
Inhibition of Pyruvate Dehydrogenase in the Heart as an Initiating Event in the Development of Diabetic Cardiomyopathy


{kind=link}
Abstract
:1. Metabolic Inflexibility and Diabetic Cardiomyopathy
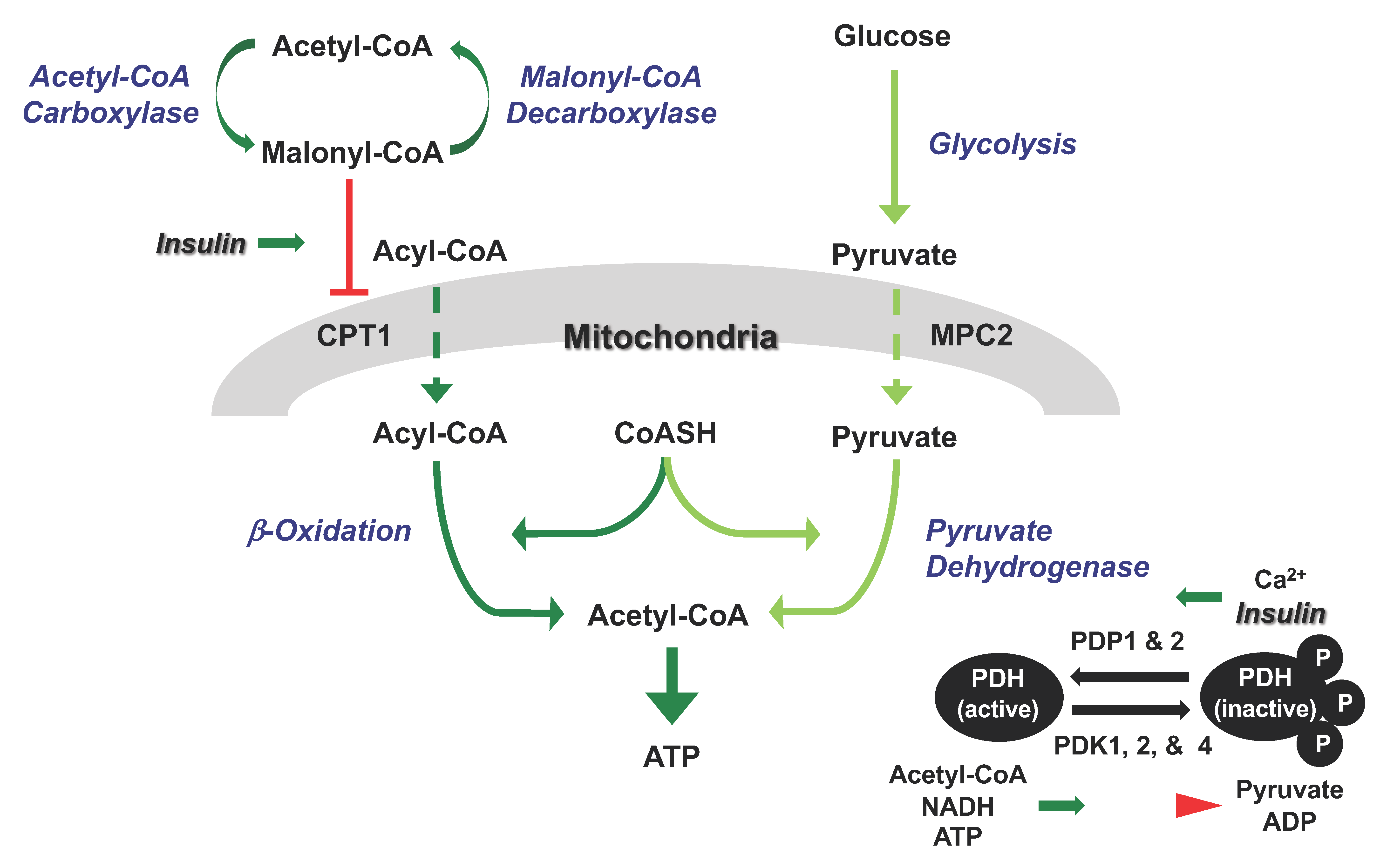
2. Regulation of Mitochondrial Fatty Acid and Glucose Oxidation in the Heart
3. Pyruvate Dehydrogenase Initiates Metabolic Inflexibility in a Model of Obesity/Type 2 Diabetes
4. Myocardial Pyruvate Oxidation in Humans with Type 2 Diabetes
5. Experimental Considerations When Analyzing Pyruvate Dehydrogenase Activity
6. Evidence from Genetic Mouse Models That Diminished Pyruvate Oxidation Can Induce Metabolic Cardiomyopathy
7. Potential Mechanisms by Which Metabolic Disruptions Drive the Development of Cardiomyopathy
8. Summary and Perspective
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Berthiaume, J.M.; Kurdys, J.G.; Muntean, D.M.; Rosca, M.G. Mitochondrial NAD(+)/NADH Redox State and Diabetic Cardiomyopathy. Antioxid. Redox Signal. 2019, 30, 375–398. [Google Scholar] [CrossRef] [Green Version]
- Jia, G.; DeMarco, V.G.; Sowers, J.R. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 2016, 12, 144–153. [Google Scholar] [CrossRef]
- Tan, Y.; Zhang, Z.; Zheng, C.; Wintergerst, K.A.; Keller, B.B.; Cai, L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: Preclinical and clinical evidence. Nat. Rev. Cardiol. 2020, 17, 585–607. [Google Scholar] [CrossRef]
- Hubert, H.B.; Feinleib, M.; McNamara, P.M.; Castelli, W.P. Obesity as an independent risk factor for cardiovascular disease: A 26-year follow-up of participants in the Framingham Heart Study. Circulation 1983, 67, 968–977. [Google Scholar] [CrossRef] [Green Version]
- Kenchaiah, S.; Evans, J.C.; Levy, D.; Wilson, P.W.; Benjamin, E.J.; Larson, M.G.; Kannel, W.B.; Vasan, R.S. Obesity and the risk of heart failure. N. Engl. J. Med. 2002, 347, 305–313. [Google Scholar] [CrossRef]
- Goodwin, G.W.; Taylor, C.S.; Taegtmeyer, H. Regulation of energy metabolism of the heart during acute increase in heart work. J. Biol. Chem. 1998, 273, 29530–29539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiraoka, T.; DeBuysere, M.; Olson, M.S. Studies of the effects of beta-adrenergic agonists on the regulation of pyruvate dehydrogenase in the perfused rat heart. J. Biol. Chem. 1980, 255, 7604–7609. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef] [PubMed]
- McGarry, J.D.; Brown, N.F. The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur. J. Biochem. 1997, 244, 1–14. [Google Scholar] [CrossRef] [PubMed]
- McGarry, J.D.; Woeltje, K.F.; Kuwajima, M.; Foster, D.W. Regulation of ketogenesis and the renaissance of carnitine palmitoyltransferase. Diabetes Metab. Rev. 1989, 5, 271–284. [Google Scholar] [CrossRef]
- Awan, M.M.; Saggerson, E.D. Malonyl-CoA metabolism in cardiac myocytes and its relevance to the control of fatty acid oxidation. Biochem. J. 1993, 295 Pt 1, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Gamble, J.; Lopaschuk, G.D. Insulin inhibition of 5′ adenosine monophosphate-activated protein kinase in the heart results in activation of acetyl coenzyme A carboxylase and inhibition of fatty acid oxidation. Metabolism 1997, 46, 1270–1274. [Google Scholar] [CrossRef] [PubMed]
- Behal, R.H.; Buxton, D.B.; Robertson, J.G.; Olson, M.S. Regulation of the pyruvate dehydrogenase multienzyme complex. Annu. Rev. Nutr. 1993, 13, 497–520. [Google Scholar] [CrossRef] [PubMed]
- Linn, T.C.; Pettit, F.H.; Hucho, F.; Reed, L.J. Alpha-keto acid dehydrogenase complexes. XI. Comparative studies of regulatory properties of the pyruvate dehydrogenase complexes from kidney, heart, and liver mitochondria. Proc. Natl. Acad. Sci. USA 1969, 64, 227–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linn, T.C.; Pettit, F.H.; Reed, L.J. Alpha-keto acid dehydrogenase complexes. X. Regulation of the activity of the pyruvate dehydrogenase complex from beef kidney mitochondria by phosphorylation and dephosphorylation. Proc. Natl. Acad. Sci. USA 1969, 62, 234–241. [Google Scholar] [CrossRef] [Green Version]
- Patel, M.S.; Korotchkina, L.G. Regulation of the pyruvate dehydrogenase complex. Biochem. Soc. Trans. 2006, 34 Pt 2, 217–222. [Google Scholar] [CrossRef]
- Rardin, M.J.; Wiley, S.E.; Naviaux, R.K.; Murphy, A.N.; Dixon, J.E. Monitoring phosphorylation of the pyruvate dehydrogenase complex. Anal. Biochem. 2009, 389, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Sugden, M.C.; Holness, M.J. Mechanisms underlying regulation of the expression and activities of the mammalian pyruvate dehydrogenase kinases. Arch. Physiol. Biochem. 2006, 112, 139–149. [Google Scholar] [CrossRef]
- Hue, L.; Taegtmeyer, H. The Randle cycle revisited: A new head for an old hat. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E578–E591. [Google Scholar] [CrossRef] [Green Version]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 1, 785–789. [Google Scholar] [CrossRef]
- Coore, H.G.; Denton, R.M.; Martin, B.R.; Randle, P.J. Regulation of adipose tissue pyruvate dehydrogenase by insulin and other hormones. Biochem. J. 1971, 125, 115–127. [Google Scholar] [CrossRef]
- Coore, H.G.; Denton, R.M.; Martin, B.R.; Randle, P.J. Effects of insulin and adrenaline on rat epididymal-fat-pad pyruvate dehydrogenase. Biochem. J. 1971, 123, 38P–39P. [Google Scholar] [CrossRef] [Green Version]
- Denton, R.M.; Coore, H.G.; Martin, B.R.; Randle, P.J. Insulin activates pyruvate dehydrogenase in rat epididymal adipose tissue. Nat. New Biol. 1971, 231, 115–116. [Google Scholar] [CrossRef]
- Denton, R.M.; Martin, B.R.; Coore, H.G.; Randle, P.J. Properties of pyruvate dehydrogenase from fat-cell mitochondria and its hormonal control. Biochem. J. 1971, 123, 39P. [Google Scholar] [CrossRef] [Green Version]
- Karwi, Q.G.; Wagg, C.S.; Altamimi, T.R.; Uddin, G.M.; Ho, K.L.; Darwesh, A.M.; Seubert, J.M.; Lopaschuk, G.D. Insulin directly stimulates mitochondrial glucose oxidation in the heart. Cardiovasc. Diabetol. 2020, 19, 207. [Google Scholar] [CrossRef]
- Kerbey, A.L.; Randle, P.J.; Cooper, R.H.; Whitehouse, S.; Pask, H.T.; Denton, R.M. Regulation of pyruvate dehydrogenase in rat heart. Mechanism of regulation of proportions of dephosphorylated and phosphorylated enzyme by oxidation of fatty acids and ketone bodies and of effects of diabetes: Role of coenzyme A, acetyl-coenzyme A and reduced and oxidized nicotinamide-adenine dinucleotide. Biochem. J. 1976, 154, 327–348. [Google Scholar] [CrossRef]
- Kobayashi, K.; Neely, J.R. Mechanism of pyruvate dehydrogenase activation by increased cardiac work. J. Mol. Cell. Cardiol. 1983, 15, 369–382. [Google Scholar] [CrossRef]
- Newman, J.D.; Armstrong, J.M.; Bornstein, J. Assay of insulin mediator activity with soluble pyruvate dehydrogenase phosphatase. Endocrinology 1985, 116, 1912–1919. [Google Scholar] [CrossRef]
- Seals, J.R.; Jarett, L. Activation of pyruvate dehydrogenase by direct addition of insulin to an isolated plasma membrane/mitochondria mixture: Evidence for generated of insulin’s second messenger in a subcellular system. Proc. Natl. Acad. Sci. USA 1980, 77, 77–81. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.I.; Mukherjee, C.; Jungas, R.L. Studies on the mechanism of activation of adipose tissue pyruvate dehydrogenase by insulin. J. Biol. Chem. 1973, 248, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.; Loffler, G.; Schirmann, A.; Wieland, O. Control of pyruvate dehydrogenase interconversion in adipose tissue by insulin. FEBS Lett. 1971, 15, 229–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abel, E.D.; Litwin, S.E.; Sweeney, G. Cardiac remodeling in obesity. Physiol. Rev. 2008, 88, 389–419. [Google Scholar] [CrossRef] [PubMed]
- Calligaris, S.D.; Lecanda, M.; Solis, F.; Ezquer, M.; Gutierrez, J.; Brandan, E.; Leiva, A.; Sobrevia, L.; Conget, P. Mice long-term high-fat diet feeding recapitulates human cardiovascular alterations: An animal model to study the early phases of diabetic cardiomyopathy. PLoS ONE 2013, 8, e60931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceylan-Isik, A.F.; Kandadi, M.R.; Xu, X.; Hua, Y.; Chicco, A.J.; Ren, J.; Nair, S. Apelin administration ameliorates high fat diet-induced cardiac hypertrophy and contractile dysfunction. J. Mol. Cell. Cardiol. 2013, 63, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Crewe, C.; Kinter, M.; Szweda, L.I. Rapid inhibition of pyruvate dehydrogenase: An initiating event in high dietary fat-induced loss of metabolic flexibility in the heart. PLoS ONE 2013, 8, e77280. [Google Scholar] [CrossRef]
- Fisher-Wellman, K.H.; Ryan, T.E.; Smith, C.D.; Gilliam, L.A.; Lin, C.T.; Reese, L.R.; Torres, M.J.; Neufer, P.D. A Direct Comparison of Metabolic Responses to High-Fat Diet in C57BL/6J and C57BL/6NJ Mice. Diabetes 2016, 65, 3249–3261. [Google Scholar] [CrossRef] [Green Version]
- Holness, M.J.; Smith, N.D.; Bulmer, K.; Hopkins, T.; Gibbons, G.F.; Sugden, M.C. Evaluation of the role of peroxisome-proliferator-activated receptor alpha in the regulation of cardiac pyruvate dehydrogenase kinase 4 protein expression in response to starvation, high-fat feeding and hyperthyroidism. Biochem. J. 2002, 364 Pt 3, 687–694. [Google Scholar] [CrossRef] [Green Version]
- Orfali, K.A.; Fryer, L.G.; Holness, M.J.; Sugden, M.C. Long-term regulation of pyruvate dehydrogenase kinase by high-fat feeding. Experiments in vivo and in cultured cardiomyocytes. FEBS Lett. 1993, 336, 501–505. [Google Scholar] [CrossRef] [Green Version]
- Ouwens, D.M.; Boer, C.; Fodor, M.; de Galan, P.; Heine, R.J.; Maassen, J.A.; Diamant, M. Cardiac dysfunction induced by high-fat diet is associated with altered myocardial insulin signalling in rats. Diabetologia 2005, 48, 1229–1237. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Cho, Y.R.; Kim, H.J.; Higashimori, T.; Danton, C.; Lee, M.K.; Dey, A.; Rothermel, B.; Kim, Y.B.; Kalinowski, A.; et al. Unraveling the temporal pattern of diet-induced insulin resistance in individual organs and cardiac dysfunction in C57BL/6 mice. Diabetes 2005, 54, 3530–3540. [Google Scholar] [CrossRef] [Green Version]
- Rindler, P.M.; Cacciola, A.; Kinter, M.; Szweda, L.I. Catalase-dependent H2O2 consumption by cardiac mitochondria and redox-mediated loss in insulin signaling. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1091–H1096. [Google Scholar] [CrossRef]
- Sverdlov, A.L.; Elezaby, A.; Qin, F.; Behring, J.B.; Luptak, I.; Calamaras, T.D.; Siwik, D.A.; Miller, E.J.; Liesa, M.; Shirihai, O.S.; et al. Mitochondrial Reactive Oxygen Species Mediate Cardiac Structural, Functional, and Mitochondrial Consequences of Diet-Induced Metabolic Heart Disease. J. Am. Heart. Assoc. 2016, 5, e002555. [Google Scholar] [CrossRef] [Green Version]
- Takimoto, E.; Kass, D.A. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 2007, 49, 241–248. [Google Scholar] [CrossRef]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovasc. Res. 2009, 81, 449–456. [Google Scholar] [CrossRef] [Green Version]
- Ussher, J.R.; Koves, T.R.; Jaswal, J.S.; Zhang, L.; Ilkayeva, O.; Dyck, J.R.; Muoio, D.M.; Lopaschuk, G.D. Insulin-stimulated cardiac glucose oxidation is increased in high-fat diet-induced obese mice lacking malonyl CoA decarboxylase. Diabetes 2009, 58, 1766–1775. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.R.; Tran, M.K.; Salazar, K.L.; Young, M.E.; Taegtmeyer, H. Western diet, but not high fat diet, causes derangements of fatty acid metabolism and contractile dysfunction in the heart of Wistar rats. Biochem. J. 2007, 406, 457–467. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Vaka, V.R.; He, X.; Booz, G.W.; Chen, J.X. High-fat diet induces cardiac remodelling and dysfunction: Assessment of the role played by SIRT3 loss. J. Cell Mol. Med. 2015, 19, 1847–1856. [Google Scholar] [CrossRef] [Green Version]
- Russo, S.B.; Baicu, C.F.; Van Laer, A.; Geng, T.; Kasiganesan, H.; Zile, M.R.; Cowart, L.A. Ceramide synthase 5 mediates lipid-induced autophagy and hypertrophy in cardiomyocytes. J. Clin. Investig. 2012, 122, 3919–3930. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Gonzalez, S.; Marin-Royo, G.; Jurado-Lopez, R.; Bartolome, M.V.; Romero-Miranda, A.; Luaces, M.; Islas, F.; Nieto, M.L.; Martinez-Martinez, E.; Cachofeiro, V. The Crosstalk between Cardiac Lipotoxicity and Mitochondrial Oxidative Stress in the Cardiac Alterations in Diet-Induced Obesity in Rats. Cells 2020, 9, 451. [Google Scholar] [CrossRef] [Green Version]
- Rider, O.J.; Apps, A.; Miller, J.; Lau, J.Y.C.; Lewis, A.J.M.; Peterzan, M.A.; Dodd, M.S.; Lau, A.Z.; Trumper, C.; Gallagher, F.A.; et al. Noninvasive In Vivo Assessment of Cardiac Metabolism in the Healthy and Diabetic Human Heart Using Hyperpolarized 13C MRI. Circ. Res. 2020, 126, 725–736. [Google Scholar] [CrossRef] [Green Version]
- Boersma, G.J.; Johansson, E.; Pereira, M.J.; Heurling, K.; Skrtic, S.; Lau, J.; Katsogiannos, P.; Panagiotou, G.; Lubberink, M.; Kullberg, J.; et al. Altered Glucose Uptake in Muscle, Visceral Adipose Tissue, and Brain Predict Whole-Body Insulin Resistance and may Contribute to the Development of Type 2 Diabetes: A Combined PET/MR Study. Horm. Metab. Res. 2018, 50, e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maki, M.; Nuutila, P.; Laine, H.; Voipio-Pulkki, L.M.; Haaparanta, M.; Solin, O.; Knuuti, J.M. Myocardial glucose uptake in patients with NIDDM and stable coronary artery disease. Diabetes 1997, 46, 1491–1496. [Google Scholar] [CrossRef] [PubMed]
- Sondergaard, H.M.; Bottcher, M.; Marie Madsen, M.; Schmitz, O.; Hansen, S.B.; Nielsen, T.T.; Botker, H.E. Impact of type 2 diabetes on myocardial insulin sensitivity to glucose uptake and perfusion in patients with coronary artery disease. J. Clin. Endocrinol. Metab. 2006, 91, 4854–4861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utriainen, T.; Takala, T.; Luotolahti, M.; Ronnemaa, T.; Laine, H.; Ruotsalainen, U.; Haaparanta, M.; Nuutila, P.; Yki-Jarvinen, H. Insulin resistance characterizes glucose uptake in skeletal muscle but not in the heart in NIDDM. Diabetologia 1998, 41, 555–559. [Google Scholar] [CrossRef] [Green Version]
- Iozzo, P.; Chareonthaitawee, P.; Dutka, D.; Betteridge, D.J.; Ferrannini, E.; Camici, P.G. Independent association of type 2 diabetes and coronary artery disease with myocardial insulin resistance. Diabetes 2002, 51, 3020–3024. [Google Scholar] [CrossRef] [Green Version]
- Dutka, D.P.; Pitt, M.; Pagano, D.; Mongillo, M.; Gathercole, D.; Bonser, R.S.; Camici, P.G. Myocardial glucose transport and utilization in patients with type 2 diabetes mellitus, left ventricular dysfunction, and coronary artery disease. J. Am. Coll. Cardiol. 2006, 48, 2225–2231. [Google Scholar] [CrossRef]
- Manchester, J.; Kong, X.; Nerbonne, J.; Lowry, O.H.; Lawrence, J.C., Jr. Glucose transport and phosphorylation in single cardiac myocytes: Rate-limiting steps in glucose metabolism. Am. J. Physiol. 1994, 266, E326–E333. [Google Scholar] [CrossRef]
- Jeoung, N.H.; Wu, P.; Joshi, M.A.; Jaskiewicz, J.; Bock, C.B.; Depaoli-Roach, A.A.; Harris, R.A. Role of pyruvate dehydrogenase kinase isoenzyme 4 (PDHK4) in glucose homoeostasis during starvation. Biochem. J. 2006, 397, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Peters, J.M.; Harris, R.A. Adaptive increase in pyruvate dehydrogenase kinase 4 during starvation is mediated by peroxisome proliferator-activated receptor alpha. Biochem. Biophys. Res. Commun. 2001, 287, 391–396. [Google Scholar] [CrossRef]
- Wu, P.; Sato, J.; Zhao, Y.; Jaskiewicz, J.; Popov, K.M.; Harris, R.A. Starvation and diabetes increase the amount of pyruvate dehydrogenase kinase isoenzyme 4 in rat heart. Biochem. J. 1998, 329 Pt 1, 197–201. [Google Scholar] [CrossRef] [Green Version]
- Crewe, C.; Schafer, C.; Lee, I.; Kinter, M.; Szweda, L.I. Regulation of Pyruvate Dehydrogenase Kinase 4 in the Heart through Degradation by the Lon Protease in Response to Mitochondrial Substrate Availability. J. Biol. Chem. 2017, 292, 305–312. [Google Scholar] [CrossRef] [Green Version]
- Schafer, C.; Young, Z.T.; Makarewich, C.A.; Elnwasany, A.; Kinter, C.; Kinter, M.; Szweda, L.I. Coenzyme A-mediated degradation of pyruvate dehydrogenase kinase 4 promotes cardiac metabolic flexibility after high-fat feeding in mice. J. Biol. Chem. 2018, 293, 6915–6924. [Google Scholar] [CrossRef] [Green Version]
- Bryson, J.M.; Cooney, G.J.; Wensley, V.R.; Blair, S.C.; Caterson, I.D. Diurnal patterns of cardiac and hepatic pyruvate dehydrogenase complex activity in gold-thioglucose-obese mice. Biochem. J. 1993, 295 Pt 3, 731–734. [Google Scholar] [CrossRef] [Green Version]
- Sidhu, S.; Gangasani, A.; Korotchkina, L.G.; Suzuki, G.; Fallavollita, J.A.; Canty, J.M.; Jr Patel, M.S. Tissue-specific pyruvate dehydrogenase complex deficiency causes cardiac hypertrophy and sudden death of weaned male mice. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H946–H952. [Google Scholar] [CrossRef] [Green Version]
- Gopal, K.; Almutairi, M.; Al Batran, R.; Eaton, F.; Gandhi, M.; Ussher, J.R. Cardiac-Specific Deletion of Pyruvate Dehydrogenase Impairs Glucose Oxidation Rates and Induces Diastolic Dysfunction. Front. Cardiovasc. Med. 2018, 5, 17. [Google Scholar] [CrossRef] [Green Version]
- Cluntun, A.A.; Badolia, R.; Lettlova, S.; Parnell, K.M.; Shankar, T.S.; Diakos, N.A.; Olson, K.A.; Taleb, I.; Tatum, S.M.; Berg, J.A.; et al. The pyruvate-lactate axis modulates cardiac hypertrophy and heart failure. Cell Metab. 2021, 33, 629–648.e10. [Google Scholar] [CrossRef]
- Fernandez-Caggiano, M.; Kamynina, A.; Francois, A.A.; Prysyazhna, O.; Eykyn, T.R.; Krasemann, S.; Crespo-Leiro, M.G.; Vieites, M.G.; Bianchi, K.; Morales, V.; et al. Mitochondrial pyruvate carrier abundance mediates pathological cardiac hypertrophy. Nat. Metab. 2020, 2, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- McCommis, K.S.; Kovacs, A.; Weinheimer, C.J.; Shew, T.M.; Koves, T.R.; Ilkayeva, O.R.; Kamm, D.R.; Pyles, K.D.; King, M.T.; Veech, R.L.; et al. Nutritional modulation of heart failure in mitochondrial pyruvate carrier-deficient mice. Nat. Metab. 2020, 2, 1232–1247. [Google Scholar] [CrossRef]
- Zhang, Y.; Taufalele, P.V.; Cochran, J.D.; Robillard-Frayne, I.; Marx, J.M.; Soto, J.; Rauckhorst, A.J.; Tayyari, F.; Pewa, A.D.; Gray, L.R.; et al. Mitochondrial pyruvate carriers are required for myocardial stress adaptation. Nat. Metab. 2020, 2, 1248–1264. [Google Scholar] [CrossRef]
- Cheng, Z.; White, M.F. Targeting Forkhead box O1 from the concept to metabolic diseases: Lessons from mouse models. Antioxid. Redox Signal. 2011, 14, 649–661. [Google Scholar] [CrossRef] [Green Version]
- Ronnebaum, S.M.; Patterson, C. The FoxO family in cardiac function and dysfunction. Annu. Rev. Physiol. 2010, 72, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Battiprolu, P.K.; Hojayev, B.; Jiang, N.; Wang, Z.V.; Luo, X.; Iglewski, M.; Shelton, J.M.; Gerard, R.D.; Rothermel, B.A.; Gillette, T.G.; et al. Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J. Clin. Investig. 2012, 122, 1109–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopal, K.; Al Batran, R.; Altamimi, T.R.; Greenwell, A.A.; Saed, C.T.; Tabatabaei Dakhili, S.A.; Dimaano, M.T.E.; Zhang, Y.; Eaton, F.; Sutendra, G.; et al. FoxO1 inhibition alleviates type 2 diabetes-related diastolic dysfunction by increasing myocardial pyruvate dehydrogenase activity. Cell Rep. 2021, 35, 108935. [Google Scholar] [CrossRef] [PubMed]
- Rider, O.J.; Francis, J.M.; Tyler, D.; Byrne, J.; Clarke, K.; Neubauer, S. Effects of weight loss on myocardial energetics and diastolic function in obesity. Int. J. Cardiovasc. Imaging 2013, 29, 1043–1050. [Google Scholar] [CrossRef]
- Le Page, L.M.; Rider, O.J.; Lewis, A.J.; Ball, V.; Clarke, K.; Johansson, E.; Carr, C.A.; Heather, L.C.; Tyler, D.J. Increasing Pyruvate Dehydrogenase Flux as a Treatment for Diabetic Cardiomyopathy: A Combined 13C Hyperpolarized Magnetic Resonance and Echocardiography Study. Diabetes 2015, 64, 2735–2743. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Jeoung, N.H.; Burgess, S.C.; Rosaaen-Stowe, K.A.; Inagaki, T.; Latif, S.; Shelton, J.M.; McAnally, J.; Bassel-Duby, R.; Harris, R.A.; et al. Overexpression of pyruvate dehydrogenase kinase 4 in heart perturbs metabolism and exacerbates calcineurin-induced cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H936–H943. [Google Scholar] [CrossRef]
- Atkinson, D.E. The energy charge of the adenylate pool as a regulatory parameter. Interaction with feedback modifiers. Biochemistry 1968, 7, 4030–4034. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.I.; Wang, Z.V.; et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019, 568, 351–356. [Google Scholar] [CrossRef]
- Tong, D.; Schiattarella, G.G.; Jiang, N.; Altamirano, F.; Szweda, P.A.; Elnwasany, A.; Lee, D.I.; Yoo, H.; Kass, D.A.; Szweda, L.I.; et al. NAD(+) Repletion Reverses Heart Failure With Preserved Ejection Fraction. Circ. Res. 2021, 128, 1629–1641. [Google Scholar] [CrossRef]
- Shao, D.; Kolwicz, S.C., Jr.; Wang, P.; Roe, N.D.; Villet, O.; Nishi, K.; Hsu, Y.A.; Flint, G.V.; Caudal, A.; Wang, W.; et al. Increasing Fatty Acid Oxidation Prevents High-Fat Diet-Induced Cardiomyopathy Through Regulating Parkin-Mediated Mitophagy. Circulation 2020, 142, 983–997. [Google Scholar] [CrossRef]
- Jeong, E.M.; Chung, J.; Liu, H.; Go, Y.; Gladstein, S.; Farzaneh-Far, A.; Lewandowski, E.D.; Dudley, S.C., Jr. Role of Mitochondrial Oxidative Stress in Glucose Tolerance, Insulin Resistance, and Cardiac Diastolic Dysfunction. J. Am. Heart Assoc. 2016, 5, e003046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rindler, P.M.; Plafker, S.M.; Szweda, L.I.; Kinter, M. High dietary fat selectively increases catalase expression within cardiac mitochondria. J. Biol. Chem. 2013, 288, 1979–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seifert, E.L.; Estey, C.; Xuan, J.Y.; Harper, M.E. Electron transport chain-dependent and -independent mechanisms of mitochondrial H2O2 emission during long-chain fatty acid oxidation. J. Biol. Chem. 2010, 285, 5748–5758. [Google Scholar] [CrossRef] [Green Version]
- Fisher-Wellman, K.H.; Neufer, P.D. Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol. Metab. 2012, 23, 142–153. [Google Scholar] [CrossRef] [Green Version]
- Guaras, A.; Perales-Clemente, E.; Calvo, E.; Acin-Perez, R.; Loureiro-Lopez, M.; Pujol, C.; Martinez-Carrascoso, I.; Nunez, E.; Garcia-Marques, F.; Rodriguez-Hernandez, M.A.; et al. The CoQH2/CoQ Ratio Serves as a Sensor of Respiratory Chain Efficiency. Cell Rep. 2016, 15, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Menendez-Montes, I.; Abdisalaam, S.; Xiao, F.; Lam, N.T.; Mukherjee, S.; Szweda, L.I.; Asaithamby, A.; Sadek, H.A. Mitochondrial fatty acid utilization increases chromatin oxidative stress in cardiomyocytes. Proc. Natl. Acad. Sci. USA 2021, 118, e2101674118. [Google Scholar] [CrossRef]
- Cardoso, A.C.; Lam, N.T.; Savla, J.J.; Nakada, Y.; Pereira, A.H.M.; Elnwasany, A.; Menendez-Montes, I.; Ensley, E.L.; Petric, U.B.; Sharma, G.; et al. Mitochondrial Substrate Utilization Regulates Cardiomyocyte Cell Cycle Progression. Nat. Metab. 2020, 2, 167–178. [Google Scholar] [CrossRef]
- Kil, I.S.; Ryu, K.W.; Lee, S.K.; Kim, J.Y.; Chu, S.Y.; Kim, J.H.; Park, S.; Rhee, S.G. Circadian Oscillation of Sulfiredoxin in the Mitochondria. Mol. Cell. 2015, 59, 651–663. [Google Scholar] [CrossRef]
- Fontaine, D.A.; Davis, D.B. Attention to Background Strain Is Essential for Metabolic Research: C57BL/6 and the International Knockout Mouse Consortium. Diabetes 2016, 65, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Kawashita, E.; Ishihara, K.; Nomoto, M.; Taniguchi, M.; Akiba, S. A comparative analysis of hepatic pathological phenotypes in C57BL/6J and C57BL/6N mouse strains in non-alcoholic steatohepatitis models. Sci. Rep. 2019, 9, 204. [Google Scholar] [CrossRef] [Green Version]
- Siersbaek, M.S.; Ditzel, N.; Hejbol, E.K.; Praestholm, S.M.; Markussen, L.K.; Avolio, F.; Li, L.; Lehtonen, L.; Hansen, A.K.; Schroder, H.D.; et al. C57BL/6J substrain differences in response to high-fat diet intervention. Sci. Rep. 2020, 10, 14052. [Google Scholar] [CrossRef] [PubMed]
- Oldford, C.; Kuksal, N.; Gill, R.; Young, A.; Mailloux, R.J. Estimation of the hydrogen peroxide producing capacities of liver and cardiac mitochondria isolated from C57BL/6N and C57BL/6J mice. Free Radic. Biol. Med. 2019, 135, 15–27. [Google Scholar] [CrossRef]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Le, Z.; Yanxiang Guo, J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef] [Green Version]
- Araki, M.; Motojima, K. Identification of ERRalpha as a specific partner of PGC-1alpha for the activation of PDK4 gene expression in muscle. FEBS J. 2006, 273, 1669–1680. [Google Scholar] [CrossRef]
- Huang, B.; Wu, P.; Bowker-Kinley, M.M.; Harris, R.A. Regulation of pyruvate dehydrogenase kinase expression by peroxisome proliferator-activated receptor-alpha ligands, glucocorticoids, and insulin. Diabetes 2002, 51, 276–283. [Google Scholar] [CrossRef] [Green Version]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elnwasany, A.; Ewida, H.A.; Szweda, P.A.; Szweda, L.I. Inhibition of Pyruvate Dehydrogenase in the Heart as an Initiating Event in the Development of Diabetic Cardiomyopathy. Antioxidants 2023, 12, 756. https://doi.org/10.3390/antiox12030756
Elnwasany A, Ewida HA, Szweda PA, Szweda LI. Inhibition of Pyruvate Dehydrogenase in the Heart as an Initiating Event in the Development of Diabetic Cardiomyopathy. Antioxidants. 2023; 12(3):756. https://doi.org/10.3390/antiox12030756
Chicago/Turabian StyleElnwasany, Abdallah, Heba A. Ewida, Pamela A. Szweda, and Luke I. Szweda. 2023. "Inhibition of Pyruvate Dehydrogenase in the Heart as an Initiating Event in the Development of Diabetic Cardiomyopathy" Antioxidants 12, no. 3: 756. https://doi.org/10.3390/antiox12030756