

A Preclinical Model for Parkinson’s Disease Based on Transcriptional Gene Activation via KEAP1/NRF2 to Develop New Antioxidant Therapies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Mechanisms Involved in Neurodegeneration in Parkinson’s Disease
2. Clinical Studies to Develop Antioxidant Therapies
3. Neurotoxins in Preclinical Models for Parkinson’s Disease
3.1. Exogenous Neurotoxins in Preclinical Models for Parkinson’s Disease
3.2. Endogenous Neurotoxins as Preclinical Model for Parkinson’s Disease
3.2.1. Neurotoxic Oligomers of Alpha-Synuclein
3.2.2. 3,4-Dihydroxyphenylacetaldehyde
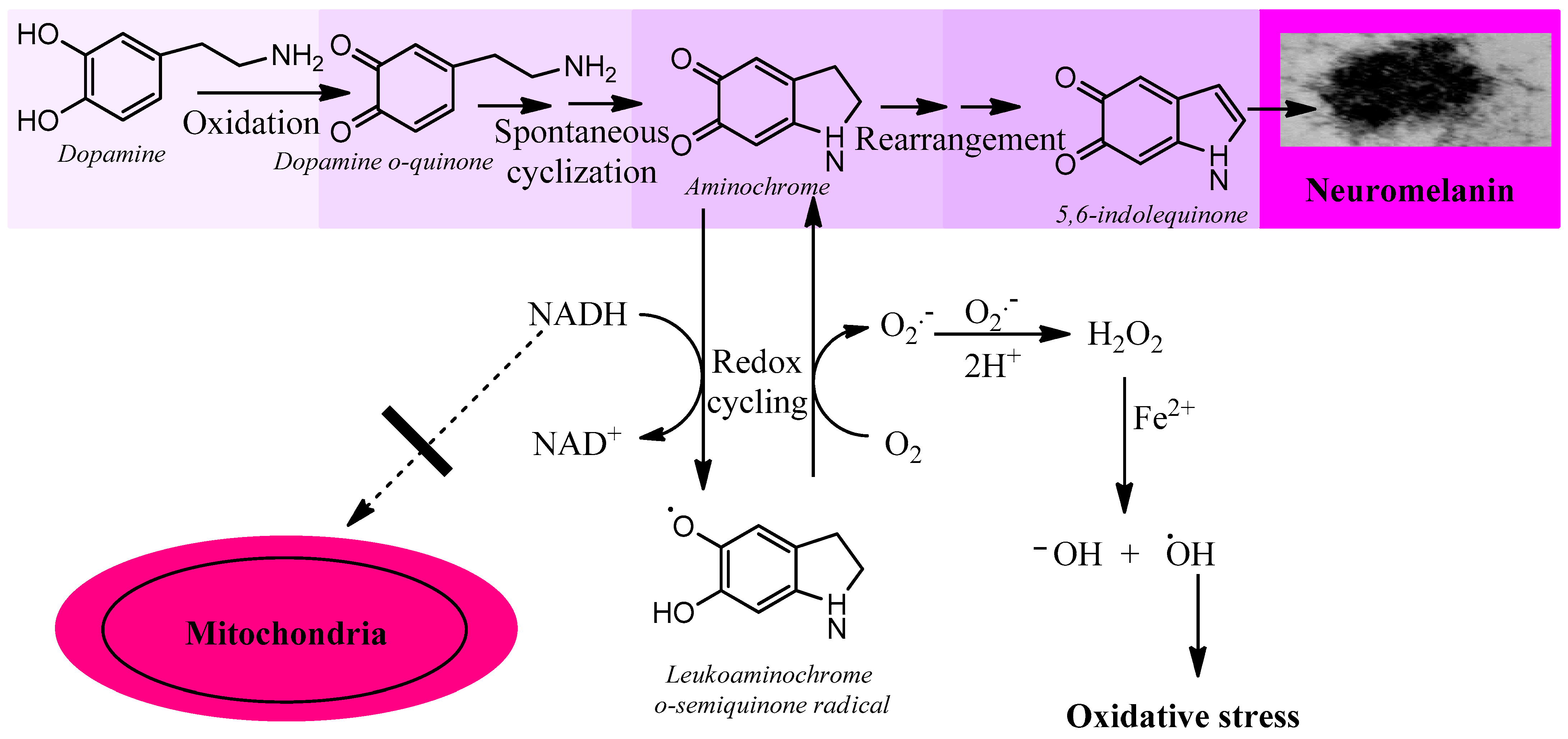
3.2.3. Aminochrome
4. Preclinical Model for Idiopathic Parkinson’s Disease
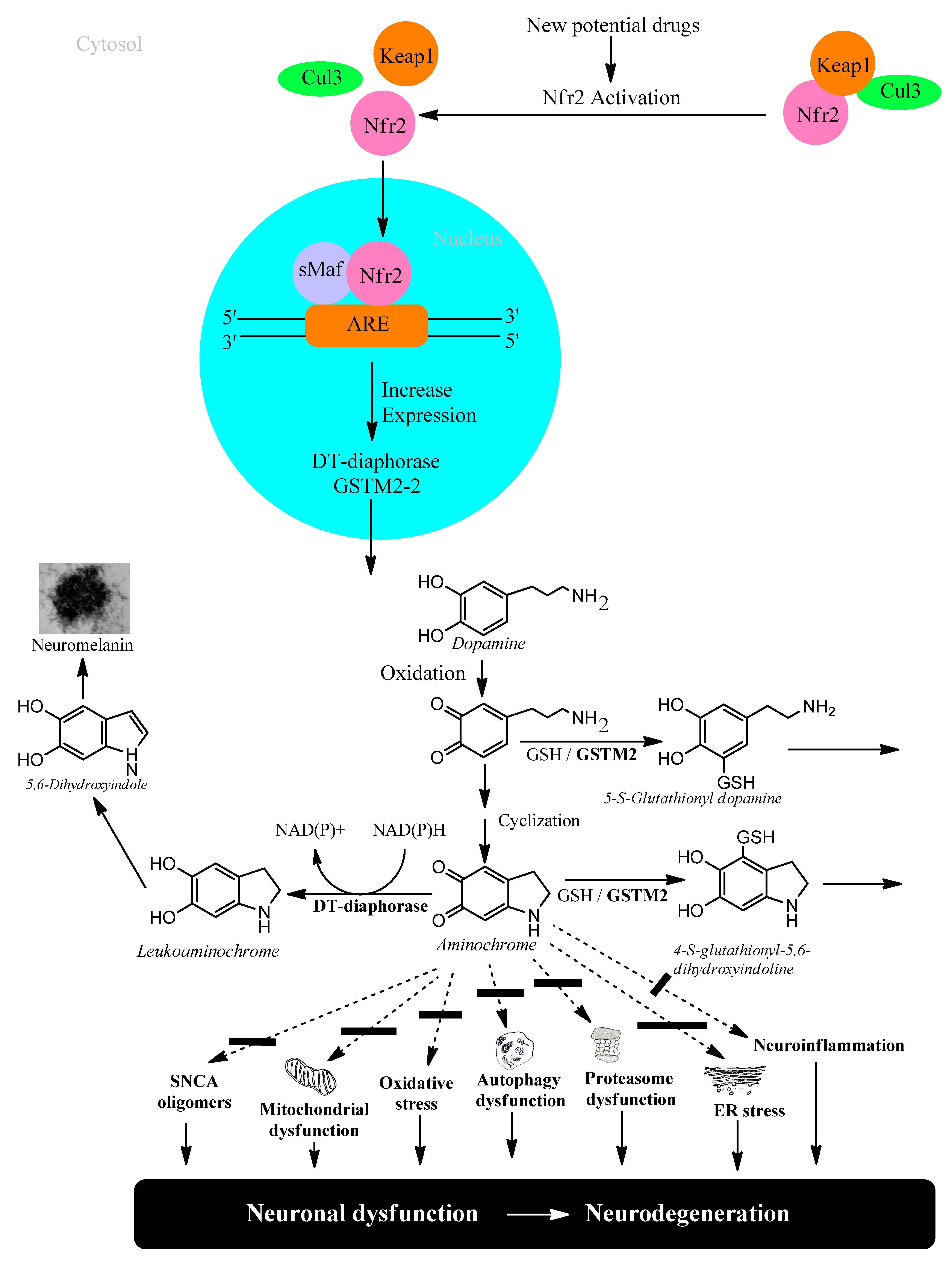
5. Transcriptional Gene Activation via KEAP1/NRF2
6. Neuroprotection against Neurodegeneration of Nigrostriatal System in Parkinson’s Disease
6.1. DT-Diaphorase
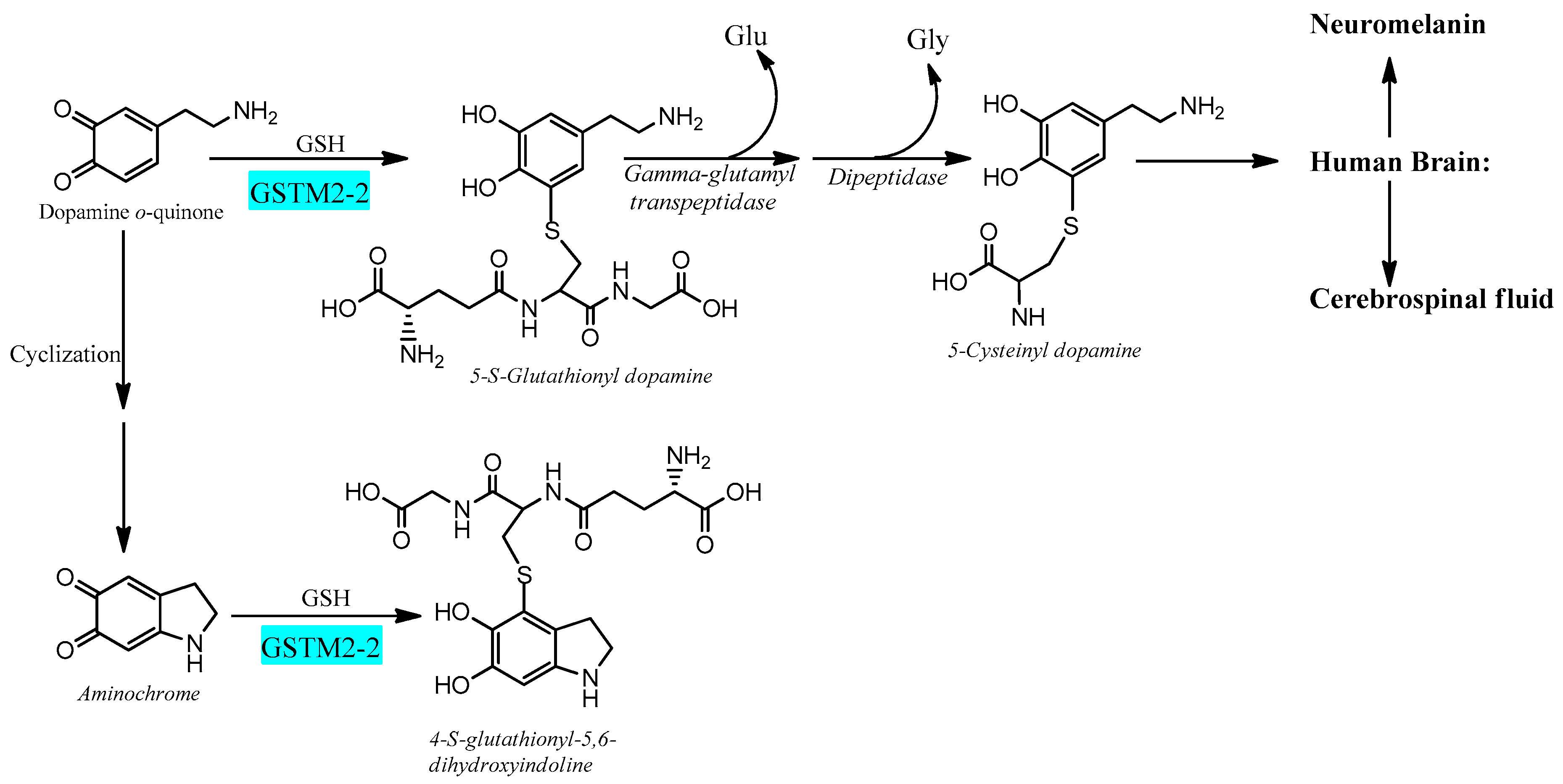
6.2. Glutathione Transferase M2-2
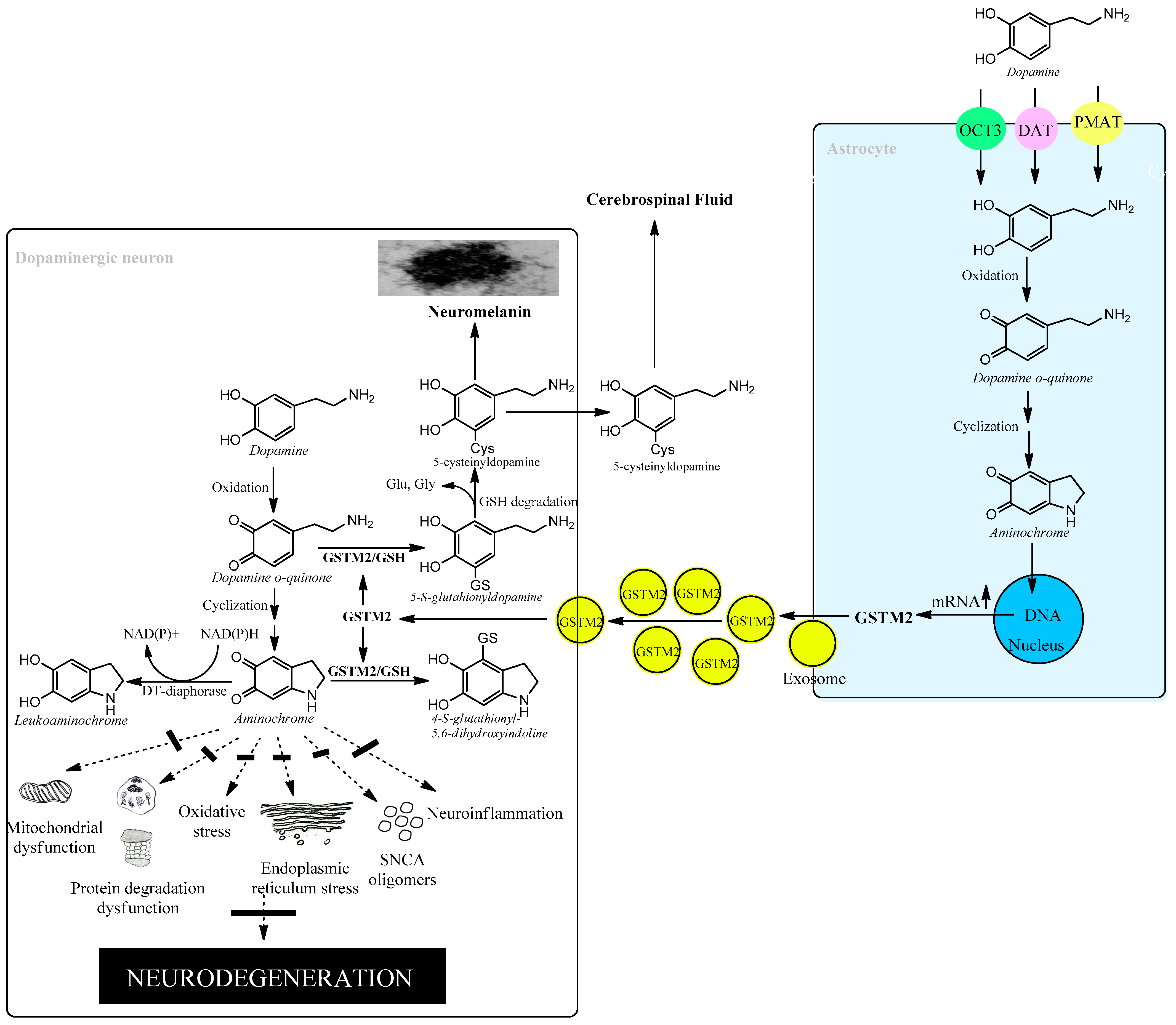
6.3. Astrocytes Protect Dopaminergic Neurons against Aminochrome Neurotoxicity
6.4. Glutathione and Oxidative Stress
7. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Balestrino, R.; Schapira, A.H.V. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology; Pathology, Genetics, a50nd Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12. [Google Scholar] [CrossRef]
- Yoo, J.M.; Lin, Y.; Heo, Y.; Lee, Y.H. Polymorphism in alpha-synuclein oligomers and its implications in toxicity under disease conditions. Front. Mol. Biosci. 2022, 9, 959425. [Google Scholar] [CrossRef]
- Day, J.O.; Mullin, S. The Genetics of Parkinson’s Disease and Implications for Clinical Practice. Genes 2021, 12, 1006. [Google Scholar] [CrossRef] [PubMed]
- Mercado, G.; Castillo, V.; Soto, P.; Sidhu, A. ER stress and Parkinson disease: Pathological inputs that converge into the secretory pathway. Brain Res. 2016, 16, 30260–30268. [Google Scholar]
- Moors, T.; Paciotti, S.; Chiasserini, D.; Calabresi, P.; Parnetti, L.; Beccari, T.; van de Berg, W.D. Lysosomal Dysfunction and α-Synuclein Aggregation in Parkinson’s Disease. Diagnostic. Links Mov. Disord. 2016, 6, 791–801. [Google Scholar] [CrossRef]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109 Pt B, 249–257. [Google Scholar] [CrossRef]
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808. [Google Scholar] [CrossRef]
- Segura-Aguilar, J. Parkinson´s disease. In Clinical Studies and Therapies in Parkinson’s Disease: Translations from Preclinical Models; Segura-Aguilar, J., Ed.; Elsevier: Cambridge, MA, USA, 2021; pp. 1–175. [Google Scholar]
- Segura-Aguilar, J.; Metodiewa, D.; Welch, C.J. Metabolic activation of dopamine o-quinones to o-semiquinones by NADPH cytochrome P450 reductase may play an important role in oxidative stress and apoptotic effects. Biochim. Biophys. Acta 1998, 1381, 1–6. [Google Scholar] [CrossRef]
- Arriagada, C.; Paris, I.; Sanchez de las Matas, M.J.; Martinez-Alvarado, P.; Cardenas, S.; Castañeda, P.; Graumann, R.; Perez-Pastene, C.; Olea-Azar, C.; Couve, E.; et al. On the neurotoxicity mechanism of leukoaminochrome o-semiquinone radical derived from dopamine oxidation: Mitochondria damage, necrosis, and hydroxyl radical formation. Neurobiol. Dis. 2004, 16, 468–477. [Google Scholar] [CrossRef]
- Musatov, A.; Robinson, N.C. Susceptibility of mitochondrial electron-transport complexes to oxidative damage. Focus on cytochrome c oxidase. Free Radic. Res. 2012, 46, 1313–1326. [Google Scholar] [CrossRef]
- Jing, L.; He, M.T.; Chang, Y.; Mehta, S.L.; He, Q.P.; Zhang, J.Z.; Li, P.A. Coenzyme Q10 protects astrocytes from ROS-induced damage through inhibition of mitochondria-mediated cell death pathway. Int. J. Biol. Sci. 2015, 11, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beal, M.F. Coenzyme Q10 administration and its potential for treatment of neurodegenerative diseases. Biofactors 1999, 9, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Park, C.G.; Park, M.; Lee, S.H.; Park, H.R.; Lim, J.; Paek, S.H.; Choy, Y.B. Intrastriatal administration of coenzyme Q10 enhances neuroprotection in a Parkinson’s disease rat model. Sci. Rep. 2020, 10, 9572. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Oakes, D.; Shoulson, I.; Henchcliffe, C.; Galpern, W.R.; Haas, R.; Juncos, J.L.; John, G.N.; Tiffini Smith, V.; Bernard, R.; et al. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: No evidence of benefit. JAMA Neurol. 2014, 71, 543–552. [Google Scholar]
- Yoritaka, A.; Kawajiri, S.; Yamamoto, Y.; Nakahara, T.; Ando, M.; Hashimoto, K.; Nagase, M.; Saito, Y.; Hattori, N. Randomized, double-blind, placebo-controlled pilot trial of reduced coenzyme Q10 for Parkinson’s disease. Parkinsonism. Relat. Disord. 2015, 21, 911–916. [Google Scholar] [CrossRef]
- Ghosh, A.; Chandran, K.; Kalivendi, S.V.; Joseph, J.; Antholine, W.E.; Hillard, C.J.; Kanthasamy, A.; Kanthasamy, A.; Kalyanaraman, B. Neuroprotection by a mitochondria-targeted drug in a Parkinson’s disease model. Free Radic. Biol. Med. 2010, 49, 1674–1684. [Google Scholar] [CrossRef] [Green Version]
- Xi, Y.; Feng, D.; Tao, K.; Wang, R.; Shi, Y.; Qin, H.; Murphy, M.P.; Yang, Q.; Zhao, G. MitoQ protects dopaminergic neurons in a 6-OHDA induced PD model by enhancing Mfn2-dependent mitochondrial fusion via activation of PGC-1α. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2859–2870. [Google Scholar] [CrossRef]
- Solesio, M.E.; Prime, T.A.; Logan, A.; Murphy, M.P.; Del Mar Arroyo-Jimenez, M.; Jordán, J.; Galindo, M.F. The mitochondria-targeted anti-oxidant MitoQ reduces aspects of mitochondrial fission in the 6-OHDA cell model of Parkinson’s disease. Biochim. Biophys. Acta 2013, 1832, 174–182. [Google Scholar] [CrossRef] [Green Version]
- Snow, B.J.; Rolfe, F.L.; Lockhart, M.M.; Frampton, C.M.; O’Sullivan, J.D.; Fung, V.; Smith, R.A.; Murphy, M.P.; Taylor, K.M.; Protect Study Group. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov. Disord. 2010, 25, 1670–1674. [Google Scholar] [CrossRef]
- Gong, L.; Zhang, Q.L.; Zhang, N.; Hua, W.Y.; Huang, Y.X.; Di, P.W.; Huang, T.; Xu, X.S.; Liu, C.F.; Hu, L.F.; et al. Neuroprotection by urate on 6-OHDA-lesioned rat model of Parkinson’s disease: Linking to Akt/GSK3β signaling pathway. J. Neurochem. 2012, 123, 876–885. [Google Scholar] [CrossRef]
- Huang, T.T.; Hao, D.L.; Wu, B.N.; Mao, L.L.; Zhang, J. Uric acid demonstrates neuroprotective effect on Parkinson’s disease mice through Nrf2-ARE signaling pathway. Biochem. Biophys. Res. Commun. 2017, 493, 1443–1449. [Google Scholar] [CrossRef]
- Crotty, G.F.; Ascherio, A.; Schwarzschild, M.A. Targeting urate to reduce oxidative stress in Parkinson disease. Exp. Neurol. 2017, 298, 210–224. [Google Scholar] [CrossRef]
- Schwarzschild, M.A.; Ascherio, A.; Casaceli, C.; Curhan, G.C.; Fitzgerald, R.; Kamp, C.; Lungu, C.; Macklin, E.A.; Marek, K.; Mozaffarian, D.; et al. Effect of Urate-Elevating Inosine on Early Parkinson Disease Progression: The SURE-PD3 Randomized Clinical Trial. JAMA 2021, 326, 926–939. [Google Scholar] [PubMed]
- Kostrzewa, R.M.; Jacobowitz, D.M. Pharmacological actions of 6-hydroxydopamine. Pharmacol. Rev. 1974, 26, 199–288. [Google Scholar]
- Simola, N.; Morelli, M.; Carta, A.R. The 6-hydroxydopamine model of Parkinson’s disease. Neurotox. Res. 2007, 11, 151–167. [Google Scholar] [CrossRef]
- Varešlija, D.; Tipton, K.F.; Davey, G.P.; McDonald, A.G. 6-Hydroxydopamine: A far from simple neurotoxin. J. Neural. Transm. 2020, 127, 213–230. [Google Scholar] [CrossRef]
- Martí, M.J.; Saura, J.; Burke, R.E.; Jackson-Lewis, V.; Jiménez, A.; Bonastre, M.; Tolosa, E. Striatal 6-hydroxydopamine induces apoptosis of nigral neurons in the adult rat. Brain Res. 2002, 20, 185–191. [Google Scholar] [CrossRef]
- Williams, A. MPTP parkinsonism. Br. Med. J. 1984, 289, 1401–1402. [Google Scholar] [CrossRef] [Green Version]
- Ni, A.; Ernst, C. Evidence That Substantia Nigra Pars Compacta Dopaminergic Neurons Are Selectively Vulnerable to Oxidative Stress Because They Are Highly Metabolically Active. Front. Cell. Neurosci. 2022, 16, 826193. [Google Scholar] [CrossRef]
- Sikorska, M.; Lanthier, P.; Miller, H.; Beyers, M.; Sodja, C.; Zurakowski, B.; Gangaraju, S.; Pandey, S.; Sandhu, J.K. Nanomicellar formulation of coenzyme Q10 (Ubisol-Q10) effectively blocks ongoing neurodegeneration in the mouse 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model: Potential use as an adjuvant treatment in Parkinson’s disease. Neurobiol. Aging 2014, 35, 2329–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Muñoz, P.; Cardenas, S.; Huenchuguala, S.; Briceño, A.; Couve, E.; Paris, I.; Segura-Aguilar, J. DT-Diaphorase Prevents Aminochrome-Induced Alpha-Synuclein Oligomer Formation and Neurotoxicity. Toxicol. Sci. 2015, 145, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.L.; Gao, J.H.; Du, T.F.; Yi, H.K.; Ma, K.L. Distribution of the Alpha-Synuclein in the Brain and the Primary Organs of the Rhesus Monkey. Appl. Biochem. Biotechnol. 2021, 193, 3187–3201. [Google Scholar] [CrossRef]
- Segura-Aguilar, J. Can we conclude a potential therapeutic action for Parkinson’s disease by using postmortem tissue and a preclinical model based on an exogenous neurotoxin? Cell Death Dis. 2018, 9, 748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakabayashi, K.; Tanji, K.; Odagiri, S.; Miki, Y.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol. Neurobiol. 2013, 47, 495–508. [Google Scholar] [CrossRef]
- Goldstein, D.S. The Catecholaldehyde Hypothesis for the Pathogenesis of Catecholaminergic Neurodegeneration: What We Know and What We Do Not Know. Int. J. Mol. Sci. 2021, 22, 5999. [Google Scholar] [CrossRef]
- Grünblatt, E.; Ruder, J.; Monoranu, C.M.; Riederer, P.; Youdim, M.B.; Mandel, S.A. Differential Alterations in Metabolism and Proteolysis-Related Proteins in Human Parkinson’s Disease Substantia Nigra. Neurotox. Res. 2018, 33, 560–568. [Google Scholar] [CrossRef]
- Biesemeier, A.; Eibl, O.; Eswara, S.; Audinot, J.N.; Wirtz, T.; Pezzoli, G.; Zucca, F.A.; Zecca, L.; Schraermeyer, U. Elemental mapping of Neuromelanin organelles of human Substantia Nigra: Correlative ultrastructural and chemical analysis by analytical transmission electron microscopy and nano-secondary ion mass spectrometry. J. Neurochem. 2016, 138, 339–353. [Google Scholar] [CrossRef]
- Engelen, M.; Vanna, R.; Bellei, C.; Zucca, F.A.; Wakamatsu, K.; Monzani, E.; Ito, S.; Casella, L.; Zecca, L. Neuromelanins of human brain have soluble and insoluble components with dolichols attached to the melanic structure. PLoS ONE 2012, 7, e48490. [Google Scholar] [CrossRef]
- Zucca, F.A.; Capucciati, A.; Bellei, C.; Sarna, M.; Sarna, T.; Monzani, E.; Casella, L.; Zecca, L. Neuromelanins in brain aging and Parkinson’s disease: Synthesis, structure, neuroinflammatory, and neurodegenerative role. IUBMB Life 2023, 75, 55–65. [Google Scholar] [CrossRef]
- Zhang, W.; Phillips, K.; Wielgus, A.R.; Liu, J.; Albertini, A.; Zucca, F.A.; Faust, R.; Qian, S.Y.; Miller, D.S.; Chignell, C.F.; et al. Neuromelanin activates microglia and induces degeneration of dopaminergic neurons: Implications for progression of Parkinson’s disease. Neurotox. Res. 2011, 19, 63–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, C.C.; Araújo, F.M.; Ferreira, R.S.; Silva, V.B.; Silva, J.H.C.; Grangeiro, M.S.; Soares, É.N.; Pereira, É.P.L.; Souza, C.S.; Costa, S.L.; et al. Aminochrome induces microglia and astrocyte activation. Toxicol. In Vitro 2017, 42, 54–60. [Google Scholar] [CrossRef]
- Aguirre, P.; Urrutia, P.; Tapia, V.; Villa, M.; Paris, I.; Segura-Aguilar, J.; Núñez, M.T. The dopamine metabolite aminochrome inhibits mitochondrial complex I and modifies the expression of iron transporters DMT1 and FPN1. Biometals 2012, 25, 795–803. [Google Scholar] [CrossRef]
- Paris, I.; Muñoz, P.; Huenchuguala, S.; Couve, E.; Sanders, L.H.; Greenamyre, J.T.; Caviedes, P.; Segura-Aguilar, J. Autophagy protects against aminochrome-induced cell death in substantia nigra-derived cell line. Toxicol. Sci. 2011, 121, 376–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, R.; Siegel, D.; Ross, D. Quinone-induced protein handling changes: Implications for major protein handling systems in quinone-mediated toxicity. Toxicol. Appl. Pharmacol. 2014, 280, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Zafar, K.S.; Inayat-Hussain, S.H.; Siegel, D.; Bao, A.; Shieh, B.; Ross, D. Overexpression of NQO1 protects human SK-N-MC neuroblastoma cells against dopamine-induced cell death. Toxicol. Lett. 2006, 166, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Huenchuguala, S.; Muñoz, P.; Zavala, P.; Villa, M.; Cuevas, C.; Ahumada, U.; Graumann, R.; Nore, B.; Couve, E.; Mannervik, B.; et al. Glutathione transferase mu 2 protects glioblastoma cells against aminochrome toxicity by preventing autophagy and lysosome dysfunction. Autophagy 2014, 10, 618–630. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, P.; Huenchuguala, S.; Paris, I.; Segura-Aguilar, J. Dopamine oxidation and autophagy. Parkinsons. Dis. 2012, 2012, 920953. [Google Scholar] [CrossRef] [Green Version]
- Segura-Aguilar, J. Dopamine oxidation to neuromelanin and neurotoxic metabolites. In Clinical Studies and Therapies in Parkinson’s Disease: Translations from Preclinical Models; Segura-Aguilar, J., Ed.; Elsevier: Cambridge, MA, USA, 2021; pp. 213–227. [Google Scholar]
- Bisaglia, M.; Mammi, S.; Bubacco, L. Kinetic and structural analysis of the early oxidation products of dopamine: Analysis of the interactions with alpha-synuclein. J. Biol. Chem. 2007, 282, 1559715-605. [Google Scholar] [CrossRef] [Green Version]
- Segura-Aguilar, J. Neuroprotective mechanisms against dopamine oxidation-dependent neurotoxicity. In Clinical Studies and Therapies in Parkinson’s Disease: Translations from Preclinical Models; Segura-Aguilar, J., Ed.; Elsevier: Cambridge, MA, USA, 2021; pp. 229–240. [Google Scholar]
- Paris, I.; Perez-Pastene, C.; Cardenas, S.; Iturriaga-Vasquez, P.; Muñoz, P.; Couve, E.; Caviedes, P.; Segura-Aguilar, J. Aminochrome induces disruption of actin, alpha-, and beta-tubulin cytoskeleton networks in substantia-nigra-derived cell line. Neurotox. Res. 2010, 18, 82–92. [Google Scholar] [CrossRef]
- Briceño, A.; Muñoz, P.; Brito, P.; Huenchuguala, S.; Segura-Aguilar, J.; Paris, I.B. Aminochrome Toxicity is Mediated by Inhibition of Microtubules Polymerization Through the Formation of Adducts with Tubulin. Neurotox. Res. 2016, 29, 381–393. [Google Scholar] [CrossRef]
- Meléndez, C.; Muñoz, P.; Segura-Aguilar, J. DT-Diaphorase Prevents Aminochrome-Induced Lysosome Dysfunction in SH-SY5Y Cells. Neurotox. Res. 2019, 35, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Herrera, A.; Muñoz, P.; Paris, I.; Díaz-Veliz, G.; Mora, S.; Inzunza, J.; Hultenby, K.; Cardenas, C.; Jaña, F.; Raisman-Vozari, R.; et al. Aminochrome induces dopaminergic neuronal dysfunction: A new animal model for Parkinson’s disease. Cell. Mol. Life Sci. 2016, 73, 3583–3597. [Google Scholar] [CrossRef] [PubMed]
- Carballo-Carbajal, I.; Laguna, A.; Romero-Giménez, J.; Cuadros, T.; Bové, J.; Martinez-Vicente, M.; Parent, A.; Gonzalez-Sepulveda, M.; Peñuelas, N.; Torra, A.; et al. Brain tyrosinase overexpression implicates age-dependent neuromelanin production in Parkinson’s disease pathogenesis. Nat. Commun. 2019, 10, 973. [Google Scholar] [CrossRef] [Green Version]
- Do, H.; Kang, E.; Yang, B.; Cha, H.J.; Choi, Y.S. A tyrosinase, mTyr-CNK, that is functionally available as a monophenol monooxygenase. Sci. Rep. 2017, 7, 17267. [Google Scholar] [CrossRef] [Green Version]
- Yamamuro, Y.; Ogura, S. Regional expression of tyrosinase in central catecholaminergic systems of colored mice. Exp. Anim. 2019, 68, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Tribl, F.; Arzberger, T.; Riederer, P.; Gerlach, M. Tyrosinase is not detected in human catecholaminergic neurons by immunohistochemistry and Western blot analysis. J. Neural. Transm. Suppl. 2007, 72, 51–55. [Google Scholar] [CrossRef]
- Zucca, F.A.; Vanna, R.; Cupaioli, F.A.; Bellei, C.; De Palma, A.; Di Silvestre, D.; Mauri, P.; Grassi, S.; Prinetti, A.; Casella, L.; et al. Neuromelanin organelles are specialized autolysosomes that accumulate undegraded proteins and lipids in aging human brain and are likely involved in Parkinson’s disease. NPJ Parkinsons. Dis. 2018, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Capucciati, A.; Zucca, F.A.; Monzani, E.; Zecca, L.; Casella, L.; Hofer, T. Interaction of Neuromelanin with Xenobiotics and Consequences for Neurodegeneration; Promising Experimental Models. Antioxidants 2021, 10, 824. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.X.; Yang, R.; Zhang, F. Role of Nrf2 in Parkinson’s Disease: Toward New Perspectives. Front. Pharmacol. 2022, 13, 919233. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Fariello, R.; Riederer, P.; Sulzer, D.; Gatti, A.; Tampellini, D. The absolute concentration of nigral neuromelanin, assayed by a new sensitive method, increases throughout the life and is dramatically decreased in Parkinson’s disease. FEBS Lett. 2002, 510, 216–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segura-Aguilar, J.; Muñoz, P.; Inzunza, J.; Varshney, M.; Nalvarte, I.; Mannervik, B. Neuroprotection against Aminochrome Neurotoxicity: Glutathione Transferase M2-2 and DT-Diaphorase. Antioxidants 2022, 11, 296. [Google Scholar] [CrossRef]
- Lozano, J.; Muñoz, P.; Nore, B.F.; Ledoux, S.; Segura-Aguilar, J. Stable expression of short interfering RNA for DT-diaphorase induces neurotoxicity. Chem. Res. Toxicol. 2010, 23, 1492–1496. [Google Scholar] [CrossRef] [PubMed]
- Huenchuguala, S.; Muñoz, P.; Graumann, R.; Paris, I.; Segura-Aguilar, J. DT-diaphorase protects astrocytes from aminochrome-induced toxicity. Neurotoxicology 2016, 55, 10–12. [Google Scholar] [CrossRef]
- Mannervik, B. Evolution of glutathione transferases and related enzymes for the protection of cells against electrophiles. Biochem. Soc. Trans. 1996, 24, 878–880. [Google Scholar] [CrossRef] [Green Version]
- Mannervik, B.; Board, P.G.; Hayes, J.D.; Listowsky, I.; Pearson, W.R. Nomenclature for mammalian soluble glutathione transferases. Methods Enzymol. 2005, 401, 1–8. [Google Scholar]
- Jakobsson, P.J.; Morgenstern, R.; Mancini, J.; Ford-Hutchinson, A.; Persson, B. Common structural features of MAPEG—A widespread superfamily of membrane associated proteins with highly divergent functions in eicosanoid and glutathione metabolism. Protein Sci. 1999, 8, 689–692. [Google Scholar] [CrossRef] [Green Version]
- Mannervik, B.; Jensson, H. Binary combinations of four protein subunits with different catalytic specificities explain the relationship between six basic glutathione S-transferases in rat liver cytosol. J. Biol. Chem. 1982, 257, 9909–9912. [Google Scholar] [CrossRef]
- Segura-Aguilar, J.; Baez, S.; Widersten, M.; Welch, C.J.; Mannervik, B. Human class Mu glutathione transferases, in particular isoenzyme M2-2, catalyze detoxication of the dopamine metabolite aminochrome. J. Biol. Chem. 1997, 272, 5727–5731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baez, S.; Segura-Aguilar, J.; Widersten, M.; Johansson, A.S.; Mannervik, B. Glutathione transferases catalyse the detoxication of oxidized metabolites (o-quinones) of catecholamines and may serve as an antioxidant system preventing degenerative cellular processes. Biochem. J. 1997, 324, 25–28. [Google Scholar] [CrossRef] [Green Version]
- Dagnino-Subiabre, A.; Cassels, B.K.; Baez, S.; Johansson, A.S.; Mannervik, B.; Segura-Aguilar, J. Glutathione transferase M2-2 catalyzes conjugation of dopamine and dopa o-quinones. Biochem. Biophys. Res. Commun. 2000, 274, 32–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, F.C.; Kuo, J.S.; Chia, L.G.; Dryhurst, G. Elevated 5-S-cysteinyldopamine/homovanillic acid ratio and reduced homovanillic acid in cerebrospinal fluid: Possible markers for and potential insights into the pathoetiology of Parkinson’s disease. J. Neural. Transm. 1996, 103, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Rosengren, E.; Linder-Eliasson, E.; Carlsson, A. Detection of 5-S-cysteinyldopamine in human brain. J. Neural. Transm. 1985, 63, 247–253. [Google Scholar] [CrossRef]
- Mosca, L.; Lendaro, E.; d’Erme, M.; Marcellini, S.; Moretti, S.; Rosei, M.A. 5-S-Cysteinyl-dopamine effect on the human dopaminergic neuroblastoma cell line SH-SY5Y. Neurochem. Int. 2006, 49, 262–269. [Google Scholar] [CrossRef]
- Cuevas, C.; Huenchuguala, S.; Muñoz, P.; Villa, M.; Paris, I.; Mannervik, B.; Segura-Aguilar, J. Glutathione transferase-M2-2 secreted from glioblastoma cell protects SH-SY5Y cells from aminochrome neurotoxicity. Neurotox. Res. 2015, 27, 217–228. [Google Scholar] [CrossRef]
- Valdes, R.; Armijo, A.; Muñoz, P.; Hultenby, K.; Hagg, A.; Inzunza, J.; Nalvarte, I.; Varshney, M.; Mannervik, B.; Segura-Aguilar, J. Cellular Trafficking of Glutathione Transferase M2-2 Between U373MG and SHSY-S7 Cells is Mediated by Exosomes. Neurotox Res. 2021, 39, 182–190. [Google Scholar] [CrossRef]
- Segura-Aguilar, J.; Mannervik, B.; Inzunza, J.; Varshney, M.; Nalvarte, I.; Muñoz, P. Astrocytes protect dopaminergic neurons against aminochrome neurotoxicity. Neural. Regen. Res. 2022, 17, 1861–1866. [Google Scholar] [CrossRef]
- Labarrere, C.A.; Kassab, G.S. Glutathione: A Samsonian life-sustaining small molecule that protects against oxidative stress, ageing and damaging inflammation. Front. Nutr. 2022, 9, 1007816. [Google Scholar] [CrossRef]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef]
- Jenner, P. Oxidative damage in neurodegenerative disease. Lancet 1994, 344, 796–798. [Google Scholar] [CrossRef] [PubMed]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol. 1994, 36, 348–355. [Google Scholar] [CrossRef]
- Aoyama, K. Glutathione in the Brain. Int. J. Mol. Sci. 2021, 22, 5010. [Google Scholar] [CrossRef]
- Lin, K.J.; Chen, S.D.; Lin, K.L.; Liou, C.-W.; Lan, M.-Y.; Chuang, Y.-C.; Wang, P.-W.; Lee, J.-J.; Wang, F.-S.; Lin, H.-Y.; et al. Iron Brain Menace: The Involvement of Ferroptosis in Parkinson Disease. Cells 2022, 11, 3829. [Google Scholar] [CrossRef]
- Bridges, R.J.; Natale, N.R.; Patel, S.A. System xc⁻ cystine/glutamate antiporter: An update on molecular pharmacology and roles within the CNS. Br. J. Pharmacol. 2012, 165, 20–34. [Google Scholar] [CrossRef] [Green Version]
- Holmay, M.J.; Terpstra, M.; Coles, L.D.; Mishra, U.; Ahlskog, M.; Öz, G.; Cloyd, J.C.; Tuite, P.J. N-Acetylcysteine boosts brain and blood glutathione in Gaucher and Parkinson diseases. Clin. Neuropharmacol. 2013, 36, 103–106. [Google Scholar] [CrossRef] [Green Version]
- Stockwell, B.R.; Jiang, X.; Gu, W. Emerging Mechanisms and Disease Relevance of Ferroptosis. Trends Cell. Biol. 2020, 30, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.L.; Yuan, L.; Li, W.; Li, J.Y. Ferroptosis in Parkinson’s disease: Glia-neuron crosstalk. Trends Mol. Med. 2022, 28, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Flohé, L. Regulatory Phenomena in the Glutathione Peroxidase Superfamily. Antioxid. Redox. Signal 2020, 33, 498–516. [Google Scholar] [CrossRef]
- Zhang, M.; An, C.; Gao, Y.; Leak, R.K.; Chen, J.; Zhang, F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog. Neurobiol. 2013, 100, 30–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayaraj, P.; Narasimhulu, C.A.; Rajagopalan, S.; Parthasarathy, S.; Desikan, R. Sesamol: A powerful functional food ingredient from sesame oil for cardioprotection. Food Funct. 2020, 11, 1198–1210. [Google Scholar] [CrossRef]
- Lai, Y.; Zeng, H.; He, M.; Qian, H.; Wu, Z.; Luo, Z.; Xue, Y.; Yao, G.; Zhang, Y. 6,8-Di-C-methyl-flavonoids with neuroprotective activities from Rhododendron fortunei. Fitoterapia 2016, 112, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Ying, L.; Wang, D.; Du, G. Analysis of Bioactive Components in the Fruit, Roots, and Leaves of Alpinia oxyphylla by UPLC-MS/MS. Evid. Based Complement. Alternat. Med. 2021, 2021, 5592518. [Google Scholar] [CrossRef] [PubMed]
- Seong Choi, K.; Shin, T.S.; Chun, J.; Ahn, G.; Jeong Han, E.; Kim, M.J.; Kim, J.B.; Kim, S.H.; Kho, K.H.; Heon Kim, D.; et al. Sargahydroquinoic acid isolated from Sargassum serratifolium as inhibitor of cellular basophils activation and passive cutaneous anaphylaxis in mice. Int. Immunopharmacol. 2022, 105, 108567. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Zhao, Y.; Liu, Y.; Zhang, S.; Zhao, W.; Liu, S.; Liu, M. Neuroprotective effect of Ginsenoside Re against neurotoxin-induced Parkinson’s disease models via induction of Nrf2. Mol. Med. Rep. 2022, 25, 215. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.Y.; Liu, G.C.; Zhang, J.X.; Wang, L.H.; Xu, C.; Yan, Z.A.; Wang, A.; Su, Y.F.; Lee, J.J.; Piao, G.C.; et al. Pharmacological Properties of Ginsenoside Re. Front. Pharmacol. 2022, 13, 754191. [Google Scholar] [CrossRef]
- Schepici, G.; Bramanti, P.; Mazzon, E. Efficacy of Sulforaphane in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8637. [Google Scholar] [CrossRef]
- Gao, Y.; Chu, S.F.; Li, J.P.; Zhang, Z.; Yan, J.Q.; Wen, Z.L.; Xia, C.Y.; Mou, Z.; Wang, Z.Z.; He, W.B.; et al. Protopanaxtriol protects against 3-nitropropionic acid-induced oxidative stress in a rat model of Huntington’s disease. Acta Pharmacol. Sin. 2015, 36, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Emran, T.B.; Islam, F.; Nath, N.; Sutradhar, H.; Das, R.; Mitra, S.; Alshahrani, M.M.; Alhasaniah, A.H.; Sharma, R. Naringin and Naringenin Polyphenols in Neurological Diseases: Understandings from a Therapeutic Viewpoint. Life 2022, 13, 99. [Google Scholar] [CrossRef]
- Ren, B.; Yuan, T.; Diao, Z.; Zhang, C.; Liu, Z.; Liu, X. Protective effects of sesamol on systemic oxidative stress-induced cognitive impairments via regulation of Nrf2/Keap1 pathway. Food Funct. 2018, 9, 5912–5924. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Chen, Y.; Wang, X.; Cui, G.; Ung, C.O.L.; Lu, J.H.; Cong, W.; Tang, B.; Lee, S.M. Oxyphylla A ameliorates cognitive deficits and alleviates neuropathology via the Akt-GSK3β and Nrf2-Keap1-HO-1 pathways in vitro and in vivo murine models of Alzheimer’s disease. J. Adv. Res. 2021, 34, 1–12. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Segura-Aguilar, J.; Mannervik, B. A Preclinical Model for Parkinson’s Disease Based on Transcriptional Gene Activation via KEAP1/NRF2 to Develop New Antioxidant Therapies. Antioxidants 2023, 12, 673. https://doi.org/10.3390/antiox12030673
Segura-Aguilar J, Mannervik B. A Preclinical Model for Parkinson’s Disease Based on Transcriptional Gene Activation via KEAP1/NRF2 to Develop New Antioxidant Therapies. Antioxidants. 2023; 12(3):673. https://doi.org/10.3390/antiox12030673
Chicago/Turabian StyleSegura-Aguilar, Juan, and Bengt Mannervik. 2023. "A Preclinical Model for Parkinson’s Disease Based on Transcriptional Gene Activation via KEAP1/NRF2 to Develop New Antioxidant Therapies" Antioxidants 12, no. 3: 673. https://doi.org/10.3390/antiox12030673