
Schizophrenia Synaptic Pathology and Antipsychotic Treatment in the Framework of Oxidative and Mitochondrial Dysfunction: Translational Highlights for the Clinics and Treatment
 , and
, and
Abstract
:1. Introduction
Aims of the Study
2. Materials and Methods
2.1. Screening and Selection Process
2.2. Search Strategy
2.3. Inclusion and Exclusion Criteria
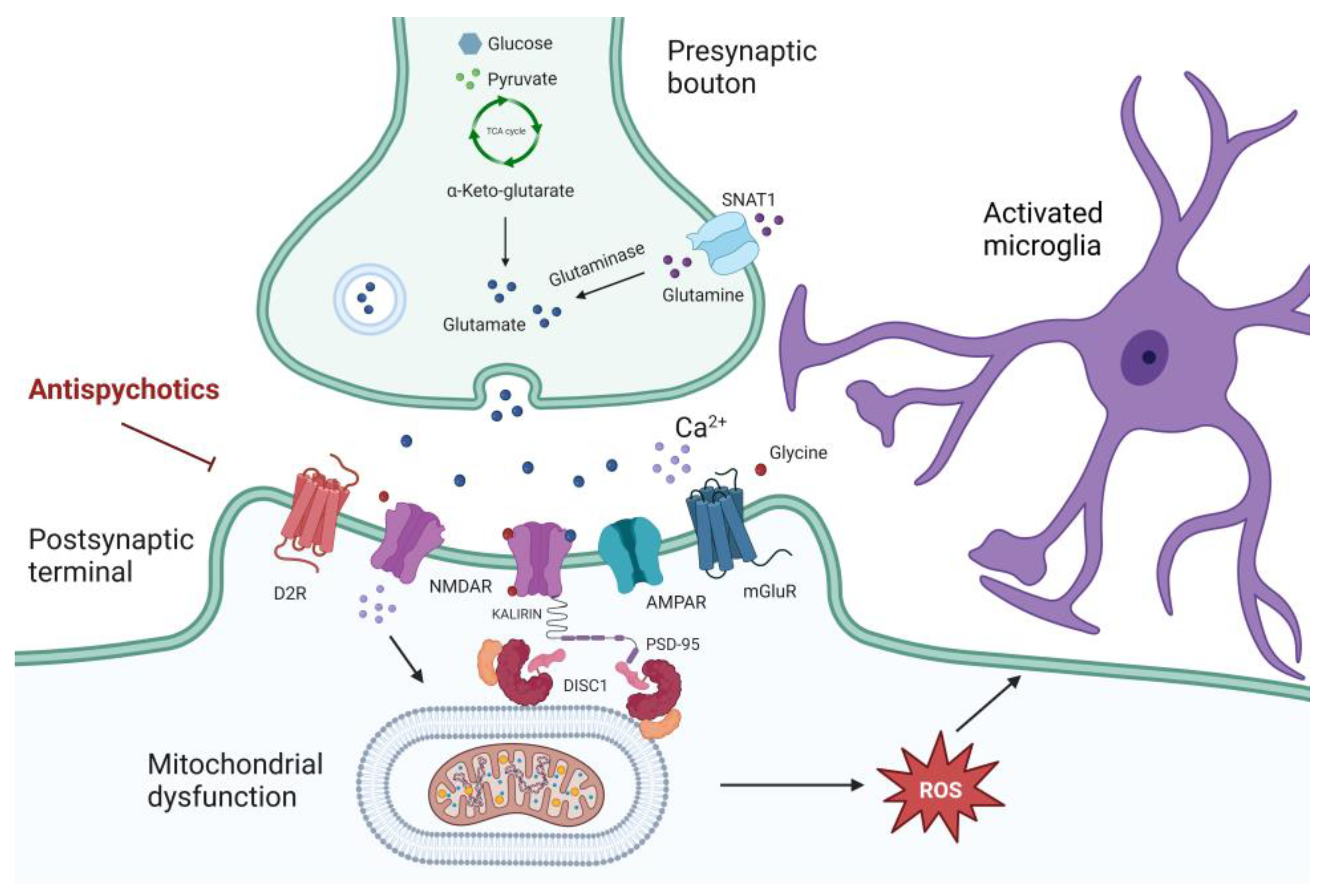
3. Molecular Abnormalities Driven by Inflammation and Oxidative Stress Relevant to Schizophrenia
3.1. Relevance of Inflammation in Schizophrenia
3.1.1. Evidence from Preclinical Studies
3.1.2. Evidence from Clinical Studies
3.2. The Interaction between Oxidative Stress and Synaptic Plasticity
3.3. Antipsychotic Modulation Induced by Inflammation and Oxidative Stress in Schizophrenia
4. General Appraisal of Antioxidant Pathway and Mitochondria Dysfunction in Schizophrenia
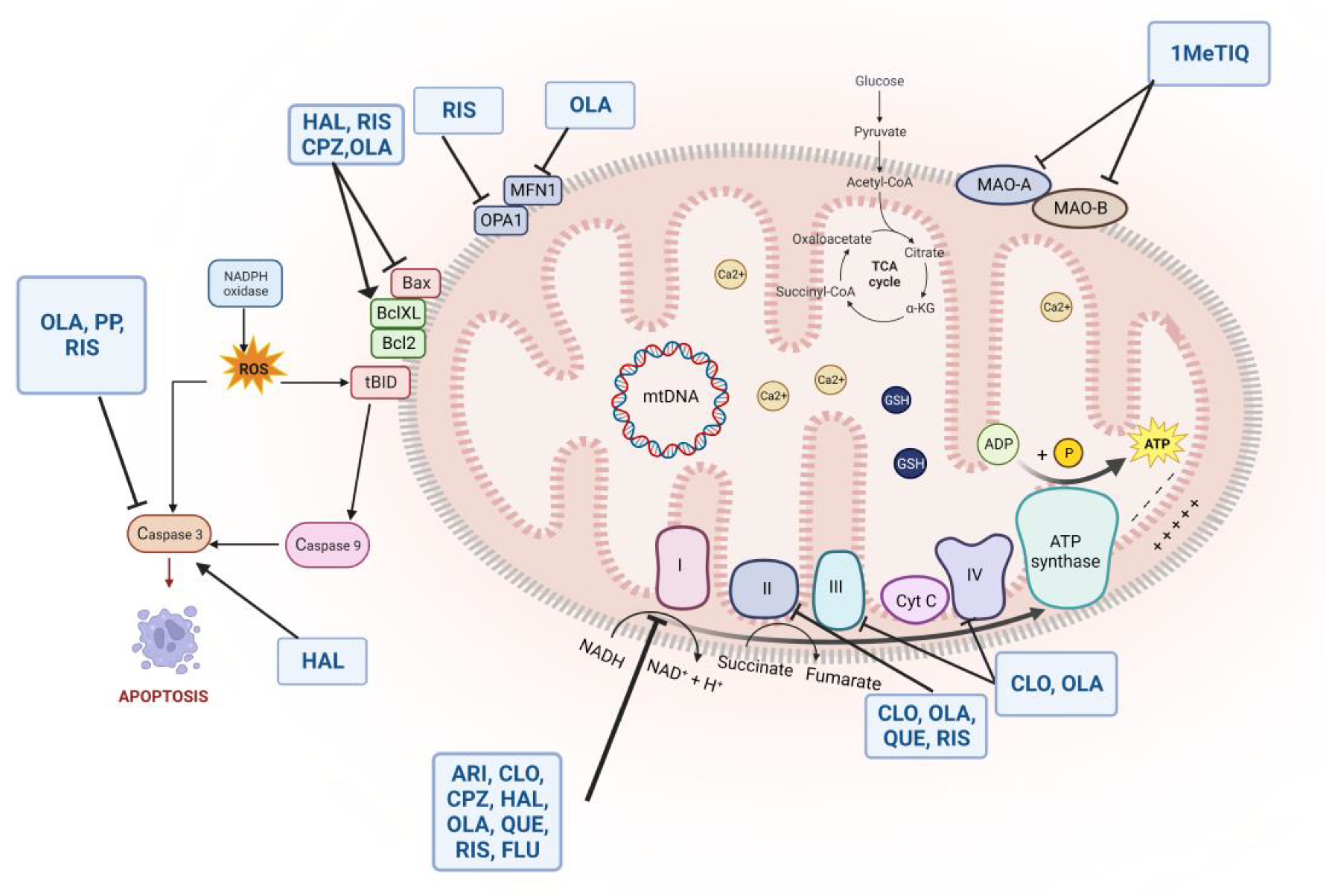
5. Antipsychotics: An Oxidant or Antioxidant Effect in Schizophrenia?
5.1. Effects of Typical Antipsychotics
5.2. Effects of Atypical Antipsychotics

{kind=link}
{kind=link}
{kind=link}
| Antipsychotics | Study Design | Model | Main Outcomes | References |
|---|---|---|---|---|
| Haloperidol | Preclinical study | S1R-knockdown mouse hippocampal HT22 cells | HAL could exert neuroprotective effects through mechanisms independent of sigma1 receptors. By the application of fluorescent probes, HAL was localized to endosomes and lysosomes and was shown to reduce the accumulation of Fe2+ and prevent the phenomena of cell death. | Hirata et al., 2022 [186] |
| In vivo preclinical study | Adult male CD1 mice | In the mouse frontal cortex, HAL was seen to decrease BDNF, GSH, and Bcl-xL levels. | Pillai et al., 2008, [188] | |
| Preclinical study | Rat primary cortical neurons and mouse hippocampal cells line HT22 | HAL resulted in a six-fold increase in the levels of ROS generated by mitochondria, together with a reduction in GSH and a concomitant elevation in intracellular Ca2+ flux in rat primary cortical neurons and mouse hippocampal cells. | Sagara 1998 [190] | |
| Preclinical study | Mice | HAL induced a decrease in GSH concentration in the cortex, striatum, and cerebellum, which was reverted by the treatment with cinnarizine. | Abdel-Salam et al., 2012 [191] | |
| Preclinical study | Human peripheral blood lymphocytes | HAL induced increased oxidative stress, reduced GSH levels, increased lipid peroxidation, and exerted genotoxic effects in human lymphocytes. | Zamani et al., 2022 [196] | |
| Haloperidol and risperidone | In vitro preclinical study | Cultures of cortical neurons of rats at E18 | HAL reduced Akt phosphorylation levels and caspase-3 activation; instead, BDNF could reverse these HAL-induced effects. Furthermore, only HAL, not RIS, could induce caspase-dependent apoptosis. | Ukai et al., 2004 [194] |
| In vitro comparative study | Cultures of astrocyte-like cells (C6 cell line) | RIS increased glutamate uptake, glutamine synthase activity, and GSH content, and high doses of HAL increased ROS. | Quincozes-Santos et al., [201] | |
| Ziprasidone, clozapine, and haloperidol | In vitro preclinical study | Human blood | ZIP and HAL but not CLO raised TBARS levels in the plasma of healthy subjects after 24 h of incubation, indicating a status of increased lipid peroxidation. | Dietrich-Muszalska et al., 2013 [212] |
| Risperidone, ziprasidone, haloperidol, aripiprazole | In vitro preclinical study | Human blood | Contrary to the oxidant effect of RIS, ZIP, or HAL, APZ had no effect on lipid peroxidation, assessed as TBARS levels in the plasma of healthy volunteers. | Dietrich-Muszalska et al., 2018 [213] |
| Haloperidol, risperidone, and paliperidone | In vitro comparative study | Human neuroblastoma SK-N-SH cells | HAL was linked to an increase in caspase-3 activity and cell death RIS and PP decreased caspase-3 activity and did not affect cell viability or cell death. | Gassó et al., 2012 [200] |
| Aripiprazole | Preclinical study | Adolescent male ICR mice | APZ was effective in enhancing cell proliferation and maturation and complexity of neuroblast dendrites in the dentate gyrus, along with decreased lipid peroxidation and increased SOD2 levels. | Chen et al., 2013 [214] |
| Chlorpromazine | Preclinical study | Rats | CPZ has neuroprotective effects through mitochondrial apoptotic pathway inhibition. Moreover, CPZ decreased the expression of cleaved caspase-3, cytochrome c, and Bax and increased the expression of Bcl-2 and TF. | Wu et al., 2016 [197] |
| Preclinical study | Rats | The long-term administration of CPZ reduced the activity of SOD. | Abdalla et al., 1994 [198] | |
| Risperidone | Clinical trial | 40 patients with SCZ | RIS decreased the initially high blood levels of SOD observed in patients with SCZ. | Zhang et al., 2003 [203] |
| In vitro preclinical study | Rat C6 cells were transfected with a plasmid encoding mouse DAO | RIS exerted a partial uncompetitive inhibition effect on human DAO and exhibited a protective effect from D-amino acid-induced cell death. | Abou El-Magd et al., 2010 [207] | |
| Olanzapine | In vitro preclinical study | mHypoA-59 hypothalamic neuronal cell line | OLA could increase ROS and mitochondrial dysfunction, triggering cytotoxic mechanisms and neural death. A possible antidote to these events is represented by NAC use. | Boz et al., 2020 [204] |
| Preclinical study | PC12 cells | In PC12 cells exposed to hydrogen peroxide, pretreatment with OLA attenuated the decrease of SOD-1 mRNA levels, while SOD enzymatic activity increased. | Wei et al., 2023 [205] | |
| Haloperidol, risperidone, clozapine, and quetiapine | Preclinical study | Cultures of peripheral mononuclear blood cells extracted from 12 patients with FEP | HAL, as well as RIS, CLO, and QTP, were all effective in raising anti-inflammatory cytokine (IL-4 and IL-10) levels. | Al-Amin et al., 2013 [199] |
| Haloperidol and olanzapine | Cross-sectional study | 50 patients with SCZ | Chronic OLA administration was associated with lower TBARS levels, which are indicators of altered lipid peroxidation, and higher antioxidant values than treatment with HAL. | Singh et al., 2008 [29] |
| Risperidone and olanzapine | Longitudinal study | 179 psychotic patients (93 of them completed the study) | Three-month treatment with RIS and OLA was associated with an increase in blood GSH, α-tocopherol, and ascorbic acid levels parallelized by a decrease in malondialdehyde concentration, a lipid peroxidation marker. | Zerin Khan et al., 2018 [31] |
| Clozapine, olanzapine, haloperidol, quetiapine, risperidone and ziprasidone | In vitro preclinical study | Human neutrophils | CLO and OLA showed to be more effective in scavenging superoxide anion on the respiratory burst and stabilizing free radicals than RIS and ZIP, probably due to the amino group part of their chemical structure, whereas QTP and HAL completely lacked antioxidant effects | Brinholi et al., 2016 [211] |
| Olanzapine and clozapine | Preclinical study | KO male mice for the Ddo | OLA, different from CLO, CPZ, and HAL, was seen to inhibit the activity of the human DASPO. | Sacchi et al., 2017 [206] |
| Perphenazine, clozapine, and risperidone | Observational study | 100 patients with SCZ | CLO showed to enhance SOD levels and reduce lipid peroxidation in comparison to PPZ and RIS. | Hendouei et al., 2018 [30] |
| Xanomeline | Preclinical study | Rat cortical neurons | Xanomeline was effective in protecting cells from OGD-induced injury and cell apoptosis, inhibiting ROS production induced by OGD, and preventing p-AKT reduction | Xin et al., 2020 [215] |
6. Oxidative System, Dopamine, and Mitochondrial Dysfunction: An Interplay Possibly Relevant for Schizophrenia and Antipsychotic Treatment
7. Mitochondrial Dysfunction, Synaptic Modulation, and Antipsychotic Treatment: Implication for Schizophrenia
8. Antipsychotic Treatment Effects on Mitochondrial Function and Intracellular Signaling
9. Antioxidants: A Possible Add-on Strategy in the Treatment of Schizophrenia?
| Type of Antioxidant | Antipsychotics | Main Outcomes | References |
|---|---|---|---|
| Allopurinol | Haloperidol (46 patients with SCZ) | There was a significant superiority over the antipsychotic alone in the treatment of positive symptoms and PANSS total score. | Akhondzadeh et al., 2005 [296] |
| Olanzapine (13 patients) Risperidone (13 patients) Clozapine (18 patients) Quetiapine (5 patients) Aripiprazole (4 patients) Haloperidol (7 patients) Patients with SCZ enrolled: 59 | Patients treated with allopurinol rated themselves as more improved than the placebo group. | Dickerson et al., 2009 [299] | |
| Ginkgo biloba | Haloperidol (109 patients with SCZ) | EGb significantly decreased SANS and SAPS scores when added to haloperidol. | Zhang et al., 2001 [300] |
| Clozapine (42 patients with TRS) | EGb, in addition to clozapine, improves negative symptoms in patients with TRS. | Doruk et al., 2008 [302] | |
| Vitamin E and EPUFAs | Haloperidol decanoate depot injection (52 patients with SCZ) | Vitamin E decreased GSSG concentration and motor delay on the overall scale of PANSS, while EPUFAs increased GSH. | Bošković et al., 2016 [303] |
| Vitamin E | Fluphenazine decanoate (40 patients with SCZ and TD) | The reduction in AIMS score induced by vitamin E in comparison to placebo. | Adler et al., 1998 [324] |
| Vitamin C | Olanzapine (10 mg/day) or quetiapine (200 mg/day) or ziprasidone (40 mg/day). Patients with SCZ enrolled: 40 | After vitamin C supplementation in patients treated with second-generation antipsychotics, blood levels of malondialdehyde and ascorbic acid were increased compared to the placebo, with an improvement in the BPRS score. | Dakhale et al., 2005 [304] |
| NAC | Clozapine (45% of participants) and olanzapine (20% of participants), other atypical antipsychotics (risperidone, quetiapine, and aripiprazole), and typical depot antipsychotics. Patients enrolled: 140 | The administration of 1 g twice daily of NAC as an add-on treatment was associated with a reduction in PANSS total, negative, and general scores as well as an improvement in akathisia. | Berk et al., 2008 [306] |
| Risperidone (up to 6 mg/d) for 8 weeks | NAC decreased the PANSS total and negative scores compared to the placebo without any difference in the frequency of adverse effects. | Farokhnia et al., 2013 [307] | |
| Chlorpromazine equivalent to 300–1000 mg (except clozapine). Patients with SCZ enrolled: 84 | NAC-treated patients exhibited an improvement in cognitive functions and ameliorated PANSS negative and positive scores, along with improvement in attention, short-term and working memory, executive functioning, and speed of processing. | Sepehrmanesh et al., 2018 [308] | |
| Clozapine, olanzapine, and other antipsychotics. Patients with SCZ enrolled: 121 | Patients with SCZ might benefit more than others from additional treatment with NAC in terms of functioning and positive symptoms. | Rapado-Castro et al., 2017 [309] | |
| Risperidone, quetiapine, clozapine, olanzapine. Patients with SCZ enrolled: 11 | NAC administration was associated with improvement in mismatch negativity, measured by auditory evoked potentials, and there was no difference in the PANSS total or subscale scores in comparison to the placebo. | Lavoie et al., 2008 [311] | |
| Quetiapine (8 patients) Clozapine (1 patient) Aripiprazole (3 patients) Amisulpride (2 patients) Risperidone (3 patients) Olanzapine (2 patients). Patients with SCZ enrolled: 20 | Six-week add-on treatment with NAC in early psychosis patients was effective in ameliorating processing speed and positive symptoms, along with an increase in white matter integrity in the fornix measured by generalized fractional anisotropy. | Klauser et al., 2018 [313] | |
| Clozapine Patients with enduring psychotic symptoms enrolled: 84 | NAC, administered at 2 g/day, was not effective in improving negative symptoms, cognition, and quality of life at 8, 24, and 52 weeks in TRS patients taking clozapine. | Neill et al., 2022 [318] |
10. Discussion
Limitations
11. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- de Bartolomeis, A.; Vellucci, L.; De Simone, G.; Mazza, B.; Barone, A.; Ciccarelli, M. Dysregulated Signaling at Postsynaptic Density: A Systematic Review and Translational Appraisal for the Pathophysiology, Clinics, and Antipsychotics′ Treatment of Schizophrenia. Cells 2023, 12, 574. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, T.X.; Hallett, P.J.; Watanabe, M.; Grant, S.G.; Isacson, O.; Yao, W.D. PSD-95 uncouples dopamine-glutamate interaction in the D1/PSD-95/NMDA receptor complex. J. Neurosci. 2009, 29, 2948–2960. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Saur, T.; Duke, A.N.; Grant, S.G.N.; Platt, D.M.; Rowlett, J.K.; Isacson, O.; Yao, W.-D. Motor Impairments, Striatal Degeneration, and Altered Dopamine-Glutamate Interplay in Mice Lacking PSD-95. J. Neurogenet. 2014, 28, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Obi-Nagata, K.; Temma, Y.; Hayashi-Takagi, A. Synaptic functions and their disruption in schizophrenia: From clinical evidence to synaptic optogenetics in an animal model. Proc. Jpn. Acad. Ser. B 2019, 95, 179–197. [Google Scholar] [CrossRef]
- De Rosa, A.; Fontana, A.; Nuzzo, T.; Garofalo, M.; Di Maio, A.; Punzo, D.; Copetti, M.; Bertolino, A.; Errico, F.; Rampino, A.; et al. Machine Learning algorithm unveils glutamatergic alterations in the post-mortem schizophrenia brain. Schizophrenia 2022, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- de Bartolomeis, A.; De Simone, G.; Ciccarelli, M.; Castiello, A.; Mazza, B.; Vellucci, L.; Barone, A. Antipsychotics-Induced Changes in Synaptic Architecture and Functional Connectivity: Translational Implications for Treatment Response and Resistance. Biomedicines 2022, 10, 3183. [Google Scholar] [CrossRef]
- Friston, K.J.; Frith, C.D. Schizophrenia: A disconnection syndrome? Clin. Neurosci. 1995, 3, 89–97. [Google Scholar]
- De Greef, R.; Maloney, A.; Gisleskog, P.O.; Schoemaker, J.; Panagides, J. Dopamine D2 Occupancy as a Biomarker for Antipsychotics: Quantifying the Relationship with Efficacy and Extrapyramidal Symptoms. AAPS J. 2011, 13, 121–130. [Google Scholar] [CrossRef]
- Breier, A.; Su, T.P.; Saunders, R.; Carson, R.E.; Kolachana, B.S.; de Bartolomeis, A.; Weinberger, D.R.; Weisenfeld, N.; Malhotra, A.K.; Eckelman, W.C.; et al. Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: Evidence from a novel positron emission tomography method. Proc. Natl. Acad. Sci. USA 1997, 94, 2569–2574. [Google Scholar] [CrossRef]
- Caravaggio, F.; Porco, N.; Kim, J.; Torres Carmona, E.; Brown, E.; Iwata, Y.; Nakajima, S.; Gerretsen, P.; Remington, G.; Graff Guerrero, A. Measuring amphetamine-induced dopamine release in humans: A comparative meta-analysis of [(11) C]-raclopride and [(11) C]-(+)-PHNO studies. Synapse 2021, 75, e22195. [Google Scholar] [CrossRef]
- Howes, O.D.; Bose, S.K.; Turkheimer, F.; Valli, I.; Egerton, A.; Valmaggia, L.R.; Murray, R.M.; McGuire, P. Dopamine synthesis capacity before onset of psychosis: A prospective [18F]-DOPA PET imaging study. Am. J. Psychiatry 2011, 168, 1311–1317. [Google Scholar] [CrossRef]
- de Bartolomeis, A.; Barone, A.; Begni, V.; Riva, M.A. Present and future antipsychotic drugs: A systematic review of the putative mechanisms of action for efficacy and a critical appraisal under a translational perspective. Pharmacol. Res. 2022, 176, 106078. [Google Scholar] [CrossRef]
- Gründer, G.; Hippius, H.; Carlsson, A. The ‘atypicality’ of antipsychotics: A concept re-examined and re-defined. Nat. Rev. Drug Discov. 2009, 8, 197–202. [Google Scholar] [CrossRef]
- Ramachandraiah, C.T.; Subramaniam, N.; Tancer, M. The story of antipsychotics: Past and present. Indian J. Psychiatry 2009, 51, 324–326. [Google Scholar] [CrossRef]
- Markowitz, J.S.; Brown, C.S.; Moore, T.R. Atypical antipsychotics. Part I: Pharmacology, pharmacokinetics, and efficacy. Ann. Pharmacother. 1999, 33, 73–85. [Google Scholar] [CrossRef]
- Hippius, H. A historical perspective of clozapine. J. Clin. Psychiatry 1999, 60 (Suppl. 12), 22–23. [Google Scholar] [PubMed]
- King, C.; Voruganti, L.N. What’s in a name? The evolution of the nomenclature of antipsychotic drugs. J. Psychiatry Neurosci. JPN 2002, 27, 168–175. [Google Scholar]
- Crilly, J. The history of clozapine and its emergence in the US market: A review and analysis. Hist. Psychiatry 2007, 18, 39–60. [Google Scholar] [CrossRef] [PubMed]
- McKenna, P.J.; Bailey, P.E. The Strange Story of Clozapine. Br. J. Psychiatry 1993, 162, 32–37. [Google Scholar] [CrossRef]
- Kapur, S.; Seeman, P. Antipsychotic agents differ in how fast they come off the dopamine D2 receptors. Implications for atypical antipsychotic action. J. Psychiatry Neurosci. 2000, 25, 161–166. [Google Scholar] [PubMed]
- Girgis, R.R.; Slifstein, M.; D′Souza, D.; Lee, Y.; Periclou, A.; Ghahramani, P.; Laszlovszky, I.; Durgam, S.; Adham, N.; Nabulsi, N.; et al. Preferential binding to dopamine D3 over D2 receptors by cariprazine in patients with schizophrenia using PET with the D3/D2 receptor ligand [(11)C]-(+)-PHNO. Psychopharmacology 2016, 233, 3503–3512. [Google Scholar] [CrossRef]
- Correll, C.U.; Demyttenaere, K.; Fagiolini, A.; Hajak, G.; Pallanti, S.; Racagni, G.; Singh, S. Cariprazine in the management of negative symptoms of schizophrenia: State of the art and future perspectives. Future Neurol. 2020, 15, FNL52. [Google Scholar] [CrossRef]
- Mailman, R.B.; Murthy, V. Third generation antipsychotic drugs: Partial agonism or receptor functional selectivity? Curr. Pharm. Des. 2010, 16, 488–501. [Google Scholar] [CrossRef]
- Scaini, G.; Quevedo, J.; Velligan, D.; Roberts, D.L.; Raventos, H.; Walss Bass, C. Second generation antipsychotic-induced mitochondrial alterations: Implications for increased risk of metabolic syndrome in patients with schizophrenia. Eur. Neuropsychopharmacol. 2018, 28, 369–380. [Google Scholar] [CrossRef]
- Steiner, J.; Martins de Souza, D.; Schiltz, K.; Sarnyai, Z.; Westphal, S.; Isermann, B.; Dobrowolny, H.; Turck, C.W.; Bogerts, B.; Bernstein, H.G.; et al. Clozapine promotes glycolysis and myelin lipid synthesis in cultured oligodendrocytes. Front. Cell. Neurosci. 2014, 8, 384. [Google Scholar] [CrossRef] [PubMed]
- Whitehurst, T.; Howes, O. The role of mitochondria in the pathophysiology of schizophrenia: A critical review of the evidence focusing on mitochondrial complex one. Neurosci. Biobehav. Rev. 2022, 132, 449–464. [Google Scholar] [CrossRef] [PubMed]
- Caruso, G.; Grasso, M.; Fidilio, A.; Tascedda, F.; Drago, F.; Caraci, F. Antioxidant Properties of Second-Generation Antipsychotics: Focus on Microglia. Pharmaceuticals 2020, 13, 457. [Google Scholar] [CrossRef] [PubMed]
- de Bartolomeis, A.; Barone, A.; Vellucci, L.; Mazza, B.; Austin, M.C.; Iasevoli, F.; Ciccarelli, M. Linking Inflammation, Aberrant Glutamate-Dopamine Interaction, and Post-synaptic Changes: Translational Relevance for Schizophrenia and Antipsychotic Treatment: A Systematic Review. Mol. Neurobiol. 2022, 59, 6460–6501. [Google Scholar] [CrossRef] [PubMed]
- Singh, O.P.; Chakraborty, I.; Dasgupta, A.; Datta, S. A comparative study of oxidative stress and interrelationship of important antioxidants in haloperidol and olanzapine treated patients suffering from schizophrenia. Indian J. Psychiatry 2008, 50, 171–176. [Google Scholar] [CrossRef]
- Hendouei, N.; Farnia, S.; Mohseni, F.; Salehi, A.; Bagheri, M.; Shadfar, F.; Barzegar, F.; Hoseini, S.D.; Charati, J.Y.; Shaki, F. Alterations in oxidative stress markers and its correlation with clinical findings in schizophrenic patients consuming perphenazine, clozapine and risperidone. Biomed. Pharmacother. 2018, 103, 965–972. [Google Scholar] [CrossRef]
- Zerin Khan, F.; Sultana, S.P.; Akhter, N.; Mosaddek, A.S.M. Effect of Olanzapine and Risperidone on Oxidative Stress in Schizophrenia Patients. Int. Biol. Biomed. J. 2018, 4, 89–97. [Google Scholar]
- Choi, K.H.; Higgs, B.W.; Weis, S.; Song, J.; Llenos, I.C.; Dulay, J.R.; Yolken, R.H.; Webster, M.J. Effects of typical and atypical antipsychotic drugs on gene expression profiles in the liver of schizophrenia subjects. BMC Psychiatry 2009, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Müller, N.; Weidinger, E.; Leitner, B.; Schwarz, M.J. The role of inflammation in schizophrenia. Front. Neurosci. 2015, 9, 372. [Google Scholar] [CrossRef] [PubMed]
- Müller, N. Inflammation in Schizophrenia: Pathogenetic Aspects and Therapeutic Considerations. Schizophr. Bull. 2018, 44, 973–982. [Google Scholar] [CrossRef]
- Parellada, E.; Gassó, P. Glutamate and microglia activation as a driver of dendritic apoptosis: A core pathophysiological mechanism to understand schizophrenia. Transl. Psychiatry 2021, 11, 271. [Google Scholar] [CrossRef]
- Anderson, G.; Berk, M.; Dodd, S.; Bechter, K.; Altamura, A.C.; Dell′osso, B.; Kanba, S.; Monji, A.; Fatemi, S.H.; Buckley, P.; et al. Immuno-inflammatory, oxidative and nitrosative stress, and neuroprogressive pathways in the etiology, course and treatment of schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 42, 1–4. [Google Scholar] [CrossRef]
- Tsai, S.Y.; Gildengers, A.G.; Hsu, J.L.; Chung, K.H.; Chen, P.H.; Huang, Y.J. Inflammation associated with volume reduction in the gray matter and hippocampus of older patients with bipolar disorder. J. Affect. Disord. 2019, 244, 60–66. [Google Scholar] [CrossRef]
- Wang, J.; Knaut, H. Chemokine signaling in development and disease. Development 2014, 141, 4199–4205. [Google Scholar] [CrossRef]
- Hindley, S.; Juurlink, B.H.; Gysbers, J.W.; Middlemiss, P.J.; Herman, M.A.; Rathbone, M.P. Nitric oxide donors enhance neurotrophin-induced neurite outgrowth through a cGMP-dependent mechanism. J. Neurosci. Res. 1997, 47, 427–439. [Google Scholar] [CrossRef]
- Ermakov, E.A.; Mednova, I.A.; Boiko, A.S.; Buneva, V.N.; Ivanova, S.A. Chemokine Dysregulation and Neuroinflammation in Schizophrenia: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 2215. [Google Scholar] [CrossRef]
- Schwarz, E.; Maukonen, J.; Hyytiäinen, T.; Kieseppä, T.; Orešič, M.; Sabunciyan, S.; Mantere, O.; Saarela, M.; Yolken, R.; Suvisaari, J. Analysis of microbiota in first episode psychosis identifies preliminary associations with symptom severity and treatment response. Schizophr. Res. 2018, 192, 398–403. [Google Scholar] [CrossRef]
- Szeligowski, T.; Yun, A.L.; Lennox, B.R.; Burnet, P.W.J. The Gut Microbiome and Schizophrenia: The Current State of the Field and Clinical Applications. Front. Psychiatry 2020, 11, 156. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef] [PubMed]
- Turano, A.; McAuley, E.M.; Muench, M.C.; Schwarz, J.M. Examining the impact of neuroimmune dysregulation on social behavior of male and female juvenile rats. Behav. Brain Res. 2021, 415, 113449. [Google Scholar] [CrossRef]
- Vidal, P.M.; Pacheco, R. The Cross-Talk Between the Dopaminergic and the Immune System Involved in Schizophrenia. Front. Pharmacol. 2020, 11, 394. [Google Scholar] [CrossRef] [PubMed]
- Trépanier, M.O.; Hopperton, K.E.; Mizrahi, R.; Mechawar, N.; Bazinet, R.P. Postmortem evidence of cerebral inflammation in schizophrenia: A systematic review. Mol. Psychiatry 2016, 21, 1009–1026. [Google Scholar] [CrossRef]
- Lodge, D.J.; Grace, A.A. Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. J. Neurosci. 2007, 27, 11424–11430. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.H.; Vyas, N.S.; Puri, B.K.; Nijran, K.S.; Al Nahhas, A. Positron emission tomography in schizophrenia: A new perspective. J. Nucl. Med. 2010, 51, 511–520. [Google Scholar] [CrossRef]
- Weinstein, J.J.; Chohan, M.O.; Slifstein, M.; Kegeles, L.S.; Moore, H.; Abi Dargham, A. Pathway-Specific Dopamine Abnormalities in Schizophrenia. Biol. Psychiatry 2017, 81, 31–42. [Google Scholar] [CrossRef]
- Panayi, M.C.; Boerner, T.; Jahans-Price, T.; Huber, A.; Sprengel, R.; Gilmour, G.; Sanderson, D.J.; Harrison, P.J.; Walton, M.E.; Bannerman, D.M. Glutamatergic dysfunction leads to a hyper-dopaminergic phenotype through deficits in short-term habituation: A mechanism for aberrant salience. Mol. Psychiatry 2023, 28, 579–587. [Google Scholar] [CrossRef] [PubMed]
- da Silva Alves, F.; Figee, M.; van Amelsvoort, T.; Veltman, D.; de Haan, L. The revised dopamine hypothesis of schizophrenia: Evidence from pharmacological MRI studies with atypical antipsychotic medication. Psychopharmacol. Bull. 2008, 41, 121–132. [Google Scholar] [PubMed]
- North, H.F.; Weissleder, C.; Fullerton, J.M.; Sager, R.; Webster, M.J.; Weickert, C.S. A schizophrenia subgroup with elevated inflammation displays reduced microglia, increased peripheral immune cell and altered neurogenesis marker gene expression in the subependymal zone. Transl. Psychiatry 2021, 11, 635. [Google Scholar] [CrossRef]
- Khandaker, G.M.; Cousins, L.; Deakin, J.; Lennox, B.R.; Yolken, R.; Jones, P.B. Inflammation and immunity in schizophrenia: Implications for pathophysiology and treatment. Lancet. Psychiatry 2015, 2, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Nasi, G.; Ahmed, T.; Rasini, E.; Fenoglio, D.; Marino, F.; Filaci, G.; Cosentino, M. Dopamine inhibits human CD8+ Treg function through D(1)-like dopaminergic receptors. J. Neuroimmunol. 2019, 332, 233–241. [Google Scholar] [CrossRef]
- Levite, M. Dopamine and T cells: Dopamine receptors and potent effects on T cells, dopamine production in T cells, and abnormalities in the dopaminergic system in T cells in autoimmune, neurological and psychiatric diseases. Acta Physiol. 2016, 216, 42–89. [Google Scholar] [CrossRef]
- Mueller, H.T.; Haroutunian, V.; Davis, K.L.; Meador Woodruff, J.H. Expression of the ionotropic glutamate receptor subunits and NMDA receptor-associated intracellular proteins in the substantia nigra in schizophrenia. Brain Research. Mol. Brain Res. 2004, 121, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Ji, E.; Boerrigter, D.; Cai, H.Q.; Lloyd, D.; Bruggemann, J.; O′Donnell, M.; Galletly, C.; Lloyd, A.; Liu, D.; Lenroot, R.; et al. Peripheral complement is increased in schizophrenia and inversely related to cortical thickness. Brain Behav. Immun. 2022, 101, 423–434. [Google Scholar] [CrossRef]
- Sellgren, C.M.; Gracias, J.; Watmuff, B.; Biag, J.D.; Thanos, J.M.; Whittredge, P.B.; Fu, T.; Worringer, K.; Brown, H.E.; Wang, J.; et al. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat. Neurosci. 2019, 22, 374–385. [Google Scholar] [CrossRef]
- Sekar, A.; Bialas, A.R.; de Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; Van Doren, V.; et al. Schizophrenia risk from complex variation of complement component 4. Nature 2016, 530, 177–183. [Google Scholar] [CrossRef]
- Comer, A.L.; Carrier, M.; Tremblay, M.È.; Cruz Martín, A. The Inflamed Brain in Schizophrenia: The Convergence of Genetic and Environmental Risk Factors That Lead to Uncontrolled Neuroinflammation. Front. Cell. Neurosci. 2020, 14, 274. [Google Scholar] [CrossRef]
- Sheridan, S.D.; Horng, J.E.; Perlis, R.H. Patient-Derived In Vitro Models of Microglial Function and Synaptic Engulfment in Schizophrenia. Biol. Psychiatry 2022, 92, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Monji, A.; Kato, T.A.; Mizoguchi, Y.; Horikawa, H.; Seki, Y.; Kasai, M.; Yamauchi, Y.; Yamada, S.; Kanba, S. Neuroinflammation in schizophrenia especially focused on the role of microglia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 42, 115–121. [Google Scholar] [CrossRef]
- Girgis, R.R.; Kumar, S.S.; Brown, A.S. The cytokine model of schizophrenia: Emerging therapeutic strategies. Biol. Psychiatry 2014, 75, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Reale, M.; Costantini, E.; Greig, N.H. Cytokine Imbalance in Schizophrenia. From Research to Clinic: Potential Implications for Treatment. Front. Psychiatry 2021, 12, 536257. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, J.K.; Lewis, D.A. Mapping the Consequences of Impaired Synaptic Plasticity in Schizophrenia through Development: An Integrative Model for Diverse Clinical Features. Trends Cogn. Sci. 2017, 21, 760–778. [Google Scholar] [CrossRef]
- Hall, J.; Trent, S.; Thomas, K.L.; O′Donovan, M.C.; Owen, M.J. Genetic risk for schizophrenia: Convergence on synaptic pathways involved in plasticity. Biol. Psychiatry 2015, 77, 52–58. [Google Scholar] [CrossRef]
- Lisman, J.E.; Coyle, J.T.; Green, R.W.; Javitt, D.C.; Benes, F.M.; Heckers, S.; Grace, A.A. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 2008, 31, 234–242. [Google Scholar] [CrossRef]
- Lewis, D.A. Cortical circuit dysfunction and cognitive deficits in schizophrenia--implications for preemptive interventions. Eur. J. Neurosci. 2012, 35, 1871–1878. [Google Scholar] [CrossRef]
- Marín, O. Interneuron dysfunction in psychiatric disorders. Nat. Rev. Neurosci. 2012, 13, 107–120. [Google Scholar] [CrossRef]
- Ertürk, A.; Wang, Y.; Sheng, M. Local pruning of dendrites and spines by caspase-3-dependent and proteasome-limited mechanisms. J. Neurosci. 2014, 34, 1672–1688. [Google Scholar] [CrossRef]
- Do, K.Q.; Cabungcal, J.H.; Frank, A.; Steullet, P.; Cuenod, M. Redox dysregulation, neurodevelopment, and schizophrenia. Curr. Opin. Neurobiol. 2009, 19, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Sawa, A.; Seidman, L.J. Is prophylactic psychiatry around the corner? combating adolescent oxidative stress for adult psychosis and schizophrenia. Neuron 2014, 83, 991–993. [Google Scholar] [CrossRef] [PubMed]
- Misiak, B.; Bartoli, F.; Carrà, G.; Stańczykiewicz, B.; Gładka, A.; Frydecka, D.; Samochowiec, J.; Jarosz, K.; Hadryś, T.; Miller, B.J. Immune-inflammatory markers and psychosis risk: A systematic review and meta-analysis. Psychoneuroendocrinology 2021, 127, 105200. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.L.; Mowry, B.J.; Pender, M.P.; Greer, J.M. Immune dysregulation and self-reactivity in schizophrenia: Do some cases of schizophrenia have an autoimmune basis? Immunol. Cell Biol. 2005, 83, 9–17. [Google Scholar] [CrossRef]
- Glantz, L.A.; Gilmore, J.H.; Lieberman, J.A.; Jarskog, L.F. Apoptotic mechanisms and the synaptic pathology of schizophrenia. Schizophr. Res. 2006, 81, 47–63. [Google Scholar] [CrossRef]
- Osimo, E.F.; Beck, K.; Reis Marques, T.; Howes, O.D. Synaptic loss in schizophrenia: A meta-analysis and systematic review of synaptic protein and mRNA measures. Mol. Psychiatry 2019, 24, 549–561. [Google Scholar] [CrossRef]
- Do, K.Q.; Cuenod, M.; Hensch, T.K. Targeting Oxidative Stress and Aberrant Critical Period Plasticity in the Developmental Trajectory to Schizophrenia. Schizophr. Bull. 2015, 41, 835–846. [Google Scholar] [CrossRef]
- Leza, J.C.; García Bueno, B.; Bioque, M.; Arango, C.; Parellada, M.; Do, K.; O′Donnell, P.; Bernardo, M. Inflammation in schizophrenia: A question of balance. Neurosci. Biobehav. Rev. 2015, 55, 612–626. [Google Scholar] [CrossRef]
- Funk, A.J.; Mielnik, C.A.; Koene, R.; Newburn, E.; Ramsey, A.J.; Lipska, B.K.; McCullumsmith, R.E. Postsynaptic Density-95 Isoform Abnormalities in Schizophrenia. Schizophr. Bull. 2017, 43, 891–899. [Google Scholar] [CrossRef]
- Barksdale, K.A.; Lahti, A.C.; Roberts, R.C. Synaptic proteins in the postmortem anterior cingulate cortex in schizophrenia: Relationship to treatment and treatment response. Neuropsychopharmacology 2014, 39, 2095–2103. [Google Scholar] [CrossRef]
- Browning, M.D.; Dudek, E.M.; Rapier, J.L.; Leonard, S.; Freedman, R. Significant reductions in synapsin but not synaptophysin specific activity in the brains of some schizophrenics. Biol. Psychiatry 1993, 34, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Catts, V.S.; Derminio, D.S.; Hahn, C.G.; Weickert, C.S. Postsynaptic density levels of the NMDA receptor NR1 subunit and PSD-95 protein in prefrontal cortex from people with schizophrenia. NPJ Schizophr. 2015, 1, 15037. [Google Scholar] [CrossRef]
- Davidsson, P.; Gottfries, J.; Bogdanovic, N.; Ekman, R.; Karlsson, I.; Gottfries, C.G.; Blennow, K. The synaptic-vesicle-specific proteins rab3a and synaptophysin are reduced in thalamus and related cortical brain regions in schizophrenic brains. Schizophr. Res. 1999, 40, 23–29. [Google Scholar] [CrossRef]
- Miyamoto, A.; Wake, H.; Moorhouse, A.J.; Nabekura, J. Microglia and synapse interactions: Fine tuning neural circuits and candidate molecules. Front. Cell. Neurosci. 2013, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef]
- Apam Castillejos, D.J.; Tendilla Beltrán, H.; Vázquez Roque, R.A.; Vázquez Hernández, A.J.; Fuentes Medel, E.; García Dolores, F.; Díaz, A.; Flores, G. Second-generation antipsychotic olanzapine attenuates behavioral and prefrontal cortex synaptic plasticity deficits in a neurodevelopmental schizophrenia-related rat model. J. Chem. Neuroanat. 2022, 125, 102166. [Google Scholar] [CrossRef]
- Boksa, P. Effects of prenatal infection on brain development and behavior: A review of findings from animal models. Brain Behav. Immun. 2010, 24, 881–897. [Google Scholar] [CrossRef] [PubMed]
- Chua, J.S.; Cowley, C.J.; Manavis, J.; Rofe, A.M.; Coyle, P. Prenatal exposure to lipopolysaccharide results in neurodevelopmental damage that is ameliorated by zinc in mice. Brain Behav. Immun. 2012, 26, 326–336. [Google Scholar] [CrossRef]
- Cannon, T.D. Microglial Activation and the Onset of Psychosis. Am. J. Psychiatry 2016, 173, 3–4. [Google Scholar] [CrossRef]
- Howes, O.D.; McCutcheon, R. Inflammation and the neural diathesis-stress hypothesis of schizophrenia: A reconceptualization. Transl. Psychiatry 2017, 7, e1024. [Google Scholar] [CrossRef]
- Glausier, J.R.; Lewis, D.A. Dendritic spine pathology in schizophrenia. Neuroscience 2013, 251, 90–107. [Google Scholar] [CrossRef]
- Smith, S.E.; Li, J.; Garbett, K.; Mirnics, K.; Patterson, P.H. Maternal immune activation alters fetal brain development through interleukin-6. J. Neurosci. 2007, 27, 10695–10702. [Google Scholar] [CrossRef]
- Pillinger, T.; D′Ambrosio, E.; McCutcheon, R.; Howes, O.D. Is psychosis a multisystem disorder? A meta-review of central nervous system, immune, cardiometabolic, and endocrine alterations in first-episode psychosis and perspective on potential models. Mol. Psychiatry 2019, 24, 776–794. [Google Scholar] [CrossRef]
- Miller, B.J.; Buckley, P.; Seabolt, W.; Mellor, A.; Kirkpatrick, B. Meta-analysis of cytokine alterations in schizophrenia: Clinical status and antipsychotic effects. Biol. Psychiatry 2011, 70, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.K.; Krebs, M.O.; Cox, D.; Guest, P.C.; Yolken, R.H.; Rahmoune, H.; Rothermundt, M.; Steiner, J.; Leweke, F.M.; van Beveren, N.J.; et al. Development of a blood-based molecular biomarker test for identification of schizophrenia before disease onset. Transl. Psychiatry 2015, 5, e601. [Google Scholar] [CrossRef] [PubMed]
- Pedrini, M.; Massuda, R.; Fries, G.R.; de Bittencourt Pasquali, M.A.; Schnorr, C.E.; Moreira, J.C.; Teixeira, A.L.; Lobato, M.I.; Walz, J.C.; Belmonte de Abreu, P.S.; et al. Similarities in serum oxidative stress markers and inflammatory cytokines in patients with overt schizophrenia at early and late stages of chronicity. J. Psychiatr. Res. 2012, 46, 819–824. [Google Scholar] [CrossRef]
- Ansari, Z.; Pawar, S.; Seetharaman, R. Neuroinflammation and oxidative stress in schizophrenia: Are these opportunities for repurposing? Postgrad. Med. 2022, 134, 187–199. [Google Scholar] [CrossRef]
- Goh, X.X.; Tang, P.Y.; Tee, S.F. Effects of antipsychotics on antioxidant defence system in patients with schizophrenia: A meta-analysis. Psychiatry Res. 2022, 309, 114429. [Google Scholar] [CrossRef]
- Yang, M.; Wang, C.; Zhao, G.; Kong, D.; Liu, L.; Yuan, S.; Chen, W.; Feng, C.; Li, Z. Comparative Analysis of the Pre- and Post-Medication Effects of Antipsychotic Agents on the Blood-Based Oxidative Stress Biomarkers in Patients with Schizophrenia: A Meta-Analysis. Curr. Neuropharmacol. 2023, 21, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhang, X.; Cong, Q.; Chen, D.; Yi, Z.; Huang, H.; Wang, C.; Li, M.; Zeng, R.; Liu, Y.; et al. miR143-3p-Mediated NRG-1-Dependent Mitochondrial Dysfunction Contributes to Olanzapine Resistance in Refractory Schizophrenia. Biol. Psychiatry 2022, 92, 419–433. [Google Scholar] [CrossRef]
- Liu, H.; Liu, H.; Jiang, S.; Su, L.; Lu, Y.; Chen, Z.; Li, X.; Li, X.; Wang, X.; Xiu, M.; et al. Sex-Specific Association between Antioxidant Defense System and Therapeutic Response to Risperidone in Schizophrenia: A Prospective Longitudinal Study. Curr. Neuropharmacol. 2022, 20, 1793–1803. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yu, R.; Gao, Y.; Li, X.; Guan, X.; Thomas, K.; Xiu, M.; Zhang, X. Antioxidant Enzymes and Weight Gain in Drug-naive First-episode Schizophrenia Patients Treated with Risperidone for 12 Weeks: A Prospective Longitudinal Study. Curr. Neuropharmacol. 2022, 20, 1774–1782. [Google Scholar] [CrossRef] [PubMed]
- Rambaud, V.; Marzo, A.; Chaumette, B. Oxidative Stress and Emergence of Psychosis. Antioxidants 2022, 11, 1870. [Google Scholar] [CrossRef] [PubMed]
- Clausen, M.V.; Hilbers, F.; Poulsen, H. The Structure and Function of the Na,K-ATPase Isoforms in Health and Disease. Front. Physiol. 2017, 8, 371. [Google Scholar] [CrossRef] [PubMed]
- Scavone, C.; Munhoz, C.D.; Kawamoto, E.M.; Glezer, I.; de Sá Lima, L.; Marcourakis, T.; Markus, R.P. Age-related changes in cyclic GMP and PKG-stimulated cerebellar Na,K-ATPase activity. Neurobiol. Aging 2005, 26, 907–916. [Google Scholar] [CrossRef]
- Corti, C.; Xuereb, J.H.; Crepaldi, L.; Corsi, M.; Michielin, F.; Ferraguti, F. Altered levels of glutamatergic receptors and Na+/K+ ATPase-α1 in the prefrontal cortex of subjects with schizophrenia. Schizophr. Res. 2011, 128, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Chávez, C.E.; Oyarzún, J.E.; Avendaño, B.C.; Mellado, L.A.; Inostroza, C.A.; Alvear, T.F.; Orellana, J.A. The Opening of Connexin 43 Hemichannels Alters Hippocampal Astrocyte Function and Neuronal Survival in Prenatally LPS-Exposed Adult Offspring. Front. Cell. Neurosci. 2019, 13, 460. [Google Scholar] [CrossRef]
- Olmos, G.; Lladó, J. Tumor necrosis factor alpha: A link between neuroinflammation and excitotoxicity. Mediat. Inflamm. 2014, 2014, 861231. [Google Scholar] [CrossRef] [PubMed]
- Allan, S.M.; Rothwell, N.J. Cytokines and acute neurodegeneration. Nat. Rev. Neurosci. 2001, 2, 734–744. [Google Scholar] [CrossRef]
- Meade, A.J.; Meloni, B.P.; Cross, J.; Bakker, A.J.; Fear, M.W.; Mastaglia, F.L.; Watt, P.M.; Knuckey, N.W. AP-1 inhibitory peptides are neuroprotective following acute glutamate excitotoxicity in primary cortical neuronal cultures. J. Neurochem. 2010, 112, 258–270. [Google Scholar] [CrossRef]
- Kwon, D.J.; Ju, S.M.; Youn, G.S.; Choi, S.Y.; Park, J. Suppression of iNOS and COX-2 expression by flavokawain A via blockade of NF-κB and AP-1 activation in RAW 264.7 macrophages. Food Chem. Toxicol. 2013, 58, 479–486. [Google Scholar] [CrossRef]
- Lopes Rocha, A.; Bezerra, T.O.; Zanotto, R.; Lages Nascimento, I.; Rodrigues, A.; Salum, C. The Antioxidant N-Acetyl-L-Cysteine Restores the Behavioral Deficits in a Neurodevelopmental Model of Schizophrenia Through a Mechanism That Involves Nitric Oxide. Front. Pharmacol. 2022, 13, 924955. [Google Scholar] [CrossRef]
- Musazzi, L.; Treccani, G.; Popoli, M. Functional and structural remodeling of glutamate synapses in prefrontal and frontal cortex induced by behavioral stress. Front. Psychiatry 2015, 6, 60. [Google Scholar] [CrossRef]
- Perez Cruz, C.; Müller Keuker, J.I.; Heilbronner, U.; Fuchs, E.; Flügge, G. Morphology of pyramidal neurons in the rat prefrontal cortex: Lateralized dendritic remodeling by chronic stress. Neural Plast. 2007, 2007, 46276. [Google Scholar] [CrossRef]
- Felger, J.C.; Cole, S.W.; Pace, T.W.; Hu, F.; Woolwine, B.J.; Doho, G.H.; Raison, C.L.; Miller, A.H. Molecular signatures of peripheral blood mononuclear cells during chronic interferon-α treatment: Relationship with depression and fatigue. Psychol. Med. 2012, 42, 1591–1603. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Knowlton, D.; Woodward, W.R.; Habecker, B.A. Regulation of noradrenergic function by inflammatory cytokines and depolarization. J. Neurochem. 2003, 86, 774–783. [Google Scholar] [CrossRef]
- Shi, W.; Meininger, C.J.; Haynes, T.E.; Hatakeyama, K.; Wu, G. Regulation of tetrahydrobiopterin synthesis and bioavailability in endothelial cells. Cell Biochem. Biophys. 2004, 41, 415–434. [Google Scholar] [CrossRef]
- Myken, A.N.; Ebdrup, B.H.; Sørensen, M.E.; Broberg, B.V.; Skjerbæk, M.W.; Glenthøj, B.Y.; Lykkesfeldt, J.; Nielsen, M.Ø. Lower Vitamin C Levels Are Associated With Less Improvement in Negative Symptoms in Initially Antipsychotic-Naïve Patients With First-Episode Psychosis. Int. J. Neuropsychopharmacol. 2022, 25, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Misiak, B.; Stańczykiewicz, B.; Kotowicz, K.; Rybakowski, J.K.; Samochowiec, J.; Frydecka, D. Cytokines and C-reactive protein alterations with respect to cognitive impairment in schizophrenia and bipolar disorder: A systematic review. Schizophr. Res. 2018, 192, 16–29. [Google Scholar] [CrossRef]
- Goldsmith, D.R.; Haroon, E.; Miller, A.H.; Strauss, G.P.; Buckley, P.F.; Miller, B.J. TNF-α and IL-6 are associated with the deficit syndrome and negative symptoms in patients with chronic schizophrenia. Schizophr. Res. 2018, 199, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Millan, M.J.; Andrieux, A.; Bartzokis, G.; Cadenhead, K.; Dazzan, P.; Fusar Poli, P.; Gallinat, J.; Giedd, J.; Grayson, D.R.; Heinrichs, M.; et al. Altering the course of schizophrenia: Progress and perspectives. Nat. Rev. Drug Discov. 2016, 15, 485–515. [Google Scholar] [CrossRef] [PubMed]
- Penzes, P.; Cahill, M.E.; Jones, K.A.; VanLeeuwen, J.E.; Woolfrey, K.M. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 2011, 14, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R. Schizophrenia: Susceptibility genes, dendritic-spine pathology and gray matter loss. Prog. Neurobiol. 2011, 95, 275–300. [Google Scholar] [CrossRef] [PubMed]
- Critchlow, H.M.; Maycox, P.R.; Skepper, J.N.; Krylova, O. Clozapine and haloperidol differentially regulate dendritic spine formation and synaptogenesis in rat hippocampal neurons. Mol. Cell. Neurosci. 2006, 32, 356–365. [Google Scholar] [CrossRef]
- Lundberg, M.; Curbo, S.; Bohman, H.; Agartz, I.; Ögren, S.O.; Patrone, C.; Mansouri, S. Clozapine protects adult neural stem cells from ketamine-induced cell death in correlation with decreased apoptosis and autophagy. Biosci. Rep. 2020, 40, BSR20193156. [Google Scholar] [CrossRef]
- Schwieler, L.; Engberg, G.; Erhardt, S. Clozapine modulates midbrain dopamine neuron firing via interaction with the NMDA receptor complex. Synapse 2004, 52, 114–122. [Google Scholar] [CrossRef] [PubMed]
- de Bartolomeis, A.; Manchia, M.; Marmo, F.; Vellucci, L.; Iasevoli, F.; Barone, A. Glycine Signaling in the Framework of Dopamine-Glutamate Interaction and Postsynaptic Density. Implications for Treatment-Resistant Schizophrenia. Front. Psychiatry 2020, 11, 369. [Google Scholar] [CrossRef]
- Mishra, A.; Reeta, K.H.; Sarangi, S.C.; Maiti, R.; Sood, M. Effect of add-on alpha lipoic acid on psychopathology in patients with treatment-resistant schizophrenia: A pilot randomized double-blind placebo-controlled trial. Psychopharmacology 2022, 239, 3525–3535. [Google Scholar] [CrossRef]
- De Lima, D.N., Jr.; Costa Filho, C.W.L.; Frota, I.J.; de Oliveira, A.L.B.; Menezes, C.E.S.; Chaves Filho, A.J.M.; Viana, G.A.; Campos, E.M.; Collares, M.; de Queiroz, M.G.R.; et al. α-Lipoic Acid as Adjunctive Treatment for Schizophrenia: A Randomized Double-Blind Study. J. Clin. Psychopharmacol. 2023, 43, 39–45. [Google Scholar] [CrossRef]
- Park, J.H.; Park, H.J.; Lee, S.E.; Kim, Y.S.; Jang, G.Y.; Han, H.D.; Jung, I.D.; Shin, K.C.; Bae, Y.M.; Kang, T.H.; et al. Repositioning of the antipsychotic drug TFP for sepsis treatment. J. Mol. Med. 2019, 97, 647–658. [Google Scholar] [CrossRef]
- Torres Rosas, R.; Yehia, G.; Peña, G.; Mishra, P.; del Rocio Thompson Bonilla, M.; Moreno Eutimio, M.A.; Arriaga Pizano, L.A.; Isibasi, A.; Ulloa, L. Dopamine mediates vagal modulation of the immune system by electroacupuncture. Nat. Med. 2014, 20, 291–295. [Google Scholar] [CrossRef]
- Muller, N.; Schwarz, M. Schizophrenia as an inflammation-mediated dysbalance of glutamatergic neurotransmission. Neurotox. Res. 2006, 10, 131–148. [Google Scholar] [CrossRef] [PubMed]
- Müller, N.; Riedel, M.; Schwarz, M.J. Psychotropic effects of COX-2 inhibitors--a possible new approach for the treatment of psychiatric disorders. Pharmacopsychiatry 2004, 37, 266–269. [Google Scholar] [CrossRef]
- Buonaguro, E.F.; Tomasetti, C.; Chiodini, P.; Marmo, F.; Latte, G.; Rossi, R.; Avvisati, L.; Iasevoli, F.; de Bartolomeis, A. Postsynaptic density protein transcripts are differentially modulated by minocycline alone or in add-on to haloperidol: Implications for treatment resistant schizophrenia. J. Psychopharmacol. 2017, 31, 406–417. [Google Scholar] [CrossRef]
- Andreazza, A.C.; Shao, L.; Wang, J.F.; Young, L.T. Mitochondrial complex I activity and oxidative damage to mitochondrial proteins in the prefrontal cortex of patients with bipolar disorder. Arch. Gen. Psychiatry 2010, 67, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Rollins, B.L.; Morgan, L.; Hjelm, B.E.; Sequeira, A.; Schatzberg, A.F.; Barchas, J.D.; Lee, F.S.; Myers, R.M.; Watson, S.J.; Akil, H.; et al. Mitochondrial Complex I Deficiency in Schizophrenia and Bipolar Disorder and Medication Influence. Complex Psychiatry 2017, 3, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Maurer, I.; Zierz, S.; Möller, H. Evidence for a mitochondrial oxidative phosphorylation defect in brains from patients with schizophrenia. Schizophr. Res. 2001, 48, 125–136. [Google Scholar] [CrossRef]
- Arion, D.; Corradi, J.P.; Tang, S.; Datta, D.; Boothe, F.; He, A.; Cacace, A.M.; Zaczek, R.; Albright, C.F.; Tseng, G.; et al. Distinctive transcriptome alterations of prefrontal pyramidal neurons in schizophrenia and schizoaffective disorder. Mol. Psychiatry 2015, 20, 1397–1405. [Google Scholar] [CrossRef]
- Altar, C.A.; Jurata, L.W.; Charles, V.; Lemire, A.; Liu, P.; Bukhman, Y.; Young, T.A.; Bullard, J.; Yokoe, H.; Webster, M.J.; et al. Deficient hippocampal neuron expression of proteasome, ubiquitin, and mitochondrial genes in multiple schizophrenia cohorts. Biol. Psychiatry 2005, 58, 85–96. [Google Scholar] [CrossRef]
- Ben Shachar, D.; Karry, R. Neuroanatomical pattern of mitochondrial complex I pathology varies between schizophrenia, bipolar disorder and major depression. PLoS ONE 2008, 3, e3676. [Google Scholar] [CrossRef]
- Marí, M.; Morales, A.; Colell, A.; García Ruiz, C.; Fernández Checa, J.C. Mitochondrial glutathione, a key survival antioxidant. Antioxid. Redox Signal. 2009, 11, 2685–2700. [Google Scholar] [CrossRef]
- Semenovich, D.S.; Plotnikov, E.Y.; Titko, O.V.; Lukiyenko, E.P.; Kanunnikova, N.P. Effects of Panthenol and N-Acetylcysteine on Changes in the Redox State of Brain Mitochondria under Oxidative Stress In Vitro. Antioxidants 2021, 10, 1699. [Google Scholar] [CrossRef]
- Palaniyappan, L.; Park, M.T.M.; Jeon, P.; Limongi, R.; Yang, K.; Sawa, A.; Théberge, J. Is There a Glutathione Centered Redox Dysregulation Subtype of Schizophrenia? Antioxidants 2021, 10, 1703. [Google Scholar] [CrossRef] [PubMed]
- Cabungcal, J.H.; Steullet, P.; Morishita, H.; Kraftsik, R.; Cuenod, M.; Hensch, T.K.; Do, K.Q. Perineuronal nets protect fast-spiking interneurons against oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 9130–9135. [Google Scholar] [CrossRef]
- Cabungcal, J.H.; Steullet, P.; Kraftsik, R.; Cuenod, M.; Do, K.Q. A developmental redox dysregulation leads to spatio-temporal deficit of parvalbumin neuron circuitry in a schizophrenia mouse model. Schizophr. Res. 2019, 213, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Fernandez Fernandez, S.; Bobo Jimenez, V.; Requejo Aguilar, R.; Gonzalez Fernandez, S.; Resch, M.; Carabias Carrasco, M.; Ros, J.; Almeida, A.; Bolaños, J.P. Hippocampal neurons require a large pool of glutathione to sustain dendrite integrity and cognitive function. Redox Biol. 2018, 19, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Grima, G.; Benz, B.; Parpura, V.; Cuénod, M.; Do, K.Q. Dopamine-induced oxidative stress in neurons with glutathione deficit: Implication for schizophrenia. Schizophr. Res. 2003, 62, 213–224. [Google Scholar] [CrossRef]
- Smith, G.A.; Lin, T.H.; Sheehan, A.E.; Van der Goes van Naters, W.; Neukomm, L.J.; Graves, H.K.; Bis Brewer, D.M.; Züchner, S.; Freeman, M.R. Glutathione S-Transferase Regulates Mitochondrial Populations in Axons through Increased Glutathione Oxidation. Neuron 2019, 103, 52–65.e56. [Google Scholar] [CrossRef]
- Herfarth, C. Working methods and tasks of the oncologic workshop of Ulm. ZFA Z. Fur Allg. 1976, 52, 1128–1130. [Google Scholar]
- Monin, A.; Baumann, P.S.; Griffa, A.; Xin, L.; Mekle, R.; Fournier, M.; Butticaz, C.; Klaey, M.; Cabungcal, J.H.; Steullet, P.; et al. Glutathione deficit impairs myelin maturation: Relevance for white matter integrity in schizophrenia patients. Mol. Psychiatry 2015, 20, 827–838. [Google Scholar] [CrossRef]
- Corcoba, A.; Steullet, P.; Duarte, J.M.; Van de Looij, Y.; Monin, A.; Cuenod, M.; Gruetter, R.; Do, K.Q. Glutathione Deficit Affects the Integrity and Function of the Fimbria/Fornix and Anterior Commissure in Mice: Relevance for Schizophrenia. Int. J. Neuropsychopharmacol. 2015, 19, pyv110. [Google Scholar] [CrossRef]
- Kilanczyk, E.; Saraswat Ohri, S.; Whittemore, S.R.; Hetman, M. Antioxidant Protection of NADPH-Depleted Oligodendrocyte Precursor Cells Is Dependent on Supply of Reduced Glutathione. ASN Neuro 2016, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Phensy, A.; Duzdabanian, H.E.; Brewer, S.; Panjabi, A.; Driskill, C.; Berz, A.; Peng, G.; Kroener, S. Antioxidant Treatment with N-acetyl Cysteine Prevents the Development of Cognitive and Social Behavioral Deficits that Result from Perinatal Ketamine Treatment. Front. Behav. Neurosci. 2017, 11, 106. [Google Scholar] [CrossRef]
- Swanepoel, T.; Möller, M.; Harvey, B.H. N-acetyl cysteine reverses bio-behavioural changes induced by prenatal inflammation, adolescent methamphetamine exposure and combined challenges. Psychopharmacology 2018, 235, 351–368. [Google Scholar] [CrossRef]
- Maas, D.A.; Eijsink, V.D.; van Hulten, J.A.; Panic, R.; De Weerd, P.; Homberg, J.R.; Vallès, A.; Nait Oumesmar, B.; Martens, G.J.M. Antioxidant treatment ameliorates prefrontal hypomyelination and cognitive deficits in a rat model of schizophrenia. Neuropsychopharmacology 2021, 46, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Cardis, R.; Cabungcal, J.H.; Dwir, D.; Do, K.Q.; Steullet, P. A lack of GluN2A-containing NMDA receptors confers a vulnerability to redox dysregulation: Consequences on parvalbumin interneurons, and their perineuronal nets. Neurobiol. Dis. 2018, 109, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Cabungcal, J.H.; Counotte, D.S.; Lewis, E.; Tejeda, H.A.; Piantadosi, P.; Pollock, C.; Calhoon, G.G.; Sullivan, E.; Presgraves, E.; Kil, J.; et al. Juvenile antioxidant treatment prevents adult deficits in a developmental model of schizophrenia. Neuron 2014, 83, 1073–1084. [Google Scholar] [CrossRef]
- Do, K.Q.; Trabesinger, A.H.; Kirsten Krüger, M.; Lauer, C.J.; Dydak, U.; Hell, D.; Holsboer, F.; Boesiger, P.; Cuénod, M. Schizophrenia: Glutathione deficit in cerebrospinal fluid and prefrontal cortex in vivo. Eur. J. Neurosci. 2000, 12, 3721–3728. [Google Scholar] [CrossRef]
- Tsugawa, S.; Noda, Y.; Tarumi, R.; Mimura, Y.; Yoshida, K.; Iwata, Y.; Elsalhy, M.; Kuromiya, M.; Kurose, S.; Masuda, F.; et al. Glutathione levels and activities of glutathione metabolism enzymes in patients with schizophrenia: A systematic review and meta-analysis. J. Psychopharmacol. 2019, 33, 1199–1214. [Google Scholar] [CrossRef]
- Das, T.K.; Javadzadeh, A.; Dey, A.; Sabesan, P.; Théberge, J.; Radua, J.; Palaniyappan, L. Antioxidant defense in schizophrenia and bipolar disorder: A meta-analysis of MRS studies of anterior cingulate glutathione. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 91, 94–102. [Google Scholar] [CrossRef]
- Wang, A.M.; Pradhan, S.; Coughlin, J.M.; Trivedi, A.; DuBois, S.L.; Crawford, J.L.; Sedlak, T.W.; Nucifora, F.C., Jr.; Nestadt, G.; Nucifora, L.G.; et al. Assessing Brain Metabolism With 7-T Proton Magnetic Resonance Spectroscopy in Patients With First-Episode Psychosis. JAMA Psychiatry 2019, 76, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Limongi, R.; Jeon, P.; Théberge, J.; Palaniyappan, L. Counteracting Effects of Glutathione on the Glutamate-Driven Excitation/Inhibition Imbalance in First-Episode Schizophrenia: A 7T MRS and Dynamic Causal Modeling Study. Antioxidants 2021, 10, 75. [Google Scholar] [CrossRef]
- Mouchlianitis, E.; Bloomfield, M.A.; Law, V.; Beck, K.; Selvaraj, S.; Rasquinha, N.; Waldman, A.; Turkheimer, F.E.; Egerton, A.; Stone, J.; et al. Treatment-Resistant Schizophrenia Patients Show Elevated Anterior Cingulate Cortex Glutamate Compared to Treatment-Responsive. Schizophr. Bull. 2016, 42, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Manchegowda, S.; Jacob, A.; Rao, N.P. Glutamate metabolites in treatment resistant schizophrenia: A meta-analysis and systematic review of (1)H-MRS studies. Psychiatry Res. Neuroimaging 2020, 300, 111080. [Google Scholar] [CrossRef] [PubMed]
- Iwata, Y.; Nakajima, S.; Plitman, E.; Truong, P.; Bani Fatemi, A.; Caravaggio, F.; Kim, J.; Shah, P.; Mar, W.; Chavez, S.; et al. Glutathione Levels and Glutathione-Glutamate Correlation in Patients With Treatment-Resistant Schizophrenia. Schizophr. Bull. Open 2021, 2, sgab006. [Google Scholar] [CrossRef]
- Flippo, K.H.; Strack, S. An emerging role for mitochondrial dynamics in schizophrenia. Schizophr. Res. 2017, 187, 26–32. [Google Scholar] [CrossRef]
- Kung, L.; Roberts, R.C. Mitochondrial pathology in human schizophrenic striatum: A postmortem ultrastructural study. Synapse 1999, 31, 67–75. [Google Scholar] [CrossRef]
- Somerville, S.M.; Lahti, A.C.; Conley, R.R.; Roberts, R.C. Mitochondria in the striatum of subjects with schizophrenia: Relationship to treatment response. Synapse 2011, 65, 215–224. [Google Scholar] [CrossRef]
- Uranova, N.A.; Vikhreva, O.V.; Rakhmanova, V.I.; Orlovskaya, D.D. Dystrophy of Oligodendrocytes and Adjacent Microglia in Prefrontal Gray Matter in Schizophrenia. Front. Psychiatry 2020, 11, 204. [Google Scholar] [CrossRef]
- Vikhreva, O.V.; Rakhmanova, V.I.; Orlovskaya, D.D.; Uranova, N.A. Ultrastructural alterations of oligodendrocytes in prefrontal white matter in schizophrenia: A post-mortem morphometric study. Schizophr. Res. 2016, 177, 28–36. [Google Scholar] [CrossRef]
- Kolomeets, N.S.; Uranova, N. Ultrastructural abnormalities of astrocytes in the hippocampus in schizophrenia and duration of illness: A postortem morphometric study. World J. Biol. Psychiatry 2010, 11, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Marchbanks, R.M.; Ryan, M.; Day, I.N.; Owen, M.; McGuffin, P.; Whatley, S.A. A mitochondrial DNA sequence variant associated with schizophrenia and oxidative stress. Schizophr. Res. 2003, 65, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Ueno, H.; Nishigaki, Y.; Kong, Q.P.; Fuku, N.; Kojima, S.; Iwata, N.; Ozaki, N.; Tanaka, M. Analysis of mitochondrial DNA variants in Japanese patients with schizophrenia. Mitochondrion 2009, 9, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Munakata, K.; Iwamoto, K.; Bundo, M.; Kato, T. Mitochondrial DNA 3243A>G mutation and increased expression of LARS2 gene in the brains of patients with bipolar disorder and schizophrenia. Biol. Psychiatry 2005, 57, 525–532. [Google Scholar] [CrossRef]
- Davies, V.J.; Hollins, A.J.; Piechota, M.J.; Yip, W.; Davies, J.R.; White, K.E.; Nicols, P.P.; Boulton, M.E.; Votruba, M. Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum. Mol. Genet. 2007, 16, 1307–1318. [Google Scholar] [CrossRef]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Chen, H.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 2007, 130, 548–562. [Google Scholar] [CrossRef]
- Bertholet, A.M.; Millet, A.M.; Guillermin, O.; Daloyau, M.; Davezac, N.; Miquel, M.C.; Belenguer, P. OPA1 loss of function affects in vitro neuronal maturation. Brain 2013, 136, 1518–1533. [Google Scholar] [CrossRef]
- Rosenfeld, M.; Brenner Lavie, H.; Ari, S.G.; Kavushansky, A.; Ben Shachar, D. Perturbation in mitochondrial network dynamics and in complex I dependent cellular respiration in schizophrenia. Biol. Psychiatry 2011, 69, 980–988. [Google Scholar] [CrossRef]
- Schumacher, J.; Jamra, R.A.; Freudenberg, J.; Becker, T.; Ohlraun, S.; Otte, A.C.; Tullius, M.; Kovalenko, S.; Bogaert, A.V.; Maier, W.; et al. Examination of G72 and D-amino-acid oxidase as genetic risk factors for schizophrenia and bipolar affective disorder. Mol. Psychiatry 2004, 9, 203–207. [Google Scholar] [CrossRef]
- Kvajo, M.; Dhilla, A.; Swor, D.E.; Karayiorgou, M.; Gogos, J.A. Evidence implicating the candidate schizophrenia/bipolar disorder susceptibility gene G72 in mitochondrial function. Mol. Psychiatry 2008, 13, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Millar, J.K.; James, R.; Christie, S.; Porteous, D.J. Disrupted in schizophrenia 1 (DISC1): Subcellular targeting and induction of ring mitochondria. Mol. Cell. Neurosci. 2005, 30, 477–484. [Google Scholar] [CrossRef]
- Norkett, R.; Modi, S.; Birsa, N.; Atkin, T.A.; Ivankovic, D.; Pathania, M.; Trossbach, S.V.; Korth, C.; Hirst, W.D.; Kittler, J.T. DISC1-dependent Regulation of Mitochondrial Dynamics Controls the Morphogenesis of Complex Neuronal Dendrites. J. Biol. Chem. 2016, 291, 613–629. [Google Scholar] [CrossRef]
- Schmidt, H.R.; Betz, R.M.; Dror, R.O.; Kruse, A.C. Structural basis for σ(1) receptor ligand recognition. Nat. Struct. Mol. Biol. 2018, 25, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K. Activation of sigma-1 receptor chaperone in the treatment of neuropsychiatric diseases and its clinical implication. J. Pharmacol. Sci. 2015, 127, 6–9. [Google Scholar] [CrossRef]
- Hirata, Y.; Oka, K.; Yamamoto, S.; Watanabe, H.; Oh Hashi, K.; Hirayama, T.; Nagasawa, H.; Takemori, H.; Furuta, K. Haloperidol Prevents Oxytosis/Ferroptosis by Targeting Lysosomal Ferrous Ions in a Manner Independent of Dopamine D2 and Sigma-1 Receptors. ACS Chem. Neurosci. 2022, 13, 2719–2727. [Google Scholar] [CrossRef]
- Nasrallah, H.A.; Chen, A.T. Multiple neurotoxic effects of haloperidol resulting in neuronal death. Ann. Clin. Psychiatry 2017, 29, 195–202. [Google Scholar]
- Pillai, A.; Veeranan Karmegam, R.; Dhandapani, K.M.; Mahadik, S.P. Cystamine prevents haloperidol-induced decrease of BDNF/TrkB signaling in mouse frontal cortex. J. Neurochem. 2008, 107, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Popgeorgiev, N.; Jabbour, L.; Gillet, G. Subcellular Localization and Dynamics of the Bcl-2 Family of Proteins. Front. Cell Dev. Biol. 2018, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Sagara, Y. Induction of reactive oxygen species in neurons by haloperidol. J. Neurochem. 1998, 71, 1002–1012. [Google Scholar] [CrossRef]
- Abdel Salam, O.M.; El Sayed El Shamarka, M.; Salem, N.A.; El Mosallamy, A.E.; Sleem, A.A. Amelioration of the haloperidol-induced memory impairment and brain oxidative stress by cinnarizine. EXCLI J. 2012, 11, 517–530. [Google Scholar] [PubMed]
- Elimadi, A.; Bouillot, L.; Sapena, R.; Tillement, J.P.; Morin, D. Dose-related inversion of cinnarizine and flunarizine effects on mitochondrial permeability transition. Eur. J. Pharmacol. 1998, 348, 115–121. [Google Scholar] [CrossRef]
- Raudenska, M.; Gumulec, J.; Babula, P.; Stracina, T.; Sztalmachova, M.; Polanska, H.; Adam, V.; Kizek, R.; Novakova, M.; Masarik, M. Haloperidol cytotoxicity and its relation to oxidative stress. Mini Rev. Med. Chem. 2013, 13, 1993–1998. [Google Scholar] [CrossRef]
- Ukai, W.; Ozawa, H.; Tateno, M.; Hashimoto, E.; Saito, T. Neurotoxic potential of haloperidol in comparison with risperidone: Implication of Akt-mediated signal changes by haloperidol. J. Neural Transm. 2004, 111, 667–681. [Google Scholar] [CrossRef] [PubMed]
- Subramanyam, B.; Rollema, H.; Woolf, T.; Castagnoli, N., Jr. Identification of a potentially neurotoxic pyridinium metabolite of haloperidol in rats. Biochem. Biophys. Res. Commun. 1990, 166, 238–244. [Google Scholar] [CrossRef]
- Zamani, E.; Ahmadi Shad, A.; Fatemi, H.; Mahboubi, S.; Motavallian, A.; Evazalipour, M. Assessment of Protective Effects of Carvacrol on Haloperidol-Induced Oxidative Stress and Genotoxicity in Human Peripheral Blood Lymphocytes. J. Toxicol. 2022, 2022, 9565881. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, A.; Li, Y.; Li, X.; Zhang, Q.; Song, W.; Wang, Y.; Ogutu, J.O.; Wang, J.; Li, J.; et al. Chlorpromazine inhibits mitochondrial apoptotic pathway via increasing expression of tissue factor. Int. J. Biochem. Cell Biol. 2016, 70, 82–91. [Google Scholar] [CrossRef]
- Abdalla, D.S.; Bechara, E.J. The effect of chlorpromazine and Li2CO3 on the superoxide dismutase and glutathione peroxidase activities of rat brain, liver and erythrocytes. Biochem. Mol. Biol. Int. 1994, 34, 1085–1090. [Google Scholar]
- Al Amin, M.M.; Nasir Uddin, M.M.; Mahmud Reza, H. Effects of antipsychotics on the inflammatory response system of patients with schizophrenia in peripheral blood mononuclear cell cultures. Clin. Psychopharmacol. Neurosci. 2013, 11, 144–151. [Google Scholar] [CrossRef]
- Gassó, P.; Mas, S.; Molina, O.; Bernardo, M.; Lafuente, A.; Parellada, E. Neurotoxic/neuroprotective activity of haloperidol, risperidone and paliperidone in neuroblastoma cells. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2012, 36, 71–77. [Google Scholar] [CrossRef]
- Quincozes Santos, A.; Bobermin, L.D.; Tonial, R.P.; Bambini Junior, V.; Riesgo, R.; Gottfried, C. Effects of atypical (risperidone) and typical (haloperidol) antipsychotic agents on astroglial functions. Eur. Arch. Psychiatry Clin. Neurosci. 2010, 260, 475–481. [Google Scholar] [CrossRef]
- Song, M.; Liu, Y.; Zhou, J.; Shi, H.; Su, X.; Shao, M.; Yang, Y.; Wang, X.; Zhao, J.; Guo, D.; et al. Potential plasma biomarker panels identification for the diagnosis of first-episode schizophrenia and monitoring antipsychotic monotherapy with the use of metabolomics analyses. Psychiatry Res. 2023, 321, 115070. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Zhou, D.F.; Cao, L.Y.; Zhang, P.Y.; Wu, G.Y.; Shen, Y.C. The effect of risperidone treatment on superoxide dismutase in schizophrenia. J. Clin. Psychopharmacol. 2003, 23, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Boz, Z.; Hu, M.; Yu, Y.; Huang, X.F. N-acetylcysteine prevents olanzapine-induced oxidative stress in mHypoA-59 hypothalamic neurons. Sci. Rep. 2020, 10, 19185. [Google Scholar] [CrossRef]
- Wei, Z.; Bai, O.; Richardson, J.S.; Mousseau, D.D.; Li, X.M. Olanzapine protects PC12 cells from oxidative stress induced by hydrogen peroxide. J. Neurosci. Res. 2003, 73, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Sacchi, S.; Novellis, V.; Paolone, G.; Nuzzo, T.; Iannotta, M.; Belardo, C.; Squillace, M.; Bolognesi, P.; Rosini, E.; Motta, Z.; et al. Olanzapine, but not clozapine, increases glutamate release in the prefrontal cortex of freely moving mice by inhibiting D-aspartate oxidase activity. Sci. Rep. 2017, 7, 46288. [Google Scholar] [CrossRef]
- Abou El Magd, R.M.; Park, H.K.; Kawazoe, T.; Iwana, S.; Ono, K.; Chung, S.P.; Miyano, M.; Yorita, K.; Sakai, T.; Fukui, K. The effect of risperidone on D-amino acid oxidase activity as a hypothesis for a novel mechanism of action in the treatment of schizophrenia. J. Psychopharmacol. 2010, 24, 1055–1067. [Google Scholar] [CrossRef]
- de Bartolomeis, A.; Vellucci, L.; Austin, M.C.; De Simone, G.; Barone, A. Rational and Translational Implications of D-Amino Acids for Treatment-Resistant Schizophrenia: From Neurobiology to the Clinics. Biomolecules 2022, 12, 909. [Google Scholar] [CrossRef]
- Hashimoto, K.; Fujita, Y.; Horio, M.; Kunitachi, S.; Iyo, M.; Ferraris, D.; Tsukamoto, T. Co-administration of a D-amino acid oxidase inhibitor potentiates the efficacy of D-serine in attenuating prepulse inhibition deficits after administration of dizocilpine. Biol. Psychiatry 2009, 65, 1103–1106. [Google Scholar] [CrossRef]
- Ferraris, D.; Duvall, B.; Ko, Y.S.; Thomas, A.G.; Rojas, C.; Majer, P.; Hashimoto, K.; Tsukamoto, T. Synthesis and biological evaluation of D-amino acid oxidase inhibitors. J. Med. Chem. 2008, 51, 3357–3359. [Google Scholar] [CrossRef]
- Brinholi, F.F.; Farias, C.C.; Bonifácio, K.L.; Higachi, L.; Casagrande, R.; Moreira, E.G.; Barbosa, D.S. Clozapine and olanzapine are better antioxidants than haloperidol, quetiapine, risperidone and ziprasidone in in vitro models. Biomed. Pharmacother. 2016, 81, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Dietrich Muszalska, A.; Kopka, J.; Kwiatkowska, A. The effects of ziprasidone, clozapine and haloperidol on lipid peroxidation in human plasma (in vitro): Comparison. Neurochem. Res. 2013, 38, 1490–1495. [Google Scholar] [CrossRef]
- Dietrich Muszalska, A.; Kolińska Łukaszuk, J. Comparative effects of aripiprazole and selected antipsychotic drugs on lipid peroxidation in plasma. Psychiatry Clin. Neurosci. 2018, 72, 329–336. [Google Scholar] [CrossRef]
- Chen, B.H.; Yan, B.C.; Park, J.H.; Ahn, J.H.; Lee, D.H.; Kim, I.H.; Cho, J.H.; Lee, J.C.; Kim, S.K.; Lee, B.; et al. Aripiprazole, an atypical antipsychotic drug, improves maturation and complexity of neuroblast dendrites in the mouse dentate gyrus via increasing superoxide dismutases. Neurochem. Res. 2013, 38, 1980–1988. [Google Scholar] [CrossRef]
- Xin, R.; Chen, Z.; Fu, J.; Shen, F.; Zhu, Q.; Huang, F. Xanomeline Protects Cortical Cells From Oxygen-Glucose Deprivation via Inhibiting Oxidative Stress and Apoptosis. Front. Physiol. 2020, 11, 656. [Google Scholar] [CrossRef]
- Ben Shachar, D. Mitochondrial dysfunction in schizophrenia: A possible linkage to dopamine. J. Neurochem. 2002, 83, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Kato, N. Mitochondrial dysfunction in bipolar disorder. Bipolar Disord. 2000, 2, 180–190. [Google Scholar] [CrossRef]
- Stefano, G.B.; Kream, R.M.; Esch, T. Mobility Coupled with Motivation Promotes Survival: The Evolution of Cognition as an Adaptive Strategy. Biology 2023, 12, 80. [Google Scholar] [CrossRef]
- Frey, B.N.; Valvassori, S.S.; Gomes, K.M.; Martins, M.R.; Dal Pizzol, F.; Kapczinski, F.; Quevedo, J. Increased oxidative stress in submitochondrial particles after chronic amphetamine exposure. Brain Res. 2006, 1097, 224–229. [Google Scholar] [CrossRef]
- Elkashef, A.M.; Al Barazi, H.; Venable, D.; Baker, I.; Hill, J.; Apud, J.; Wyatt, R.J. Dopamine effect on the mitochondria potential in B lymphocytes of schizophrenic patients and normal controls. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2002, 26, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Seybolt, S.E. Is it time to reassess alpha lipoic acid and niacinamide therapy in schizophrenia? Med. Hypotheses 2010, 75, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.W.; Smith, K.L.; Roberts, R.C.; Perez Costas, E.; Melendez Ferro, M. Assessment of cytochrome C oxidase dysfunction in the substantia nigra/ventral tegmental area in schizophrenia. PLoS ONE 2014, 9, e100054. [Google Scholar] [CrossRef] [PubMed]
- Seeman, P. Dopamine D2 receptors as treatment targets in schizophrenia. Clin. Schizophr. Relat. Psychoses 2010, 4, 56–73. [Google Scholar] [CrossRef] [PubMed]
- Uludag, K.; Wang, D.M.; Zhang, X.Y. Tardive Dyskinesia Development, Superoxide Dismutase Levels, and Relevant Genetic Polymorphisms. Oxidative Med. Cell. Longev. 2022, 2022, 5748924. [Google Scholar] [CrossRef]
- Górny, M.; Bilska Wilkosz, A.; Iciek, M.; Hereta, M.; Kamińska, K.; Kamińska, A.; Chwatko, G.; Rogóż, Z.; Lorenc Koci, E. Alterations in the Antioxidant Enzyme Activities in the Neurodevelopmental Rat Model of Schizophrenia Induced by Glutathione Deficiency during Early Postnatal Life. Antioxidants 2020, 9, 538. [Google Scholar] [CrossRef]
- Lech, M.A.; Leśkiewicz, M.; Kamińska, K.; Rogóż, Z.; Lorenc Koci, E. Glutathione Deficiency during Early Postnatal Development Causes Schizophrenia-Like Symptoms and a Reduction in BDNF Levels in the Cortex and Hippocampus of Adult Sprague-Dawley Rats. Int. J. Mol. Sci. 2021, 22, 6171. [Google Scholar] [CrossRef]
- Wąsik, A.; Białoń, M.; Żarnowska, M.; Antkiewicz Michaluk, L. Comparison of the effects of 1MeTIQ and olanzapine on performance in the elevated plus maze test and monoamine metabolism in the brain after ketamine treatment. Pharmacol. Biochem. Behav. 2019, 181, 17–27. [Google Scholar] [CrossRef]
- Antkiewicz Michaluk, L.; Ossowska, K.; Romańska, I.; Michaluk, J.; Vetulani, J. 3-Methoxytyramine, an extraneuronal dopamine metabolite plays a physiological role in the brain as an inhibitory regulator of catecholaminergic activity. Eur. J. Pharmacol. 2008, 599, 32–35. [Google Scholar] [CrossRef]
- Iasevoli, F.; Buonaguro, E.F.; Avagliano, C.; Barone, A.; Eramo, A.; Vellucci, L.; de Bartolomeis, A. The Effects of Antipsychotics on the Synaptic Plasticity Gene Homer1a Depend on a Combination of Their Receptor Profile, Dose, Duration of Treatment, and Brain Regions Targeted. Int. J. Mol. Sci. 2020, 21, 5555. [Google Scholar] [CrossRef]
- Li, S.; Sheng, Z.H. Energy matters: Presynaptic metabolism and the maintenance of synaptic transmission. Nat. Rev. Neurosci. 2022, 23, 4–22. [Google Scholar] [CrossRef]
- Devine, M.J.; Kittler, J.T. Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 2018, 19, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Bu, M.; Farrer, M.J.; Khoshbouei, H. Dynamic control of the dopamine transporter in neurotransmission and homeostasis. Npj Park. Dis. 2021, 7, 22. [Google Scholar] [CrossRef]
- Masoud, S.T.; Vecchio, L.M.; Bergeron, Y.; Hossain, M.M.; Nguyen, L.T.; Bermejo, M.K.; Kile, B.; Sotnikova, T.D.; Siesser, W.B.; Gainetdinov, R.R.; et al. Increased expression of the dopamine transporter leads to loss of dopamine neurons, oxidative stress and l-DOPA reversible motor deficits. Neurobiol. Dis. 2015, 74, 66–75. [Google Scholar] [CrossRef]
- Naoi, M.; Maruyama, W.; Shamoto-Nagai, M. Neuroprotective Function of Rasagiline and Selegiline, Inhibitors of Type B Monoamine Oxidase, and Role of Monoamine Oxidases in Synucleinopathies. Int. J. Mol. Sci. 2022, 23, 11059. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Andreazza, A.C. The relationship between oxidative stress and post-translational modification of the dopamine transporter in bipolar disorder. Expert Rev. Neurother. 2012, 12, 849–859. [Google Scholar] [CrossRef]
- Amato, D.; Kruyer, A.; Samaha, A.N.; Heinz, A. Hypofunctional Dopamine Uptake and Antipsychotic Treatment-Resistant Schizophrenia. Front. Psychiatry 2019, 10, 314. [Google Scholar] [CrossRef]
- Chestnykh, D.A.; Amato, D.; Kornhuber, J.; Müller, C.P. Pharmacotherapy of schizophrenia: Mechanisms of antipsychotic accumulation, therapeutic action and failure. Behav. Brain Res. 2021, 403, 113144. [Google Scholar] [CrossRef] [PubMed]
- Amato, D.; Canneva, F.; Cumming, P.; Maschauer, S.; Groos, D.; Dahlmanns, J.K.; Grömer, T.W.; Chiofalo, L.; Dahlmanns, M.; Zheng, F.; et al. A dopaminergic mechanism of antipsychotic drug efficacy, failure, and failure reversal: The role of the dopamine transporter. Mol. Psychiatry 2020, 25, 2101–2118. [Google Scholar] [CrossRef]
- Howes, O.D.; Kapur, S. The dopamine hypothesis of schizophrenia: Version III--the final common pathway. Schizophr. Bull. 2009, 35, 549–562. [Google Scholar] [CrossRef]
- Afshari, P.; Yao, W.D.; Middleton, F.A. Reduced Slc1a1 expression is associated with neuroinflammation and impaired sensorimotor gating and cognitive performance in mice: Implications for schizophrenia. PLoS ONE 2017, 12, e0183854. [Google Scholar] [CrossRef]
- Yu, H.; Yan, H.; Wang, L.; Li, J.; Tan, L.; Deng, W.; Chen, Q.; Yang, G.; Zhang, F.; Lu, T.; et al. Five novel loci associated with antipsychotic treatment response in patients with schizophrenia: A genome-wide association study. Lancet. Psychiatry 2018, 5, 327–338. [Google Scholar] [CrossRef]
- Forder, J.P.; Tymianski, M. Postsynaptic mechanisms of excitotoxicity: Involvement of postsynaptic density proteins, radicals, and oxidant molecules. Neuroscience 2009, 158, 293–300. [Google Scholar] [CrossRef]
- De Bartolomeis, A.; Tomasetti, C. Calcium-Dependent Networks in Dopamine–Glutamate Interaction: The Role of Postsynaptic Scaffolding Proteins. Mol. Neurobiol. 2012, 46, 275–296. [Google Scholar] [CrossRef]
- Chiba, S.; Hashimoto, R.; Hattori, S.; Yohda, M.; Lipska, B.; Weinberger, D.R.; Kunugi, H. Effect of antipsychotic drugs on DISC1 and dysbindin expression in mouse frontal cortex and hippocampus. J. Neural Transm. 2006, 113, 1337–1346. [Google Scholar] [CrossRef]
- Weng, Y.-T.; Chien, T.; Kuan, I.-I.; Chern, Y. The TRAX, DISC1, and GSK3 complex in mental disorders and therapeutic interventions. J. Biomed. Sci. 2018, 25, 71. [Google Scholar] [CrossRef] [PubMed]
- Freyberg, Z.; Ferrando, S.J.; Javitch, J.A. Roles of the Akt/GSK-3 and Wnt signaling pathways in schizophrenia and antipsychotic drug action. Am. J. Psychiatry 2010, 167, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Hu, M.; Xie, Y.; Yu, Y.; Jaaro Peled, H.; Huang, X.F. Aripiprazole and haloperidol protect neurite lesions via reducing excessive D2R-DISC1 complex formation. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 92, 59–69. [Google Scholar] [CrossRef] [PubMed]
- de Bartolomeis, A.; Barone, A.; Buonaguro, E.F.; Tomasetti, C.; Vellucci, L.; Iasevoli, F. The Homer1 family of proteins at the crossroad of dopamine-glutamate signaling: An emerging molecular "Lego" in the pathophysiology of psychiatric disorders. A systematic review and translational insight. Neurosci. Biobehav. Rev. 2022, 136, 104596. [Google Scholar] [CrossRef]
- Barone, A.; De Simone, G.; Ciccarelli, M.; Buonaguro, E.F.; Tomasetti, C.; Eramo, A.; Vellucci, L.; de Bartolomeis, A. A Postsynaptic Density Immediate Early Gene-Based Connectome Analysis of Acute NMDAR Blockade and Reversal Effect of Antipsychotic Administration. Int. J. Mol. Sci. 2023, 24, 4372. [Google Scholar] [CrossRef]
- Barone, A.; Signoriello, S.; Latte, G.; Vellucci, L.; Giordano, G.; Avagliano, C.; Buonaguro, E.F.; Marmo, F.; Tomasetti, C.; Iasevoli, F.; et al. Modulation of glutamatergic functional connectivity by a prototypical antipsychotic: Translational inference from a postsynaptic density immediate-early gene-based network analysis. Behav. Brain Res. 2021, 404, 113160. [Google Scholar] [CrossRef]
- Nepliouev, I.; Zhang, Z.S.; Stiber, J.A. Effect of oxidative stress on homer scaffolding proteins. PLoS ONE 2011, 6, e26128. [Google Scholar] [CrossRef]
- El Rawas, R.; Amaral, I.M.; Hofer, A. The Anti-social Brain in Schizophrenia: A Role of CaMKII? Front. Psychiatry 2022, 13, 868244. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.S.; Rauniyar, N.; Sakaue, F.; Lam, T.T.; Williams, K.R.; Nairn, A.C. Development of Targeted Mass Spectrometry-Based Approaches for Quantitation of Proteins Enriched in the Postsynaptic Density (PSD). Proteomes 2019, 7, 12. [Google Scholar] [CrossRef]
- Okabe, S. Molecular anatomy of the postsynaptic density. Mol. Cell. Neurosci. 2007, 34, 503–518. [Google Scholar] [CrossRef] [PubMed]
- Matas, E.; William, D.J.F.; Toro, C.T. Abnormal expression of post-synaptic proteins in prefrontal cortex of patients with schizophrenia. Neurosci. Lett. 2021, 745, 135629. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.N.; Cook, S.G.; Allen, H.F.; Crosby, K.C.; Singh, T.; Coultrap, S.J.; Bayer, K.U. Characterization of six CaMKIIα variants found in patients with schizophrenia. iScience 2021, 24, 103184. [Google Scholar] [CrossRef]
- Yabuki, Y.; Wu, L.; Fukunaga, K. Cognitive enhancer ST101 improves schizophrenia-like behaviors in neonatal ventral hippocampus-lesioned rats in association with improved CaMKII/PKC pathway. J. Pharmacol. Sci. 2019, 140, 263–272. [Google Scholar] [CrossRef]
- Frankland, P.W.; O′Brien, C.; Ohno, M.; Kirkwood, A.; Silva, A.J. Alpha-CaMKII-dependent plasticity in the cortex is required for permanent memory. Nature 2001, 411, 309–313. [Google Scholar] [CrossRef]
- Choi, Y.; Chen, H.V.; Lipton, S.A. Three pairs of cysteine residues mediate both redox and zn2+ modulation of the nmda receptor. J. Neurosci. 2001, 21, 392–400. [Google Scholar] [CrossRef]
- Köhr, G.; Eckardt, S.; Lüddens, H.; Monyer, H.; Seeburg, P.H. NMDA receptor channels: Subunit-specific potentiation by reducing agents. Neuron 1994, 12, 1031–1040. [Google Scholar] [CrossRef]
- Bodhinathan, K.; Kumar, A.; Foster, T.C. Intracellular redox state alters NMDA receptor response during aging through Ca2+/calmodulin-dependent protein kinase II. J. Neurosci. 2010, 30, 1914–1924. [Google Scholar] [CrossRef] [PubMed]
- Celano, E.; Tiraboschi, E.; Consogno, E.; D′Urso, G.; Mbakop, M.P.; Gennarelli, M.; de Bartolomeis, A.; Racagni, G.; Popoli, M. Selective regulation of presynaptic calcium/calmodulin-dependent protein kinase II by psychotropic drugs. Biol. Psychiatry 2003, 53, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Amiri, S.; Jafari Sabet, M.; Keyhanfar, F.; Falak, R.; Shabani, M.; Rezayof, A. Hippocampal and prefrontal cortical NMDA receptors mediate the interactive effects of olanzapine and lithium in memory retention in rats: The involvement of CAMKII-CREB signaling pathways. Psychopharmacology 2020, 237, 1383–1396. [Google Scholar] [CrossRef]
- Rushlow, W.J.; Seah, C.; Sutton, L.P.; Bjelica, A.; Rajakumar, N. Antipsychotics affect multiple calcium calmodulin dependent proteins. Neuroscience 2009, 161, 877–886. [Google Scholar] [CrossRef]
- Ninan, I.; Jardemark, K.E.; Liang, X.; Wang, R.Y. Calcium/calmodulin-dependent kinase II is involved in the facilitating effect of clozapine on NMDA- and electrically evoked responses in the medial prefrontal cortical pyramidal cells. Synapse 2003, 47, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Naisbitt, S.; Kim, E.; Tu, J.C.; Xiao, B.; Sala, C.; Valtschanoff, J.; Weinberg, R.J.; Worley, P.F.; Sheng, M. Shank, a Novel Family of Postsynaptic Density Proteins that Binds to the NMDA Receptor/PSD-95/GKAP Complex and Cortactin. Neuron 1999, 23, 569–582. [Google Scholar] [CrossRef]
- Brenman, J.E.; Chao, D.S.; Gee, S.H.; McGee, A.W.; E Craven, S.; Santillano, D.R.; Wu, Z.; Huang, F.; Xia, H.; Peters, M.F.; et al. Interaction of Nitric Oxide Synthase with the Postsynaptic Density Protein PSD-95 and α1-Syntrophin Mediated by PDZ Domains. Cell 1996, 84, 757–767. [Google Scholar] [CrossRef]
- Freudenberg, F.; Alttoa, A.; Reif, A. Neuronal nitric oxide synthase (NOS1) and its adaptor, NOS1AP, as a genetic risk factors for psychiatric disorders. Genes Brain Behav. 2015, 14, 46–63. [Google Scholar] [CrossRef]
- Svane, K.C.; Asis, E.K.; Omelchenko, A.; Kunnath, A.J.; Brzustowicz, L.M.; Silverstein, S.M.; Firestein, B.L. d-Serine administration affects nitric oxide synthase 1 adaptor protein and DISC1 expression in sex-specific manner. Mol. Cell. Neurosci. 2018, 89, 20–32. [Google Scholar] [CrossRef]
- Bar Yosef, T.; Hussein, W.; Yitzhaki, O.; Damri, O.; Givon, L.; Marom, C.; Gurman, V.; Levine, J.; Bersudsky, Y.; Agam, G.; et al. Mitochondrial function parameters as a tool for tailored drug treatment of an individual with psychosis: A proof of concept study. Sci. Rep. 2020, 10, 12258. [Google Scholar] [CrossRef]
- Ben Shachar, D. Mitochondrial multifaceted dysfunction in schizophrenia; complex I as a possible pathological target. Schizophr. Res. 2017, 187, 3–10. [Google Scholar] [CrossRef]
- Bergman, O.; Ben Shachar, D. Mitochondrial Oxidative Phosphorylation System (OXPHOS) Deficits in Schizophrenia: Possible Interactions with Cellular Processes. Can. J. Psychiatry 2016, 61, 457–469. [Google Scholar] [CrossRef]
- Burkhardt, C.; Kelly, J.P.; Lim, Y.H.; Filley, C.M.; Parker, W.D., Jr. Neuroleptic medications inhibit complex I of the electron transport chain. Ann. Neurol. 1993, 33, 512–517. [Google Scholar] [CrossRef]
- Maurer, I.; Möller, H.J. Inhibition of complex I by neuroleptics in normal human brain cortex parallels the extrapyramidal toxicity of neuroleptics. Mol. Cell. Biochem. 1997, 174, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Balijepalli, S.; Boyd, M.R.; Ravindranath, V. Inhibition of mitochondrial complex I by haloperidol: The role of thiol oxidation. Neuropharmacology 1999, 38, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Modica Napolitano, J.S.; Lagace, C.J.; Brennan, W.A.; Aprille, J.R. Differential effects of typical and atypical neuroleptics on mitochondrial function in vitro. Arch. Pharmacal Res. 2003, 26, 951–959. [Google Scholar] [CrossRef]
- Casademont, J.; Garrabou, G.; Miró, O.; López, S.; Pons, A.; Bernardo, M.; Cardellach, F. Neuroleptic treatment effect on mitochondrial electron transport chain: Peripheral blood mononuclear cells analysis in psychotic patients. J. Clin. Psychopharmacol. 2007, 27, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Balijepalli, S.; Kenchappa, R.S.; Boyd, M.R.; Ravindranath, V. Protein thiol oxidation by haloperidol results in inhibition of mitochondrial complex I in brain regions: Comparison with atypical antipsychotics. Neurochem. Int. 2001, 38, 425–435. [Google Scholar] [CrossRef]
- Prince, J.A.; Yassin, M.S.; Oreland, L. Neuroleptic-induced mitochondrial enzyme alterations in the rat brain. J. Pharmacol. Exp. Ther. 1997, 280, 261–267. [Google Scholar]
- Whatley, S.A.; Curti, D.; Das Gupta, F.; Ferrier, I.N.; Jones, S.; Taylor, C.; Marchbanks, R.M. Superoxide, neuroleptics and the ubiquinone and cytochrome b5 reductases in brain and lymphocytes from normals and schizophrenic patients. Mol. Psychiatry 1998, 3, 227–237. [Google Scholar] [CrossRef]
- Barrientos, A.; Marín, C.; Miró, O.; Casademont, J.; Gómez, M.; Nunes, V.; Tolosa, E.; Urbano Márquez, A.; Cardellach, F. Biochemical and molecular effects of chronic haloperidol administration on brain and muscle mitochondria of rats. J. Neurosci. Res. 1998, 53, 475–481. [Google Scholar] [CrossRef]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.G.; Liu, Z.G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Peng, Z.; Zhou, Y.; Wang, J.; Lin, X.; Dong, X.; Liu, X.; Jiang, J.; Jiang, Y.; Li, L. Quetiapine induces myocardial necroptotic cell death through bidirectional regulation of cannabinoid receptors. Toxicol. Lett. 2019, 313, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Xuan, Y.; Yan, G.; Wu, R.; Huang, Q.; Li, X.; Xu, H. The cuprizone-induced changes in (1)H-MRS metabolites and oxidative parameters in C57BL/6 mouse brain: Effects of quetiapine. Neurochem. Int. 2015, 90, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Unsal, C.; Albayrak, Y.; Albayrak, N.; Kuloglu, M.; Hashimoto, K. Reduced serum paraoxonase 1 (PON1) activity in patients with schizophrenia treated with olanzapine but not quetiapine. Neuropsychiatr. Dis. Treat. 2013, 9, 1545–1552. [Google Scholar] [CrossRef]
- Arzuk, E.; Karakuş, F.; Orhan, H. Bioactivation of clozapine by mitochondria of the murine heart: Possible cause of cardiotoxicity. Toxicology 2021, 447, 152628. [Google Scholar] [CrossRef]
- Elmorsy, E.; Alelwani, W.; Kattan, S.; Babteen, N.; Alnajeebi, A.; Ghulam, J.; Mosad, S. Antipsychotics inhibit the mitochondrial bioenergetics of pancreatic beta cells isolated from CD1 mice. Basic Clin. Pharmacol. Toxicol. 2021, 128, 154–168. [Google Scholar] [CrossRef]
- Huang, C.H.; Fu, S.H.; Hsu, S.; Huang, Y.Y.; Chen, S.T.; Hsu, B.R. High-fat diet aggravates islet beta-cell toxicity in mice treated with clozapine. Chang Gung Med. J. 2012, 35, 318–322. [Google Scholar] [CrossRef]
- Madhu, L.N.; Kodali, M.; Attaluri, S.; Shuai, B.; Melissari, L.; Rao, X.; Shetty, A.K. Melatonin improves brain function in a model of chronic Gulf War Illness with modulation of oxidative stress, NLRP3 inflammasomes, and BDNF-ERK-CREB pathway in the hippocampus. Redox Biol. 2021, 43, 101973. [Google Scholar] [CrossRef]
- Ľupták, M.; Fišar, Z.; Hroudová, J. Effect of Novel Antipsychotics on Energy Metabolism-In Vitro Study in Pig Brain Mitochondria. Mol. Neurobiol. 2021, 58, 5548–5563. [Google Scholar] [CrossRef]
- Chan, S.T.; McCarthy, M.J.; Vawter, M.P. Psychiatric drugs impact mitochondrial function in brain and other tissues. Schizophr. Res. 2020, 217, 136–147. [Google Scholar] [CrossRef]
- Ji, B.; La, Y.; Gao, L.; Zhu, H.; Tian, N.; Zhang, M.; Yang, Y.; Zhao, X.; Tang, R.; Ma, G.; et al. A comparative proteomics analysis of rat mitochondria from the cerebral cortex and hippocampus in response to antipsychotic medications. J. Proteome Res. 2009, 8, 3633–3641. [Google Scholar] [CrossRef] [PubMed]
- Ji, B.; Zhang, Z.; Zhang, M.; Zhu, H.; Zhou, K.; Yang, J.; Li, Y.; Sun, L.; Feng, G.; Wang, Y.; et al. Differential expression profiling of the synaptosome proteome in a rat model of antipsychotic resistance. Brain Res. 2009, 1295, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Vadodaria, K.C.; Lenkei, Z.; Kato, T.; Gage, F.H.; Marchetto, M.C.; Santos, R. Mitochondria, Metabolism, and Redox Mechanisms in Psychiatric Disorders. Antioxid. Redox Signal. 2019, 31, 275–317. [Google Scholar] [CrossRef]
- Magalhães, P.V.; Dean, O.; Andreazza, A.C.; Berk, M.; Kapczinski, F. Antioxidant treatments for schizophrenia. Cochrane Database Syst. Rev. 2016, 2, Cd008919. [Google Scholar] [CrossRef] [PubMed]
- Akhondzadeh, S.; Safarcherati, A.; Amini, H. Beneficial antipsychotic effects of allopurinol as add-on therapy for schizophrenia: A double blind, randomized and placebo controlled trial. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2005, 29, 253–259. [Google Scholar] [CrossRef]
- Brunstein, M.G.; Ghisolfi, E.S.; Ramos, F.L.; Lara, D.R. A clinical trial of adjuvant allopurinol therapy for moderately refractory schizophrenia. J. Clin. Psychiatry 2005, 66, 213–219. [Google Scholar] [CrossRef]
- Weiser, M.; Gershon, A.A.; Rubinstein, K.; Petcu, C.; Ladea, M.; Sima, D.; Podea, D.; Keefe, R.S.; Davis, J.M. A randomized controlled trial of allopurinol vs. placebo added on to antipsychotics in patients with schizophrenia or schizoaffective disorder. Schizophr. Res. 2012, 138, 35–38. [Google Scholar] [CrossRef]
- Dickerson, F.B.; Stallings, C.R.; Origoni, A.E.; Sullens, A.; Khushalani, S.; Sandson, N.; Yolken, R.H. A double-blind trial of adjunctive allopurinol for schizophrenia. Schizophr. Res. 2009, 109, 66–69. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Zhou, D.F.; Zhang, P.Y.; Wu, G.Y.; Su, J.M.; Cao, L.Y. A double-blind, placebo-controlled trial of extract of Ginkgo biloba added to haloperidol in treatment-resistant patients with schizophrenia. J. Clin. Psychiatry 2001, 62, 878–883. [Google Scholar] [CrossRef]
- Luo, H.C.; Shen, Y.C.; Meng, F.Q. Therapeutic effect of shuxuening combining neuroleptics for the treatment of chronic schizophrenia–a double blind study. Chin. J. Integr. Tradit. West. Med. 1997, 17, 139–142. [Google Scholar]
- Doruk, A.; Uzun, O.; Ozşahin, A. A placebo-controlled study of extract of ginkgo biloba added to clozapine in patients with treatment-resistant schizophrenia. Int. Clin. Psychopharmacol. 2008, 23, 223–227. [Google Scholar] [CrossRef]
- Bošković, M.; Vovk, T.; Koprivšek, J.; Plesničar, B.K.; Grabnar, I. Vitamin E and essential polyunsaturated fatty acids supplementation in schizophrenia patients treated with haloperidol. Nutr. Neurosci. 2016, 19, 156–161. [Google Scholar] [CrossRef]
- Dakhale, G.N.; Khanzode, S.D.; Khanzode, S.S.; Saoji, A. Supplementation of vitamin C with atypical antipsychotics reduces oxidative stress and improves the outcome of schizophrenia. Psychopharmacology 2005, 182, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Bentsen, H.; Osnes, K.; Refsum, H.; Solberg, D.K.; Bøhmer, T. A randomized placebo-controlled trial of an omega-3 fatty acid and vitamins E+C in schizophrenia. Transl. Psychiatry 2013, 3, e335. [Google Scholar] [CrossRef]
- Berk, M.; Copolov, D.; Dean, O.; Lu, K.; Jeavons, S.; Schapkaitz, I.; Anderson Hunt, M.; Judd, F.; Katz, F.; Katz, P.; et al. N-acetyl cysteine as a glutathione precursor for schizophrenia--a double-blind, randomized, placebo-controlled trial. Biol. Psychiatry 2008, 64, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Farokhnia, M.; Azarkolah, A.; Adinehfar, F.; Khodaie Ardakani, M.R.; Hosseini, S.M.; Yekehtaz, H.; Tabrizi, M.; Rezaei, F.; Salehi, B.; Sadeghi, S.M.; et al. N-acetylcysteine as an adjunct to risperidone for treatment of negative symptoms in patients with chronic schizophrenia: A randomized, double-blind, placebo-controlled study. Clin. Neuropharmacol. 2013, 36, 185–192. [Google Scholar] [CrossRef]
- Sepehrmanesh, Z.; Heidary, M.; Akasheh, N.; Akbari, H.; Heidary, M. Therapeutic effect of adjunctive N-acetyl cysteine (NAC) on symptoms of chronic schizophrenia: A double-blind, randomized clinical trial. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 82, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Rapado Castro, M.; Dodd, S.; Bush, A.I.; Malhi, G.S.; Skvarc, D.R.; On, Z.X.; Berk, M.; Dean, O.M. Cognitive effects of adjunctive N-acetyl cysteine in psychosis. Psychol. Med. 2017, 47, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Rapado Castro, M.; Berk, M.; Venugopal, K.; Bush, A.I.; Dodd, S.; Dean, O.M. Towards stage specific treatments: Effects of duration of illness on therapeutic response to adjunctive treatment with N-acetyl cysteine in schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2015, 57, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, S.; Murray, M.M.; Deppen, P.; Knyazeva, M.G.; Berk, M.; Boulat, O.; Bovet, P.; Bush, A.I.; Conus, P.; Copolov, D.; et al. Glutathione precursor, N-acetyl-cysteine, improves mismatch negativity in schizophrenia patients. Neuropsychopharmacology 2008, 33, 2187–2199. [Google Scholar] [CrossRef] [PubMed]
- Carmeli, C.; Knyazeva, M.G.; Cuénod, M.; Do, K.Q. Glutathione precursor N-acetyl-cysteine modulates EEG synchronization in schizophrenia patients: A double-blind, randomized, placebo-controlled trial. PLoS ONE 2012, 7, e29341. [Google Scholar] [CrossRef] [PubMed]
- Klauser, P.; Xin, L.; Fournier, M.; Griffa, A.; Cleusix, M.; Jenni, R.; Cuenod, M.; Gruetter, R.; Hagmann, P.; Conus, P.; et al. N-acetylcysteine add-on treatment leads to an improvement of fornix white matter integrity in early psychosis: A double-blind randomized placebo-controlled trial. Transl. Psychiatry 2018, 8, 220. [Google Scholar] [CrossRef] [PubMed]
- Mullier, E.; Roine, T.; Griffa, A.; Xin, L.; Baumann, P.S.; Klauser, P.; Cleusix, M.; Jenni, R.; Alemàn Gómez, Y.; Gruetter, R.; et al. N-Acetyl-Cysteine Supplementation Improves Functional Connectivity Within the Cingulate Cortex in Early Psychosis: A Pilot Study. Int. J. Neuropsychopharmacol. 2019, 22, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Knolle, F.; Ermakova, A.O.; Justicia, A.; Fletcher, P.C.; Bunzeck, N.; Düzel, E.; Murray, G.K. Brain responses to different types of salience in antipsychotic naïve first episode psychosis: An fMRI study. Transl. Psychiatry 2018, 8, 196. [Google Scholar] [CrossRef]
- Breier, A.; Liffick, E.; Hummer, T.A.; Vohs, J.L.; Yang, Z.; Mehdiyoun, N.F.; Visco, A.C.; Metzler, E.; Zhang, Y.; Francis, M.M. Effects of 12-month, double-blind N-acetyl cysteine on symptoms, cognition and brain morphology in early phase schizophrenia spectrum disorders. Schizophr. Res. 2018, 199, 395–402. [Google Scholar] [CrossRef]
- Barone, A.; De Prisco, M.; Altavilla, B.; Avagliano, C.; Balletta, R.; Buonaguro, E.F.; Ciccarelli, M.; D′Ambrosio, L.; Giordano, S.; Latte, G.; et al. Disorganization domain as a putative predictor of Treatment Resistant Schizophrenia (TRS) diagnosis: A machine learning approach. J. Psychiatr. Res. 2022, 155, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Neill, E.; Rossell, S.L.; Yolland, C.; Meyer, D.; Galletly, C.; Harris, A.; Siskind, D.; Berk, M.; Bozaoglu, K.; Dark, F.; et al. N-Acetylcysteine (NAC) in Schizophrenia Resistant to Clozapine: A Double-Blind, Randomized, Placebo-Controlled Trial Targeting Negative Symptoms. Schizophr. Bull. 2022, 48, 1263–1272. [Google Scholar] [CrossRef]
- Andrade, C. Antipsychotic Augmentation With N-Acetylcysteine for Patients With Schizophrenia. J. Clin. Psychiatry 2022, 83, 43016. [Google Scholar] [CrossRef]
- Zhang, W.F.; Tan, Y.L.; Zhang, X.Y.; Chan, R.C.; Wu, H.R.; Zhou, D.F. Extract of Ginkgo biloba treatment for tardive dyskinesia in schizophrenia: A randomized, double-blind, placebo-controlled trial. J. Clin. Psychiatry 2011, 72, 615–621. [Google Scholar] [CrossRef]
- Dorevitch, A.; Kalian, M.; Shlafman, M.; Lerner, V. Treatment of long-term tardive dyskinesia with vitamin E. Biol. Psychiatry 1997, 41, 114–116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Zhou, D.F.; Cao, L.Y.; Xu, C.Q.; Chen, D.C.; Wu, G.Y. The effect of vitamin E treatment on tardive dyskinesia and blood superoxide dismutase: A double-blind placebo-controlled trial. J. Clin. Psychopharmacol. 2004, 24, 83–86. [Google Scholar] [CrossRef]
- Adler, L.A.; Peselow, E.; Rotrosen, J.; Duncan, E.; Lee, M.; Rosenthal, M.; Angrist, B. Vitamin E treatment of tardive dyskinesia. Am. J. Psychiatry 1993, 150, 1405–1407. [Google Scholar] [CrossRef] [PubMed]
- Adler, L.A.; Edson, R.; Lavori, P.; Peselow, E.; Duncan, E.; Rosenthal, M.; Rotrosen, J. Long-term treatment effects of vitamin E for tardive dyskinesia. Biol. Psychiatry 1998, 43, 868–872. [Google Scholar] [CrossRef] [PubMed]
- Schneider Thoma, J.; Chalkou, K.; Dörries, C.; Bighelli, I.; Ceraso, A.; Huhn, M.; Siafis, S.; Davis, J.M.; Cipriani, A.; Furukawa, T.A.; et al. Comparative efficacy and tolerability of 32 oral and long-acting injectable antipsychotics for the maintenance treatment of adults with schizophrenia: A systematic review and network meta-analysis. Lancet 2022, 399, 824–836. [Google Scholar] [CrossRef]
- Jahan, M.S.; Tsuzuki, T.; Ito, T.; Bhuiyan, M.E.R.; Takahashi, I.; Takamatsu, H.; Kumanogoh, A.; Negishi, T.; Yukawa, K. PlexinA1-deficient mice exhibit decreased cell density and augmented oxidative stress in parvalbumin-expressing interneurons in the medial prefrontal cortex. IBRO Neurosci. Rep. 2022, 13, 500–512. [Google Scholar] [CrossRef]
- Park, S.U.; Ferrer, J.V.; Javitch, J.A.; Kuhn, D.M. Peroxynitrite inactivates the human dopamine transporter by modification of cysteine 342: Potential mechanism of neurotoxicity in dopamine neurons. J. Neurosci. 2002, 22, 4399–4405. [Google Scholar] [CrossRef]
- Talukdar, P.M.; Abdul, F.; Maes, M.; Binu, V.S.; Venkatasubramanian, G.; Kutty, B.M.; Debnath, M. Maternal Immune Activation Causes Schizophrenia-like Behaviors in the Offspring through Activation of Immune-Inflammatory, Oxidative and Apoptotic Pathways, and Lowered Antioxidant Defenses and Neuroprotection. Mol. Neurobiol. 2020, 57, 4345–4361. [Google Scholar] [CrossRef]
- Pandurangi, A.K.; Buckley, P.F. Inflammation, Antipsychotic Drugs, and Evidence for Effectiveness of Anti-inflammatory Agents in Schizophrenia. Curr. Top. Behav. Neurosci. 2020, 44, 227–244. [Google Scholar] [CrossRef]
- MacDowell, K.S.; Martín Hernández, D.; Ulecia Morón, C.; Bris, Á.G.; Madrigal, J.L.M.; García Bueno, B.; Caso, J.R. Paliperidone attenuates chronic stress-induced changes in the expression of inflammasomes-related protein in the frontal cortex of male rats. Int. Immunopharmacol. 2021, 90, 107217. [Google Scholar] [CrossRef]
- Scheiber, C.; Schulz, T.; Schneider, J.M.; Bechter, K.; Schneider, E.M. Old and New Biomarkers for Infection, Inflammation, and Autoimmunity in Treatment-Resistant Affective and Schizophrenic Spectrum Disorders. Pharmaceuticals 2022, 15, 299. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Cao, T.; Cai, H. Peripheral biomarkers of treatment-resistant schizophrenia: Genetic, inflammation and stress perspectives. Front. Pharmacol. 2022, 13, 1005702. [Google Scholar] [CrossRef] [PubMed]
- Buosi, P.; Borghi, F.A.; Lopes, A.M.; Facincani, I.D.S.; Fernandes Ferreira, R.; Oliveira Brancati, C.I.F.; do Carmo, T.S.; Souza, D.R.S.; da Silva, D.G.H.; de Almeida, E.A.; et al. Oxidative stress biomarkers in treatment-responsive and treatment-resistant schizophrenia patients. Trends Psychiatry Psychother. 2021, 43, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Weiden, P.J.; Breier, A.; Kavanagh, S.; Miller, A.C.; Brannan, S.K.; Paul, S.M. Antipsychotic Efficacy of KarXT (Xanomeline-Trospium): Post Hoc Analysis of Positive and Negative Syndrome Scale Categorical Response Rates, Time Course of Response, and Symptom Domains of Response in a Phase 2 Study. J. Clin. Psychiatry 2022, 83, 40913. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Simone, G.; Mazza, B.; Vellucci, L.; Barone, A.; Ciccarelli, M.; de Bartolomeis, A. Schizophrenia Synaptic Pathology and Antipsychotic Treatment in the Framework of Oxidative and Mitochondrial Dysfunction: Translational Highlights for the Clinics and Treatment. Antioxidants 2023, 12, 975. https://doi.org/10.3390/antiox12040975
De Simone G, Mazza B, Vellucci L, Barone A, Ciccarelli M, de Bartolomeis A. Schizophrenia Synaptic Pathology and Antipsychotic Treatment in the Framework of Oxidative and Mitochondrial Dysfunction: Translational Highlights for the Clinics and Treatment. Antioxidants. 2023; 12(4):975. https://doi.org/10.3390/antiox12040975
Chicago/Turabian StyleDe Simone, Giuseppe, Benedetta Mazza, Licia Vellucci, Annarita Barone, Mariateresa Ciccarelli, and Andrea de Bartolomeis. 2023. "Schizophrenia Synaptic Pathology and Antipsychotic Treatment in the Framework of Oxidative and Mitochondrial Dysfunction: Translational Highlights for the Clinics and Treatment" Antioxidants 12, no. 4: 975. https://doi.org/10.3390/antiox12040975