia 2022, 70, 2032–2044. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, B.; Daneshgar, N.; Lee, H.-C.; Song, L.-S.; Dai, D.-F. Mitochondrial Oxidative Stress Mediates Bradyarrhythmia in Leigh Syndrome Mitochondrial Disease Mice. Antioxidants 2023, 12, 1001. https://doi.org/10.3390/antiox12051001
Chen B, Daneshgar N, Lee H-C, Song L-S, Dai D-F. Mitochondrial Oxidative Stress Mediates Bradyarrhythmia in Leigh Syndrome Mitochondrial Disease Mice. Antioxidants. 2023; 12(5):1001. https://doi.org/10.3390/antiox12051001
Chicago/Turabian StyleChen, Biyi, Nastaran Daneshgar, Hsiang-Chun Lee, Long-Sheng Song, and Dao-Fu Dai. 2023. "Mitochondrial Oxidative Stress Mediates Bradyarrhythmia in Leigh Syndrome Mitochondrial Disease Mice" Antioxidants 12, no. 5: 1001. https://doi.org/10.3390/antiox12051001