
Shikonin Induces ROS-Dependent Apoptosis Via Mitochondria Depolarization and ER Stress in Adult T Cell Leukemia/Lymphoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Reagents
2.3. MTT Assay
2.4. Annexin V and Propidium Iodide (PI) Staining
2.5. Cell Viability Assay
2.6. Intracellular ROS Measurement
2.7. Mitochondrial Membrane Potential (Δψ) Measurement
2.8. Protein Extraction and Western Blot Analysis
2.9. Xenograft Mouse Model
2.10. Statistical Analysis
3. Results
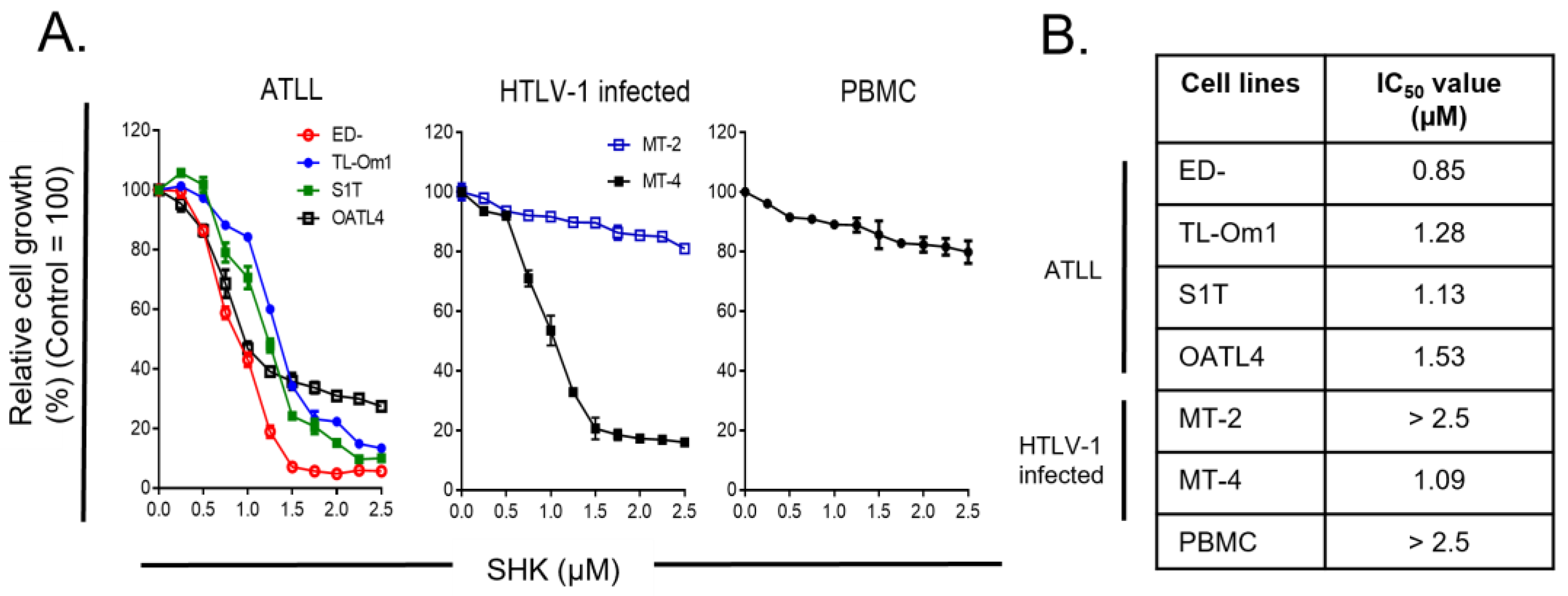
3.1. Antiproliferative Effects of SHK on ATLL Cells with Little Impacts on PBMC
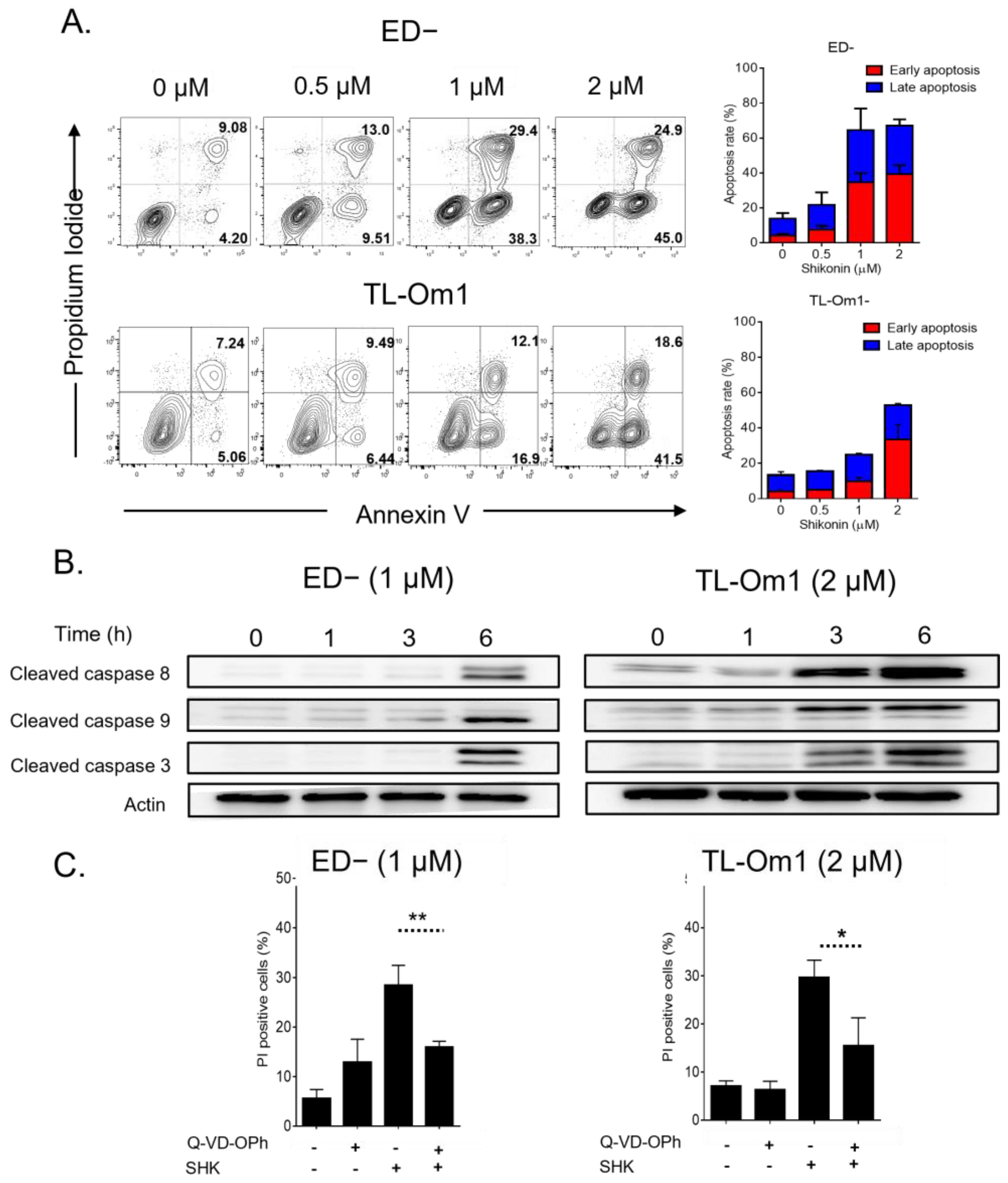
3.2. SHK Mediated Caspase-Dependent Apoptosis in ATL Cells
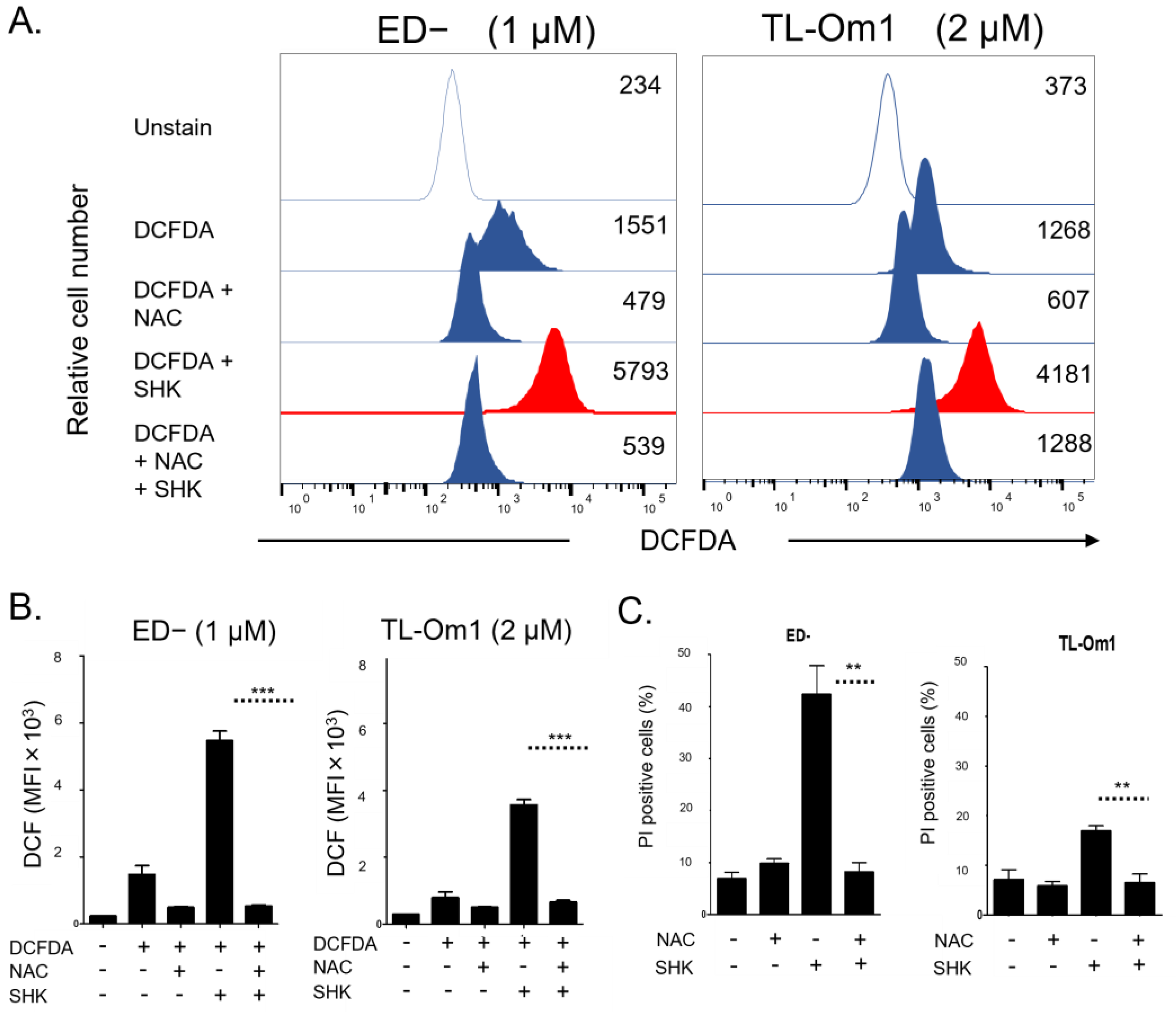
3.3. Accumulation of Intracellular ROS Involved in SHK-Induced ATLL Cell Death
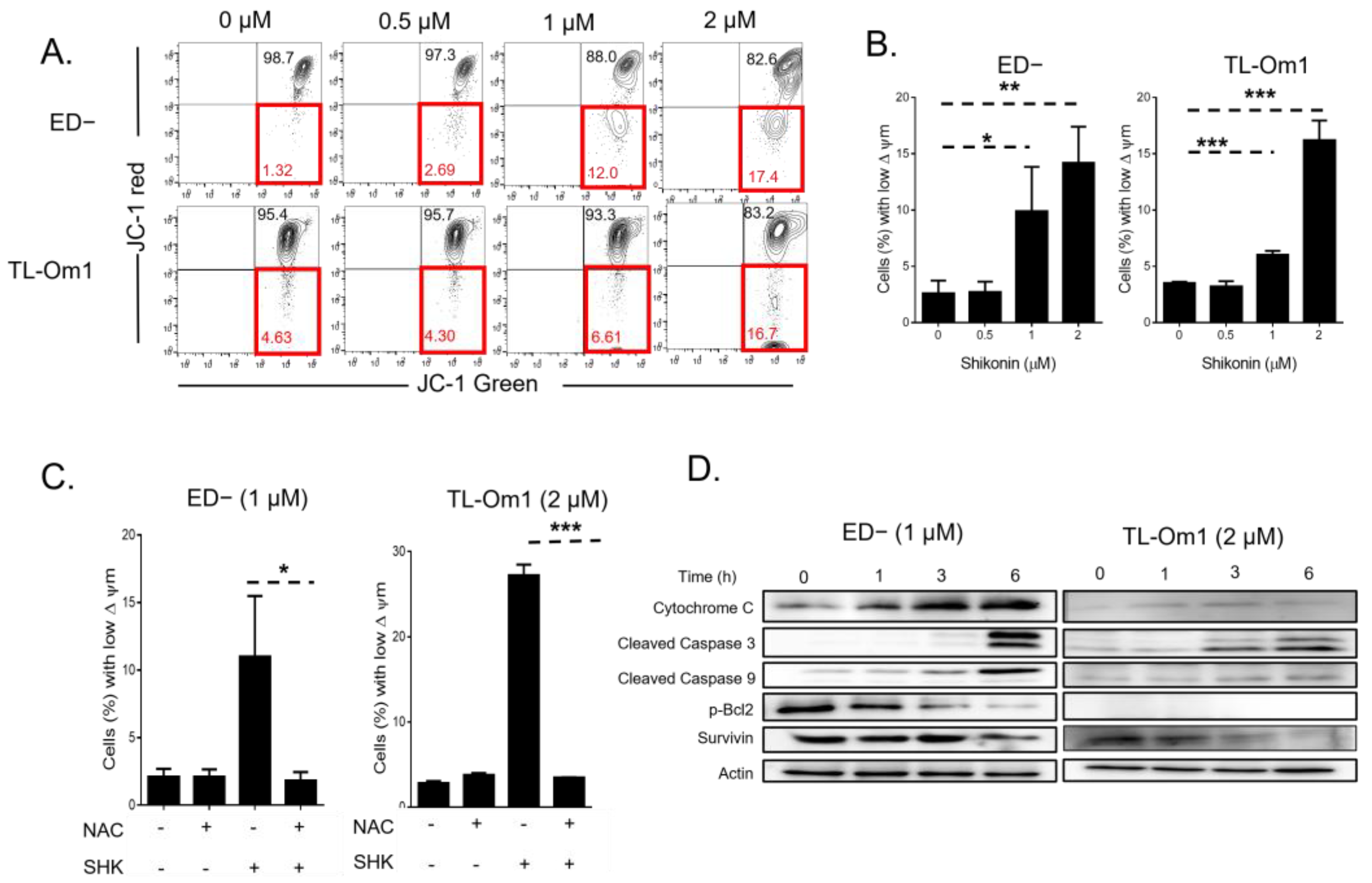
3.4. ROS Are Upstream of SHK Mediated Mitochondria Depolarization
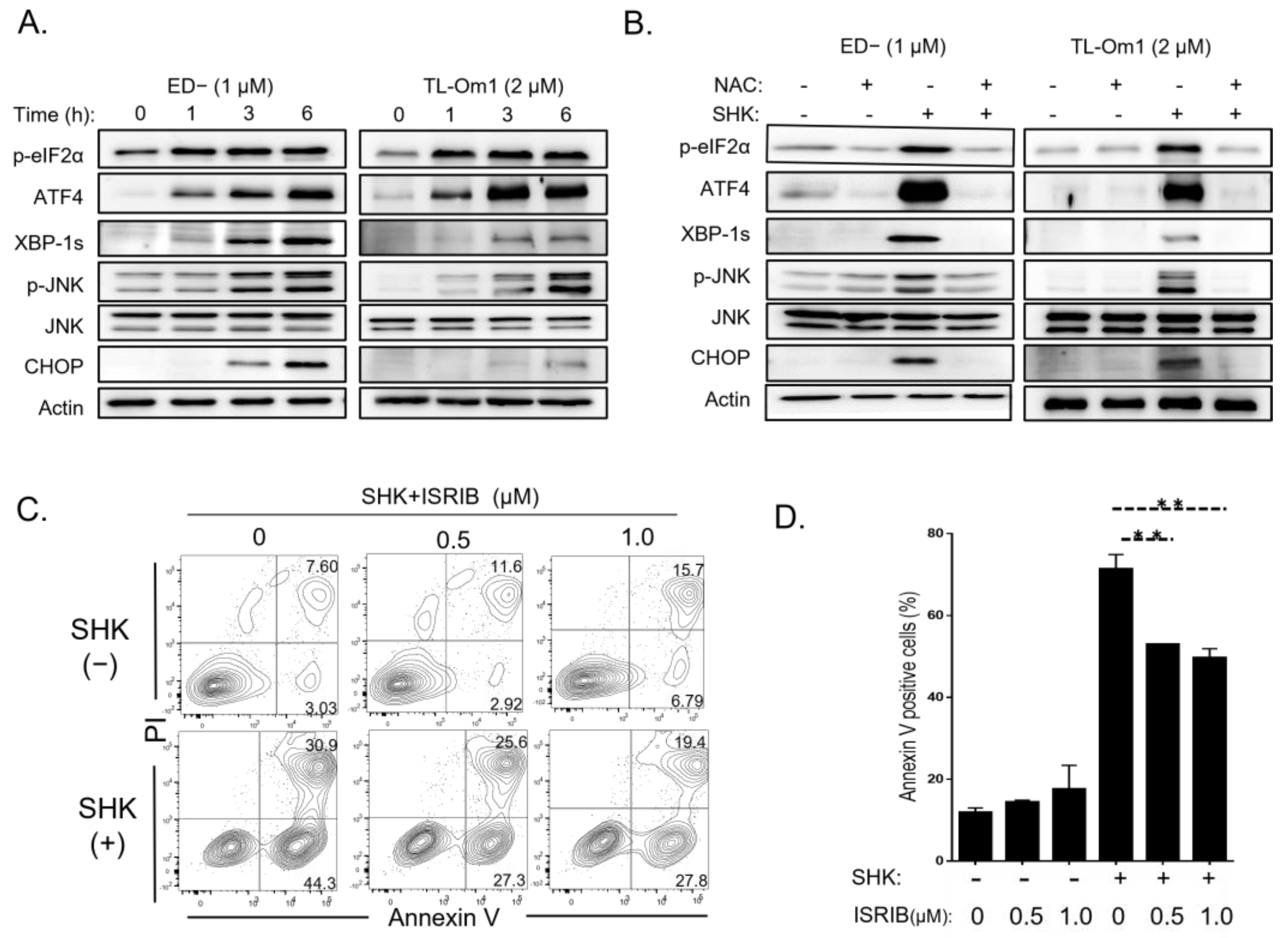
3.5. ROS Induced by SHK Activates ER Stress and Induces Apoptosis
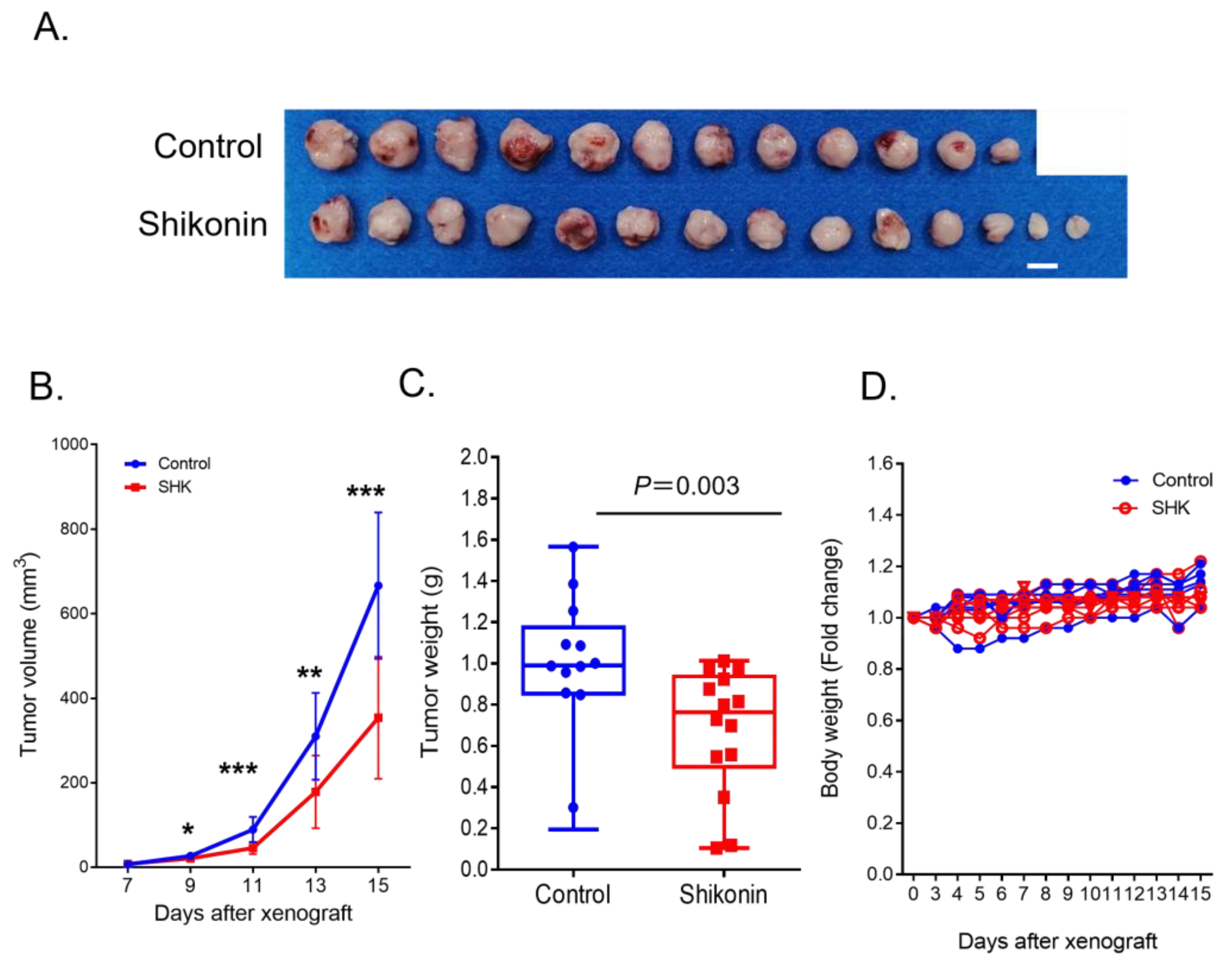
3.6. SHK Suppressed ATLL Cell Growth in an ATLL-Xenografted Mouse Model
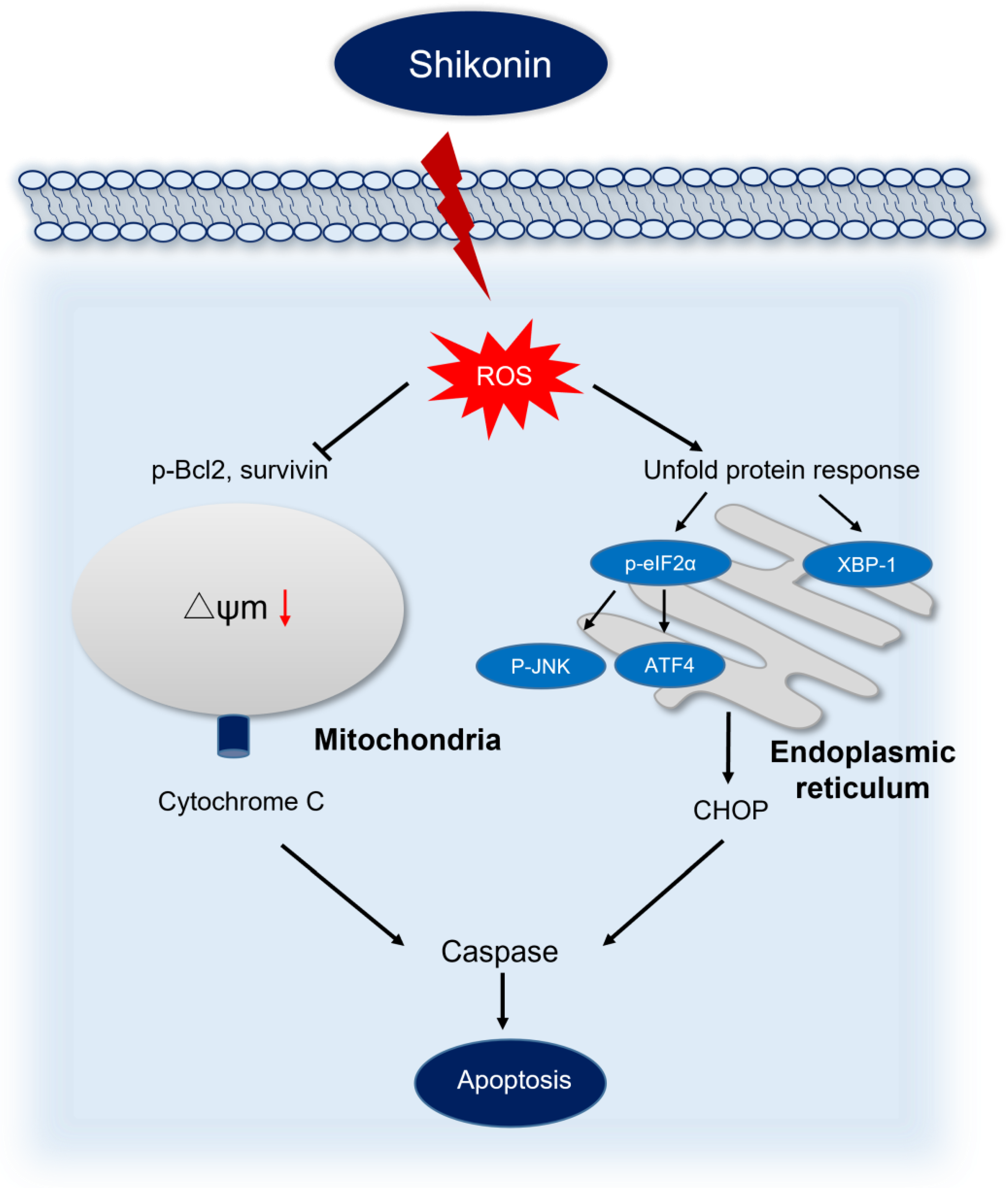
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nosaka, K.; Matsuoka, M. Adult T-cell leukemia-lymphoma as a viral disease: Subtypes based on viral aspects. Cancer Sci. 2021, 112, 1688–1694. [Google Scholar] [CrossRef]
- El Hajj, H.; Tsukasaki, K.; Cheminant, M.; Bazarbachi, A.; Watanabe, T.; Hermine, O. Novel Treatments of Adult T Cell Leukemia Lymphoma. Front. Microbiol. 2020, 11, 1062. [Google Scholar] [CrossRef] [PubMed]
- Cook, L.B.; Phillips, A.A. How I treat adult T-cell leukemia/lymphoma. Blood 2021, 137, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Hermine, O.; Ramos, J.C.; Tobinai, K. A Review of New Findings in Adult T-cell Leukemia-Lymphoma: A Focus on Current and Emerging Treatment Strategies. Adv. Ther. 2018, 35, 135–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuya, H.; Cook, L.B.M.; Rowan, A.G.; Satou, Y.; Taylor, G.P.; Bangham, C.R.M. Phosphatidylinositol 3-kinase-delta (PI3K-delta) is a potential therapeutic target in adult T-cell leukemia-lymphoma. Biomark. Res. 2018, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.B.; Harhaj, E.W. HTLV-1 tax stabilizes MCL-1 via TRAF6-dependent K63-linked polyubiquitination to promote cell survival and transformation. PLoS Pathog. 2014, 10, e1004458. [Google Scholar] [CrossRef]
- Macaire, H.; Riquet, A.; Moncollin, V.; Biemont-Trescol, M.C.; Duc Dodon, M.; Hermine, O.; Debaud, A.L.; Mahieux, R.; Mesnard, J.M.; Pierre, M.; et al. Tax protein-induced expression of antiapoptotic Bfl-1 protein contributes to survival of human T-cell leukemia virus type 1 (HTLV-1)-infected T-cells. J. Biol. Chem. 2012, 287, 21357–21370. [Google Scholar] [CrossRef] [Green Version]
- Tanaka-Nakanishi, A.; Yasunaga, J.; Takai, K.; Matsuoka, M. HTLV-1 bZIP factor suppresses apoptosis by attenuating the function of FoxO3a and altering its localization. Cancer Res. 2014, 74, 188–200. [Google Scholar] [CrossRef] [Green Version]
- Houssein, M.; Khalil, M.; Fatfat, M.; Gali-Muhtasib, H. Apoptosis as a mechanism for the treatment of adult T cell leukemia: Promising drugs from benchside to bedside. Drug Discov. Today 2020, 25, 1189–1197. [Google Scholar] [CrossRef]
- Cook, L.B.; Fuji, S.; Hermine, O.; Bazarbachi, A.; Ramos, J.C.; Ratner, L.; Horwitz, S.; Fields, P.; Tanase, A.; Bumbea, H.; et al. Revised Adult T-Cell Leukemia-Lymphoma International Consensus Meeting Report. J. Clin. Oncol. 2019, 37, 677–687. [Google Scholar] [CrossRef]
- Yoshie, O. CCR4 as a Therapeutic Target for Cancer Immunotherapy. Cancers 2021, 13, 5542. [Google Scholar] [CrossRef]
- Illian, D.N.; Hafiz, I.; Meila, O.; Utomo, A.R.H.; Nuryawan, A.; Siregar, G.A.; Basyuni, M. Current Status, Distribution, and Future Directions of Natural Products against Colorectal Cancer in Indonesia: A Systematic Review. Molecules 2021, 26, 4984. [Google Scholar] [CrossRef] [PubMed]
- Andujar, I.; Rios, J.L.; Giner, R.M.; Recio, M.C. Pharmacological properties of shikonin—A review of literature since 2002. Planta Med. 2013, 79, 1685–1697. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; He, J.; Song, X.; Tan, L.; Wang, M.; Jiang, P.; Li, Y.; Cao, Z.; Peng, C. Pharmacological properties and derivatives of shikonin-A review in recent years. Pharmacol. Res. 2019, 149, 104463. [Google Scholar] [CrossRef]
- Sun, Q.; Gong, T.; Liu, M.; Ren, S.; Yang, H.; Zeng, S.; Zhao, H.; Chen, L.; Ming, T.; Meng, X.; et al. Shikonin, a naphthalene ingredient: Therapeutic actions, pharmacokinetics, toxicology, clinical trials and pharmaceutical researches. Phytomedicine 2022, 94, 153805. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Yang, Z.; Wang, X.; Tao, R.; Zhou, Y. TRAIL Enhances Shikonin Induced Apoptosis through ROS/JNK Signaling in Cholangiocarcinoma Cells. Cell. Physiol. Biochem. 2017, 42, 1073–1086. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.M.; Kariya, R.; Boonnate, P.; Kawaguchi, A.; Okada, S. Induction of apoptosis by Shikonin through ROS-mediated intrinsic and extrinsic apoptotic pathways in primary effusion lymphoma. Transl. Oncol. 2021, 14, 101006. [Google Scholar] [CrossRef]
- Duan, D.; Zhang, B.; Yao, J.; Liu, Y.; Fang, J. Shikonin targets cytosolic thioredoxin reductase to induce ROS-mediated apoptosis in human promyelocytic leukemia HL-60 cells. Free Radic. Biol. Med. 2014, 70, 182–193. [Google Scholar] [CrossRef]
- Zhang, F.Y.; Hu, Y.; Que, Z.Y.; Wang, P.; Liu, Y.H.; Wang, Z.H.; Xue, Y.X. Shikonin Inhibits the Migration and Invasion of Human Glioblastoma Cells by Targeting Phosphorylated beta-Catenin and Phosphorylated PI3K/Akt: A Potential Mechanism for the Anti-Glioma Efficacy of a Traditional Chinese Herbal Medicine. Int. J. Mol. Sci. 2015, 16, 23823–23848. [Google Scholar] [CrossRef] [PubMed]
- Mora-Molina, R.; Stohr, D.; Rehm, M.; Lopez-Rivas, A. cFLIP downregulation is an early event required for endoplasmic reticulum stress-induced apoptosis in tumor cells. Cell Death Dis. 2022, 13, 111. [Google Scholar] [CrossRef]
- Chaiyarit, S.; Thongboonkerd, V. Comparative analyses of cell disruption methods for mitochondrial isolation in high-throughput proteomics study. Anal. Biochem. 2009, 394, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Ono, A.; Hattori, S.; Kariya, R.; Iwanaga, S.; Taura, M.; Harada, H.; Suzu, S.; Okada, S. Comparative study of human hematopoietic cell engraftment into BALB/c and C57BL/6 strain of rag-2/jak3 double-deficient mice. J. Biomed. Biotechnol. 2011, 2011, 539748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attia, M.A.; Weiss, D.W. Immunology of spontaneous mammary carcinomas in mice. V. Acquired tumor resistance and enhancement in strain A mice infected with mammary tumor virus. Cancer Res. 1966, 26, 1787–1800. [Google Scholar]
- Harada, H.; Suzu, S.; Ito, T.; Okada, S. Selective expansion and engraftment of human CD16+ NK cells in NOD/SCID mice. Eur. J. Immunol. 2005, 35, 3599–3609. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Cai, A.; Chen, G.; Xi, H.; Wu, X.; Cui, J.; Zhang, K.; Zhao, X.; Yu, J.; Wei, B.; et al. Shikonin induces mitochondria-mediated apoptosis and enhances chemotherapeutic sensitivity of gastric cancer through reactive oxygen species. Sci. Rep. 2016, 6, 38267. [Google Scholar] [CrossRef] [Green Version]
- Gara, R.K.; Srivastava, V.K.; Duggal, S.; Bagga, J.K.; Bhatt, M.; Sanyal, S.; Mishra, D.P. Shikonin selectively induces apoptosis in human prostate cancer cells through the endoplasmic reticulum stress and mitochondrial apoptotic pathway. J. Biomed. Sci. 2015, 22, 26. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Zhang, C.; Ren, A.; Li, T.; Jin, R.; Li, G.; Gu, X.; Shi, R.; Zhao, Y. Shikonin Suppresses Skin Carcinogenesis via Inhibiting Cell Proliferation. PLoS ONE 2015, 10, e0126459. [Google Scholar] [CrossRef]
- Sweeney, S.R.; Collins, M.; Pandey, R.; Chiou, J.; Lodi, A.; Tiziani, S. Identification of a synergistic combination of dimethylaminoparthenolide and shikonin alters metabolism and inhibits proliferation of pediatric precursor-B cell acute lymphoblastic leukemia. Mol. Carcinog. 2020, 59, 399–411. [Google Scholar] [CrossRef]
- Wang, F.; Mayca Pozo, F.; Tian, D.; Geng, X.; Yao, X.; Zhang, Y.; Tang, J. Shikonin Inhibits Cancer Through P21 Upregulation and Apoptosis Induction. Front. Pharmacol. 2020, 11, 861. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, B.; Huang, Z.; Zhao, D.W.; Zeng, Q. Shikonin Inhibites Migration and Invasion of Thyroid Cancer Cells by Downregulating DNMT1. Med. Sci. Monit. 2018, 24, 661–670. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Gao, Q.; Li, W.; Zhu, L.; Shang, Q.; Feng, S.; Jia, J.; Jia, Q.; Shen, S.; Su, Z. Shikonin inhibits cancer cell cycling by targeting Cdc25s. BMC Cancer 2019, 19, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Zhu, Y.; Hu, J.; Jiang, L.; Li, L.; Jia, S.; Zen, K. Shikonin Inhibits Tumor Growth in Mice by Suppressing Pyruvate Kinase M2-mediated Aerobic Glycolysis. Sci. Rep. 2018, 8, 14517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Chen, Z.; Ni, F.; Ye, X.; Qian, W. Shikonin overcomes drug resistance and induces necroptosis by regulating the miR-92a-1-5p/MLKL axis in chronic myeloid leukemia. Aging 2020, 12, 17662–17680. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhuang, J.; Song, W.; Shen, W.; Wu, W.; Shen, H.; Han, S. Mitochondria-associated endoplasmic reticulum membrane: Overview and inextricable link with cancer. J. Cell. Mol. Med. 2023, 27, 906–919. [Google Scholar] [CrossRef]
- Kim, T.W. Nodakenin Induces ROS-Dependent Apoptotic Cell Death and ER Stress in Radioresistant Breast Cancer. Antioxidants 2023, 12, 492. [Google Scholar] [CrossRef]
- Peng, S.Y.; Tang, J.Y.; Lan, T.H.; Shiau, J.P.; Chen, K.L.; Jeng, J.H.; Yen, C.Y.; Chang, H.W. Oxidative-Stress-Mediated ER Stress Is Involved in Regulating Manoalide-Induced Antiproliferation in Oral Cancer Cells. Int. J. Mol. Sci. 2023, 24, 3987. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.L.; Yu, C.I.; Lee, T.H.; Chuang, J.M.; Han, K.F.; Lin, C.S.; Huang, W.P.; Chen, J.Y.; Chen, C.Y.; Lin, M.Y.; et al. Plumbagin induces the apoptosis of drug-resistant oral cancer in vitro and in vivo through ROS-mediated endoplasmic reticulum stress and mitochondrial dysfunction. Phytomedicine 2023, 111, 154655. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Cui, J.; Zhang, K.; Xi, H.; Cai, A.; Li, J.; Gao, Y.; Hu, C.; Liu, Y.; Lu, Y.; et al. Shikonin induces ROS-based mitochondria-mediated apoptosis in colon cancer. Oncotarget 2017, 8, 109094–109106. [Google Scholar] [CrossRef] [Green Version]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boonnate, P.; Kariya, R.; Okada, S. Shikonin Induces ROS-Dependent Apoptosis Via Mitochondria Depolarization and ER Stress in Adult T Cell Leukemia/Lymphoma. Antioxidants 2023, 12, 864. https://doi.org/10.3390/antiox12040864
Boonnate P, Kariya R, Okada S. Shikonin Induces ROS-Dependent Apoptosis Via Mitochondria Depolarization and ER Stress in Adult T Cell Leukemia/Lymphoma. Antioxidants. 2023; 12(4):864. https://doi.org/10.3390/antiox12040864
Chicago/Turabian StyleBoonnate, Piyanard, Ryusho Kariya, and Seiji Okada. 2023. "Shikonin Induces ROS-Dependent Apoptosis Via Mitochondria Depolarization and ER Stress in Adult T Cell Leukemia/Lymphoma" Antioxidants 12, no. 4: 864. https://doi.org/10.3390/antiox12040864