
Oxidative Stress and Inflammation in Acute and Chronic Lung Injuries
 ,
,  ,
, 
 and
and 
Abstract
:1. Introduction
2. Oxidative Stress and Inflammation in Asthma
2.1. The Role of Oxidative Stress in Asthma Pathophysiology
2.2. Most Common Experimental Models of Asthma for the Study of Oxidative Stress
2.3. Therapeutic Strategies in the Treatment of Asthma
2.4. Natural Products and Bioactives
2.5. Diet and Supplements
2.6. Enzyme Mimetics and Bioactives
3. Oxidative Stress and Inflammation in COPD and Emphysema
3.1. Proteolytic Imbalance
3.2. Redox Imbalance
3.3. Experimental Models of Pulmonary Emphysema and COPD
3.4. Antioxidant Therapeutic Strategies for COPD
3.5. Effectiveness of Synthetic Antioxidants in Emphysema/COPD
3.6. Natural Antioxidants Effective against Emphysema/COPD
4. Oxidative Stress and Inflammation in Idiopathic Pulmonary Fibrosis
5. Oxidative Stress and Inflammation in Hyperoxia
5.1. Stress and Oxidative Damage Induced by Hyperoxia
5.2. Relationship between Oxidative Stress and Inflammation in Hyperoxia
6. Oxidative Stress and Inflammation in Sepsis
6.1. Sepsis and Acute Lung Injury/Acute Respiratory Distress Syndrome
6.2. Antioxidant Treatment in Sepsis
7. Oxidative Stress and Inflammation in Ventilator-Induced Lung Injury (VILI)
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schluger, N.W.; Koppaka, R. Lung disease in a global context. A call for public health action. Ann. Am. Thorac. Soc. 2014, 11, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, J.; Kiley, J.; Bateman, E.D.; Viegi, G.; Cruz, A.A.; Khaltaev, N.; Ait Khaled, N.; Baena-Cagnani, C.E.; Barreto, M.L.; Billo, N.; et al. Prioritised research agenda for prevention and control of chronic respiratory diseases. Eur. Respir. J. 2010, 36, 995–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, Z.J.; Hoffmann, B.; Morawska, L.; Adams, M.; Furman, E.; Yorgancioglu, A.; Greenbaum, D.; Neira, M.; Brunekreef, B.; Forastiere, F.; et al. Air pollution and COVID-19: Clearing the air and charting a post-pandemic course: A joint workshop report of ERS, ISEE, HEI and WHO. Eur. Respir. J. 2021, 58, 2101063. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Ciminieri, C.; Hu, Q.; Lehmann, M.; Konigshoff, M.; Gosens, R. WNT Signalling in Lung Physiology and Pathology. Handb. Exp. Pharmacol. 2021, 269, 305–336. [Google Scholar] [CrossRef]
- Lammi, M.R.; Baughman, R.P.; Birring, S.S.; Russell, A.M.; Ryu, J.H.; Scholand, M.; Distler, O.; LeSage, D.; Sarver, C.; Antoniou, K.; et al. Outcome Measures for Clinical Trials in Interstitial Lung Diseases. Curr. Respir. Med. Rev. 2015, 11, 163–174. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Zhao, S.; Huang, H.; Wang, W.; Zhang, T.; Zhou, Z.; Feng, Q.; Liang, J.; Xiao, Z.; Hui, Z.; et al. Treatment outcomes of patients with stage III non-small cell lung cancer and interstitial lung diseases receiving intensity-modulated radiation therapy: A single-center experience of 85 cases. Thorac. Cancer 2022, 13, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Pratt, C.A.; Brown, A.G.M.; Dixit, S.; Farmer, N.; Natarajan, A.; Boyington, J.; Shi, S.; Lu, Q.; Cotton, P. Perspectives on Precision Nutrition Research in Heart, Lung, and Blood Diseases and Sleep Disorders. Adv. Nutr. 2022, 13, 1402–1414. [Google Scholar] [CrossRef]
- Brown, A.G.M.; Desvigne-Nickens, P.M.; Redmond, N.; Barnes, V.I.; Campo, R.A. National Heart, Lung, and Blood Institute: Social Determinants of Health Research, Fiscal Year 2008-2020. Am. J. Prev. Med. 2022, 63, 85–92. [Google Scholar] [CrossRef]
- Andreadis, A.A.; Hazen, S.L.; Comhair, S.A.; Erzurum, S.C. Oxidative and nitrosative events in asthma. Free Radic. Biol. Med. 2003, 35, 213–225. [Google Scholar] [CrossRef]
- Sinyor, B.; Concepcion Perez, L. Pathophysiology Of Asthma. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2022. [Google Scholar]
- Donnelly, L.E.; Barnes, P.J. Chemokine receptors as therapeutic targets in chronic obstructive pulmonary disease. Trends Pharmacol. Sci. 2006, 27, 546–553. [Google Scholar] [CrossRef]
- Kudo, M.; Khalifeh Soltani, S.M.; Sakuma, S.A.; McKleroy, W.; Lee, T.H.; Woodruff, P.G.; Lee, J.W.; Huang, K.; Chen, C.; Arjomandi, M.; et al. Mfge8 suppresses airway hyperresponsiveness in asthma by regulating smooth muscle contraction. Proc. Natl. Acad. Sci. USA 2013, 110, 660–665. [Google Scholar] [CrossRef] [Green Version]
- Rahman, I.; Morrison, D.; Donaldson, K.; MacNee, W. Systemic oxidative stress in asthma, COPD, and smokers. Am. J. Respir. Crit. Care Med. 1996, 154, 1055–1060. [Google Scholar] [CrossRef]
- Jaakkola, M.S.; Ieromnimon, A.; Jaakkola, J.J. Are atopy and specific IgE to mites and molds important for adult asthma? J. Allergy Clin. Immunol. 2006, 117, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Maneechotesuwan, K.; Yao, X.; Ito, K.; Jazrawi, E.; Usmani, O.S.; Adcock, I.M.; Barnes, P.J. Suppression of GATA-3 nuclear import and phosphorylation: A novel mechanism of corticosteroid action in allergic disease. PLoS Med. 2009, 6, e1000076. [Google Scholar] [CrossRef] [Green Version]
- Chapman, D.G.; Irvin, C.G. Mechanisms of airway hyper-responsiveness in asthma: The past, present and yet to come. Clin. Exp. Allergy 2015, 45, 706–719. [Google Scholar] [CrossRef] [Green Version]
- O’Byrne, P.M.; Gauvreau, G.M.; Brannan, J.D. Provoked models of asthma: What have we learnt? Clin. Exp. Allergy 2009, 39, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Boulet, L.P. Airway remodeling in asthma: Update on mechanisms and therapeutic approaches. Curr. Opin. Pulm. Med. 2018, 24, 56–62. [Google Scholar] [CrossRef]
- Bergeron, C.; Boulet, L.P. Structural changes in airway diseases: Characteristics, mechanisms, consequences, and pharmacologic modulation. Chest 2006, 129, 1068–1087. [Google Scholar] [CrossRef]
- An, S.S.; Bai, T.R.; Bates, J.H.; Black, J.L.; Brown, R.H.; Brusasco, V.; Chitano, P.; Deng, L.; Dowell, M.; Eidelman, D.H.; et al. Airway smooth muscle dynamics: A common pathway of airway obstruction in asthma. Eur. Respir. J. 2007, 29, 834–860. [Google Scholar] [CrossRef]
- Busse, W.W. The relationship of airway hyperresponsiveness and airway inflammation: Airway hyperresponsiveness in asthma: Its measurement and clinical significance. Chest 2010, 138, 4S–10S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosoki, K.; Nakamura, A.; Kainuma, K.; Sugimoto, M.; Nagao, M.; Hiraguchi, Y.; Tanida, H.; Tokuda, R.; Wada, H.; Nobori, T.; et al. Differential activation of eosinophils by bacteria associated with asthma. Int. Arch. Allergy Immunol. 2013, 161 (Suppl. 2), 16–22. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Samoszuk, M.K.; Comhair, S.A.; Thomassen, M.J.; Farver, C.F.; Dweik, R.A.; Kavuru, M.S.; Erzurum, S.C.; Hazen, S.L. Eosinophils generate brominating oxidants in allergen-induced asthma. J. Clin. Investig. 2000, 105, 1455–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, M.J.; Hawkins, C.L. The Role of Myeloperoxidase in Biomolecule Modification, Chronic Inflammation, and Disease. Antioxid. Redox Signal. 2020, 32, 957–981. [Google Scholar] [CrossRef] [Green Version]
- Hampton, M.B.; Kettle, A.J.; Winterbourn, C.C. Inside the neutrophil phagosome: Oxidants, myeloperoxidase, and bacterial killing. Blood 1998, 92, 3007–3017. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.G.; Garg, M.L.; Blake, R.J.; Simpson, J.L.; Gibson, P.G. Oxidized vitamin E and glutathione as markers of clinical status in asthma. Clin. Nutr. 2008, 27, 579–586. [Google Scholar] [CrossRef]
- Shanmugasundaram, K.R.; Kumar, S.S.; Rajajee, S. Excessive free radical generation in the blood of children suffering from asthma. Clin. Chim. Acta 2001, 305, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Tiotiu, A.; Zounemat Kermani, N.; Badi, Y.; Pavlidis, S.; Hansbro, P.M.; Guo, Y.K.; Chung, K.F.; Adcock, I.M.; team, U.B.c.p. Sputum macrophage diversity and activation in asthma: Role of severity and inflammatory phenotype. Allergy 2021, 76, 775–788. [Google Scholar] [CrossRef]
- Slauch, J.M. How does the oxidative burst of macrophages kill bacteria? Still an open question. Mol. Microbiol. 2011, 80, 580–583. [Google Scholar] [CrossRef] [Green Version]
- Chung, K.F. Cytokines in chronic obstructive pulmonary disease. Eur. Respir. J. Suppl. 2001, 34, 50s–59s. [Google Scholar] [CrossRef] [Green Version]
- Chung, K.F.; Barnes, P.J. Cytokines in asthma. Thorax 1999, 54, 825–857. [Google Scholar] [CrossRef] [Green Version]
- Yuan, B.; Tang, W.H.; Lu, L.J.; Zhou, Y.; Zhu, H.Y.; Zhou, Y.L.; Zhang, H.H.; Hu, C.Y.; Xu, G.Y. TLR4 upregulates CBS expression through NF-kappaB activation in a rat model of irritable bowel syndrome with chronic visceral hypersensitivity. World J. Gastroenterol. 2015, 21, 8615–8628. [Google Scholar] [CrossRef]
- Brune, B.; Dehne, N.; Grossmann, N.; Jung, M.; Namgaladze, D.; Schmid, T.; von Knethen, A.; Weigert, A. Redox control of inflammation in macrophages. Antioxid. Redox Signal. 2013, 19, 595–637. [Google Scholar] [CrossRef] [Green Version]
- Morshed, M.; Yousefi, S.; Stockle, C.; Simon, H.U.; Simon, D. Thymic stromal lymphopoietin stimulates the formation of eosinophil extracellular traps. Allergy 2012, 67, 1127–1137. [Google Scholar] [CrossRef]
- Silveira, J.S.; Antunes, G.L.; Kaiber, D.B.; da Costa, M.S.; Marques, E.P.; Ferreira, F.S.; Gassen, R.B.; Breda, R.V.; Wyse, A.T.S.; Pitrez, P.; et al. Reactive oxygen species are involved in eosinophil extracellular traps release and in airway inflammation in asthma. J. Cell. Physiol. 2019, 234, 23633–23646. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Ichikawa, T.; Numakura, T.; Yamada, M.; Koarai, A.; Fujino, N.; Murakami, K.; Yamanaka, S.; Sasaki, Y.; Kyogoku, Y.; et al. PGC-1alpha regulates airway epithelial barrier dysfunction induced by house dust mite. Respir. Res. 2021, 22, 63. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.S.; Chung, K.F. Blood neutrophil activation markers in severe asthma: Lack of inhibition by prednisolone therapy. Respir. Res. 2006, 7, 59. [Google Scholar] [CrossRef] [Green Version]
- McGovern, T.K.; Chen, M.; Allard, B.; Larsson, K.; Martin, J.G.; Adner, M. Neutrophilic oxidative stress mediates organic dust-induced pulmonary inflammation and airway hyperresponsiveness. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L155–L165. [Google Scholar] [CrossRef] [Green Version]
- Grabcanovic-Musija, F.; Obermayer, A.; Stoiber, W.; Krautgartner, W.D.; Steinbacher, P.; Winterberg, N.; Bathke, A.C.; Klappacher, M.; Studnicka, M. Neutrophil extracellular trap (NET) formation characterises stable and exacerbated COPD and correlates with airflow limitation. Respir. Res. 2015, 16, 59. [Google Scholar] [CrossRef] [Green Version]
- Pham, D.L.; Ban, G.Y.; Kim, S.H.; Shin, Y.S.; Ye, Y.M.; Chwae, Y.J.; Park, H.S. Neutrophil autophagy and extracellular DNA traps contribute to airway inflammation in severe asthma. Clin. Exp. Allergy 2017, 47, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Comhair, S.A.; Erzurum, S.C. Redox control of asthma: Molecular mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 2010, 12, 93–124. [Google Scholar] [CrossRef] [Green Version]
- Sahiner, U.M.; Birben, E.; Erzurum, S.; Sackesen, C.; Kalayci, O. Oxidative stress in asthma. World Allergy Organ. J. 2011, 4, 151–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Auerbach, A.; Hernandez, M.L. The effect of environmental oxidative stress on airway inflammation. Curr. Opin. Allergy Clin. Immunol. 2012, 12, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Nadeem, A.; Masood, A.; Siddiqui, N. Oxidant--antioxidant imbalance in asthma: Scientific evidence, epidemiological data and possible therapeutic options. Ther. Adv. Respir. Dis. 2008, 2, 215–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasti, B.; Liu, S.K.; Xiang, X.D. Role of Epigenetics in the Pathogenesis, Treatment, Prediction, and Cellular Transformation of Asthma. Mediat. Inflamm. 2021, 2021, 9412929. [Google Scholar] [CrossRef]
- Alati, R.; Al Mamun, A.; O’Callaghan, M.; Najman, J.M.; Williams, G.M. In utero and postnatal maternal smoking and asthma in adolescence. Epidemiology 2006, 17, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.L.; Ho, S.M. Environmental epigenetics and asthma: Current concepts and call for studies. Am. J. Respir. Crit. Care Med. 2008, 177, 567–573. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.Y.; Ho, S.M. Epigenetic reprogramming and imprinting in origins of disease. Rev. Endocr. Metab. Disord. 2007, 8, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, Y.; Bossley, C.; Gupta, A.; Akashi, K.; Tsartsali, L.; Mercado, N.; Barnes, P.J.; Bush, A.; Ito, K. Passive smoking impairs histone deacetylase-2 in children with severe asthma. Chest 2014, 145, 305–312. [Google Scholar] [CrossRef] [Green Version]
- Setiawan, H.; Nagaoka, K.; Kubo, M.; Fujikura, Y.; Ogino, K. Involvement of Xanthine Oxidoreductase-related Oxidative Stress in a Dermatophagoides farinae-induced Asthma Model of NC/Nga Mice. Acta Med. Okayama 2016, 70, 175–182. [Google Scholar] [CrossRef]
- Hew, M.; Bhavsar, P.; Torrego, A.; Meah, S.; Khorasani, N.; Barnes, P.J.; Adcock, I.; Chung, K.F. Relative corticosteroid insensitivity of peripheral blood mononuclear cells in severe asthma. Am. J. Respir. Crit. Care Med. 2006, 174, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Fabbri, L.; Peters, S.P.; Pavord, I.; Wenzel, S.E.; Lazarus, S.C.; Macnee, W.; Lemaire, F.; Abraham, E. Allergic rhinitis, asthma, airway biology, and chronic obstructive pulmonary disease in AJRCCM in 2004. Am. J. Respir. Crit. Care Med. 2005, 171, 686–698. [Google Scholar] [CrossRef] [Green Version]
- Gilliland, F.D.; Gauderman, W.J.; Vora, H.; Rappaport, E.; Dubeau, L. Effects of glutathione-S-transferase M1, T1, and P1 on childhood lung function growth. Am. J. Respir. Crit. Care Med. 2002, 166, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Alam, J.; Venkatesan, M.I.; Eiguren-Fernandez, A.; Schmitz, D.; Di Stefano, E.; Slaughter, N.; Killeen, E.; Wang, X.; Huang, A.; et al. Nrf2 is a key transcription factor that regulates antioxidant defense in macrophages and epithelial cells: Protecting against the proinflammatory and oxidizing effects of diesel exhaust chemicals. J. Immunol. 2004, 173, 3467–3481. [Google Scholar] [CrossRef] [Green Version]
- Oberdorster, G. Pulmonary effects of inhaled ultrafine particles. Int. Arch. Occup. Environ. Health 2001, 74, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bowler, R.P.; Crapo, J.D. Oxidative stress in airways: Is there a role for extracellular superoxide dismutase? Am. J. Respir. Crit. Care Med. 2002, 166, S38–S43. [Google Scholar] [CrossRef] [PubMed]
- Comhair, S.A.; Bhathena, P.R.; Dweik, R.A.; Kavuru, M.; Erzurum, S.C. Rapid loss of superoxide dismutase activity during antigen-induced asthmatic response. Lancet 2000, 355, 624. [Google Scholar] [CrossRef]
- Comhair, S.A.; Bhathena, P.R.; Farver, C.; Thunnissen, F.B.; Erzurum, S.C. Extracellular glutathione peroxidase induction in asthmatic lungs: Evidence for redox regulation of expression in human airway epithelial cells. FASEB J. 2001, 15, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.Y.; Jedlicka, A.E.; Reddy, S.P.; Kensler, T.W.; Yamamoto, M.; Zhang, L.Y.; Kleeberger, S.R. Role of NRF2 in protection against hyperoxic lung injury in mice. Am. J. Respir. Cell Mol. Biol. 2002, 26, 175–182. [Google Scholar] [CrossRef]
- Itoh, K.; Tong, K.I.; Yamamoto, M. Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic. Biol. Med. 2004, 36, 1208–1213. [Google Scholar] [CrossRef]
- Jaiswal, A.K. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic. Biol. Med. 2004, 36, 1199–1207. [Google Scholar] [CrossRef]
- Plantinga, M.; Guilliams, M.; Vanheerswynghels, M.; Deswarte, K.; Branco-Madeira, F.; Toussaint, W.; Vanhoutte, L.; Neyt, K.; Killeen, N.; Malissen, B.; et al. Conventional and monocyte-derived CD11b(+) dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity 2013, 38, 322–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debeuf, N.; Haspeslagh, E.; van Helden, M.; Hammad, H.; Lambrecht, B.N. Mouse Models of Asthma. Curr. Protoc. Mouse Biol. 2016, 6, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Birrell, M.A.; Van Oosterhout, A.J.; Belvisi, M.G. Do the current house dust mite-driven models really mimic allergic asthma? Eur. Respir. J. 2010, 36, 1220–1221. [Google Scholar] [CrossRef] [PubMed]
- Saglani, S.; Lloyd, C.M. Novel concepts in airway inflammation and remodelling in asthma. Eur. Respir. J. 2015, 46, 1796–1804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehiro, A.; Lahn, M.; Makela, M.J.; Dakhama, A.; Fujita, M.; Joetham, A.; Mason, R.J.; Born, W.; Gelfand, E.W. Tumor necrosis factor-alpha negatively regulates airway hyperresponsiveness through gamma-delta T cells. Am. J. Respir. Crit. Care Med. 2001, 164, 2229–2238. [Google Scholar] [CrossRef] [PubMed]
- Goplen, N.; Karim, M.Z.; Liang, Q.; Gorska, M.M.; Rozario, S.; Guo, L.; Alam, R. Combined sensitization of mice to extracts of dust mite, ragweed, and Aspergillus species breaks through tolerance and establishes chronic features of asthma. J. Allergy Clin. Immunol. 2009, 123, 925–932. [Google Scholar] [CrossRef] [Green Version]
- Nesi, R.T.; Barroso, M.V.; Souza Muniz, V.; de Arantes, A.C.; Martins, M.A.; Brito Gitirana, L.; Neves, J.S.; Benjamim, C.F.; Lanzetti, M.; Valenca, S.S. Pharmacological modulation of reactive oxygen species (ROS) improves the airway hyperresponsiveness by shifting the Th1 response in allergic inflammation induced by ovalbumin. Free Radic. Res. 2017, 51, 708–722. [Google Scholar] [CrossRef]
- Chang, Y.S.; Kim, Y.K.; Jeon, S.G.; Kim, S.H.; Kim, S.S.; Park, H.W.; Min, K.U.; Kim, Y.Y.; Cho, S.H. Influence of the Adjuvants and Genetic Background on the Asthma Model Using Recombinant Der f 2 in Mice. Immune Netw. 2013, 13, 295–300. [Google Scholar] [CrossRef] [Green Version]
- Gorska, M.M. Mouse Models of Asthma. Methods Mol. Biol. 2018, 1809, 351–362. [Google Scholar] [CrossRef]
- Riccioni, G.; Barbara, M.; Bucciarelli, T.; di Ilio, C.; D’Orazio, N. Antioxidant vitamin supplementation in asthma. Ann. Clin. Lab. Sci. 2007, 37, 96–101. [Google Scholar]
- Papiris, S.A.; Manali, E.D.; Kolilekas, L.; Triantafillidou, C.; Tsangaris, I. Acute severe asthma: New approaches to assessment and treatment. Drugs 2009, 69, 2363–2391. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Burgos, E.; Gomez-Serranillos, M.P. Terpene compounds in nature: A review of their potential antioxidant activity. Curr. Med. Chem. 2012, 19, 5319–5341. [Google Scholar] [CrossRef]
- Hollman, P.C.; Katan, M.B. Health effects and bioavailability of dietary flavonols. Free Radic. Res. 1999, 31 (Suppl. 1), S75–S80. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Holvoet, S.; Mercenier, A. Dietary polyphenols in the prevention and treatment of allergic diseases. Clin. Exp. Allergy 2011, 41, 1346–1359. [Google Scholar] [CrossRef]
- Sakoda, C.P.P.; de Toledo, A.C.; Perini, A.; Pinheiro, N.M.; Hiyane, M.I.; Grecco, S.D.S.; de Fatima Lopes Calvo Tiberio, I.; Camara, N.O.S.; de Arruda Martins, M.; Lago, J.H.G.; et al. Sakuranetin reverses vascular peribronchial and lung parenchyma remodeling in a murine model of chronic allergic pulmonary inflammation. Acta Histochem. 2016, 118, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Toledo, A.C.; Sakoda, C.P.; Perini, A.; Pinheiro, N.M.; Magalhaes, R.M.; Grecco, S.; Tiberio, I.F.; Camara, N.O.; Martins, M.A.; Lago, J.H.; et al. Flavonone treatment reverses airway inflammation and remodelling in an asthma murine model. Br. J. Pharmacol. 2013, 168, 1736–1749. [Google Scholar] [CrossRef] [Green Version]
- Hertog, M.G.; Feskens, E.J.; Hollman, P.C.; Katan, M.B.; Kromhout, D. Dietary antioxidant flavonoids and risk of coronary heart disease: The Zutphen Elderly Study. Lancet 1993, 342, 1007–1011. [Google Scholar] [CrossRef]
- Manach, C.; Texier, O.; Morand, C.; Crespy, V.; Regerat, F.; Demigne, C.; Remesy, C. Comparison of the bioavailability of quercetin and catechin in rats. Free Radic. Biol. Med. 1999, 27, 1259–1266. [Google Scholar] [CrossRef]
- Amalia, P.M.; Possa, M.N.; Augusto, M.C.; Francisca, L.S. Quercetin prevents oxidative stress in cirrhotic rats. Dig. Dis. Sci. 2007, 52, 2616–2621. [Google Scholar] [CrossRef]
- Formica, J.V.; Regelson, W. Review of the biology of Quercetin and related bioflavonoids. Food Chem. Toxicol. 1995, 33, 1061–1080. [Google Scholar] [CrossRef]
- Park, C.; So, H.S.; Shin, C.H.; Baek, S.H.; Moon, B.S.; Shin, S.H.; Lee, H.S.; Lee, D.W.; Park, R. Quercetin protects the hydrogen peroxide-induced apoptosis via inhibition of mitochondrial dysfunction in H9c2 cardiomyoblast cells. Biochem. Pharmacol. 2003, 66, 1287–1295. [Google Scholar] [CrossRef]
- Townsend, E.A.; Emala, C.W., Sr. Quercetin acutely relaxes airway smooth muscle and potentiates beta-agonist-induced relaxation via dual phosphodiesterase inhibition of PLCbeta and PDE4. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L396–L403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, J.; Moreno, J.J. Effect of resveratrol, a natural polyphenolic compound, on reactive oxygen species and prostaglandin production. Biochem. Pharmacol. 2000, 59, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Kode, A.; Rajendrasozhan, S.; Caito, S.; Yang, S.R.; Megson, I.L.; Rahman, I. Resveratrol induces glutathione synthesis by activation of Nrf2 and protects against cigarette smoke-mediated oxidative stress in human lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L478–L488. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; He, F.; Chen, L.; Li, Q.; Jin, S.; Zheng, H.; Lin, J.; Zhang, H.; Ma, S.; Mei, J.; et al. Resveratrol inhibits pulmonary fibrosis by regulating miR-21 through MAPK/AP-1 pathways. Biomed. Pharmacother. 2018, 105, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Li, T.; Li, J.H.; Miao, S.Y.; Xiao, X.Z. The Effects of Resveratrol on Inflammation and Oxidative Stress in a Rat Model of Chronic Obstructive Pulmonary Disease. Molecules 2017, 22, 1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.L.; Zhang, H.G.; Xu, Y.L.; Gao, Y.H.; Yang, X.J.; Hao, X.Q.; Li, X.H. Resveratrol inhibits right ventricular hypertrophy induced by monocrotaline in rats. Clin. Exp. Pharmacol. Physiol. 2010, 37, 150–155. [Google Scholar] [CrossRef]
- Alharris, E.; Mohammed, A.; Alghetaa, H.; Zhou, J.; Nagarkatti, M.; Nagarkatti, P. The Ability of Resveratrol to Attenuate Ovalbumin-Mediated Allergic Asthma Is Associated With Changes in Microbiota Involving the Gut-Lung Axis, Enhanced Barrier Function and Decreased Inflammation in the Lungs. Front. Immunol. 2022, 13, 805770. [Google Scholar] [CrossRef]
- Zhang, H.; Shih, A.; Rinna, A.; Forman, H.J. Resveratrol and 4-hydroxynonenal act in concert to increase glutamate cysteine ligase expression and glutathione in human bronchial epithelial cells. Arch. Biochem. Biophys. 2009, 481, 110–115. [Google Scholar] [CrossRef] [Green Version]
- Andre, D.M.; Calixto, M.C.; Sollon, C.; Alexandre, E.C.; Leiria, L.O.; Tobar, N.; Anhe, G.F.; Antunes, E. Therapy with resveratrol attenuates obesity-associated allergic airway inflammation in mice. Int. Immunopharmacol. 2016, 38, 298–305. [Google Scholar] [CrossRef]
- Koumpagioti, D.; Boutopoulou, B.; Moriki, D.; Priftis, K.N.; Douros, K. Does Adherence to the Mediterranean Diet Have a Protective Effect against Asthma and Allergies in Children? A Systematic Review. Nutrients 2022, 14, 1618. [Google Scholar] [CrossRef] [PubMed]
- Vassilopoulou, E.; Guibas, G.V.; Papadopoulos, N.G. Mediterranean-Type Diets as a Protective Factor for Asthma and Atopy. Nutrients 2022, 14, 1825. [Google Scholar] [CrossRef]
- Niewiem, M.; Grzybowska-Chlebowczyk, U. Intestinal Barrier Permeability in Allergic Diseases. Nutrients 2022, 14, 1893. [Google Scholar] [CrossRef]
- Gilliland, F.D.; Berhane, K.T.; Li, Y.F.; Gauderman, W.J.; McConnell, R.; Peters, J. Children’s lung function and antioxidant vitamin, fruit, juice, and vegetable intake. Am. J. Epidemiol. 2003, 158, 576–584. [Google Scholar] [CrossRef] [Green Version]
- Morabia, A.; Sorenson, A.; Kumanyika, S.K.; Abbey, H.; Cohen, B.H.; Chee, E. Vitamin A, cigarette smoking, and airway obstruction. Am. Rev. Respir. Dis. 1989, 140, 1312–1316. [Google Scholar] [CrossRef] [PubMed]
- Goth, F.E.M.; Schmidt, B.J.; Juul, K.; Albertsen, P.; Agertoft, L.; Jorgensen, I.M. Cohort profile: The vitamin A and D and nitric oxide (AD-ON) observational cohort on lung development and symptoms in premature and mature children in North Zealand, Denmark. BMJ Open 2022, 12, e054952. [Google Scholar] [CrossRef]
- Feng, L.; Sun, F.; Chen, Y.; Athari, S.S.; Chen, X. Studying the Effects of Vitamin A on the Severity of Allergic Rhinitis and Asthma. Iran J. Allergy Asthma Immunol. 2021, 20, 648–692. [Google Scholar] [CrossRef]
- Shorey-Kendrick, L.E.; McEvoy, C.T.; O’Sullivan, S.M.; Milner, K.; Vuylsteke, B.; Tepper, R.S.; Haas, D.M.; Park, B.; Gao, L.; Vu, A.; et al. Impact of vitamin C supplementation on placental DNA methylation changes related to maternal smoking: Association with gene expression and respiratory outcomes. Clin. Epigenet. 2021, 13, 177. [Google Scholar] [CrossRef] [PubMed]
- Abuhajar, S.M.; Taleb, M.H.; Ellulu, M.S. Vitamin C deficiency and risk of metabolic complications among adults with chronic respiratory diseases: A case-control study. Clin. Nutr. ESPEN 2021, 43, 448–455. [Google Scholar] [CrossRef]
- Cerqua, I.; Neukirch, K.; Terlizzi, M.; Granato, E.; Caiazzo, E.; Cicala, C.; Ialenti, A.; Capasso, R.; Werz, O.; Sorrentino, R.; et al. A vitamin E long-chain metabolite and the inspired drug candidate alpha-amplexichromanol relieve asthma features in an experimental model of allergen sensitization. Pharmacol. Res. 2022, 181, 106250. [Google Scholar] [CrossRef]
- Jiang, Q.; Im, S.; Wagner, J.G.; Hernandez, M.L.; Peden, D.B. Gamma-tocopherol, a major form of vitamin E in diets: Insights into antioxidant and anti-inflammatory effects, mechanisms, and roles in disease management. Free Radic. Biol. Med. 2022, 178, 347–359. [Google Scholar] [CrossRef]
- Fadjare Frempong, T.; Owusu Boadi, N.; Badu, M. Optimization of extraction conditions for polyphenols from the stem bark of Funtumia elastica (Funtum) utilizing response surface methodology. AAS Open Res. 2021, 4, 46. [Google Scholar] [CrossRef]
- Chojnacka, K.; Lewandowska, U. The influence of polyphenol-rich extracts on the production of pro-inflammatory mediators in macrophages. J. Physiol. Pharmacol. 2021, 72, 167–176. [Google Scholar] [CrossRef]
- Ni, J.; Wei, J.; Wu, T. Vitamin A for non-measles pneumonia in children. Cochrane Database Syst. Rev. 2005, 2005, CD003700. [Google Scholar] [CrossRef] [PubMed]
- Kucukbay, H.; Yakinci, C.; Kucukbay, F.Z.; Turgut, M. Serum vitamin A and beta-carotene levels in children with recurrent acute respiratory infections and diarrhoea in Malatya. J. Trop. Pediatr. 1997, 43, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Mayne, S.T. Beta-carotene, carotenoids, and disease prevention in humans. FASEB J. 1996, 10, 690–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Macias, H.; Romieu, I. Effects of antioxidant supplements and nutrients on patients with asthma and allergies. J. Allergy Clin. Immunol. 2014, 133, 1237–1244; quiz 1245. [Google Scholar] [CrossRef] [PubMed]
- Cook-Mills, J.M.; Abdala-Valencia, H.; Hartert, T. Two faces of vitamin E in the lung. Am. J. Respir. Crit. Care Med. 2013, 188, 279–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagdic, A.; Sener, O.; Bulucu, F.; Karadurmus, N.; Ozel, H.E.; Yamanel, L.; Tasci, C.; Naharci, I.; Ocal, R.; Aydin, A. Oxidative stress status and plasma trace elements in patients with asthma or allergic rhinitis. Allergol. Immunopathol. (Madr) 2011, 39, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Ginter, E.; Simko, V. Deficiency of vitamin D and vitamin C in the pathogenesis of bronchial asthma. Bratisl. Lek. Listy 2016, 117, 305–307. [Google Scholar] [CrossRef] [Green Version]
- Lan, N.; Luo, G.; Yang, X.; Cheng, Y.; Zhang, Y.; Wang, X.; Wang, X.; Xie, T.; Li, G.; Liu, Z.; et al. 25-Hydroxyvitamin D3-deficiency enhances oxidative stress and corticosteroid resistance in severe asthma exacerbation. PLoS ONE 2014, 9, e111599. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, H.; Sun, X.; Ren, L. The protective role of vitamin D3 in a murine model of asthma via the suppression of TGF-beta/Smad signaling and activation of the Nrf2/HO-1 pathway. Mol. Med. Rep. 2016, 14, 2389–2396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.Y.; Crapo, J.D. Inhibition of airway inflammation and hyperreactivity by an antioxidant mimetic. Free Radic. Biol. Med. 2002, 33, 379–386. [Google Scholar] [CrossRef]
- Larsen, G.L.; White, C.W.; Takeda, K.; Loader, J.E.; Nguyen, D.D.; Joetham, A.; Groner, Y.; Gelfand, E.W. Mice that overexpress Cu/Zn superoxide dismutase are resistant to allergen-induced changes in airway control. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L350–L359. [Google Scholar] [CrossRef]
- Jozsef, L.; Filep, J.G. Selenium-containing compounds attenuate peroxynitrite-mediated NF-kappaB and AP-1 activation and interleukin-8 gene and protein expression in human leukocytes. Free Radic. Biol. Med. 2003, 35, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Steinkamp-Fenske, K.; Bollinger, L.; Xu, H.; Yao, Y.; Horke, S.; Forstermann, U.; Li, H. Reciprocal regulation of endothelial nitric-oxide synthase and NADPH oxidase by betulinic acid in human endothelial cells. J. Pharmacol. Exp. Ther. 2007, 322, 836–842. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.T.; Yang, C.M. Role of NADPH oxidase/ROS in pro-inflammatory mediators-induced airway and pulmonary diseases. Biochem. Pharmacol. 2012, 84, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Sheran, J.; Smith, N.; Fonseca, S.; Lee, A.J. AKL1, a botanical mixture for the treatment of asthma: A randomised, double-blind, placebo-controlled, cross-over study. BMC Pulm. Med. 2007, 7, 4. [Google Scholar] [CrossRef] [Green Version]
- Stefanska, J.; Sarniak, A.; Wlodarczyk, A.; Sokolowska, M.; Pniewska, E.; Doniec, Z.; Nowak, D.; Pawliczak, R. Apocynin reduces reactive oxygen species concentrations in exhaled breath condensate in asthmatics. Exp. Lung Res. 2012, 38, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Hikichi, M.; Mizumura, K.; Maruoka, S.; Gon, Y. Pathogenesis of chronic obstructive pulmonary disease (COPD) induced by cigarette smoke. J. Thorac. Dis. 2019, 11, S2129–S2140. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, J.; Meng, Y.; Adcock, I.M.; Yao, X. Role of inflammatory cells in airway remodeling in COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 2018, 13, 3341–3348. [Google Scholar] [CrossRef] [Green Version]
- Hirota, N.; Martin, J.G. Mechanisms of airway remodeling. Chest 2013, 144, 1026–1032. [Google Scholar] [CrossRef]
- Rahman, I.; Adcock, I.M. Oxidative stress and redox regulation of lung inflammation in COPD. Eur. Respir. J. 2006, 28, 219–242. [Google Scholar] [CrossRef]
- Kirkham, P.A.; Barnes, P.J. Oxidative stress in COPD. Chest 2013, 144, 266–273. [Google Scholar] [CrossRef] [PubMed]
- McGuinness, A.J.; Sapey, E. Oxidative Stress in COPD: Sources, Markers, and Potential Mechanisms. J. Clin. Med. 2017, 6, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vij, N.; Chandramani-Shivalingappa, P.; Van Westphal, C.; Hole, R.; Bodas, M. Cigarette smoke-induced autophagy impairment accelerates lung aging, COPD-emphysema exacerbations and pathogenesis. Am. J. Physiol. Cell Physiol. 2018, 314, C73–C87. [Google Scholar] [CrossRef] [Green Version]
- Dunsmore, S.E. Treatment of COPD: A matrix perspective. Int. J. Chron. Obstruct. Pulmon. Dis. 2008, 3, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Stolz, D.; Leeming, D.J.; Kristensen, J.H.E.; Karsdal, M.A.; Boersma, W.; Louis, R.; Milenkovic, B.; Kostikas, K.; Blasi, F.; Aerts, J.; et al. Systemic Biomarkers of Collagen and Elastin Turnover Are Associated With Clinically Relevant Outcomes in COPD. Chest 2017, 151, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Elkington, P.T.; Friedland, J.S. Matrix metalloproteinases in destructive pulmonary pathology. Thorax 2006, 61, 259–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, C.A. Proteinases and oxidants as targets in the treatment of chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2005, 2, 373–385. [Google Scholar] [CrossRef]
- Birkedal-Hansen, H. Proteolytic remodeling of extracellular matrix. Curr. Opin. Cell Biol. 1995, 7, 728–735. [Google Scholar] [CrossRef]
- Bagdonas, E.; Raudoniute, J.; Bruzauskaite, I.; Aldonyte, R. Novel aspects of pathogenesis and regeneration mechanisms in COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 2015, 10, 995–1013. [Google Scholar] [CrossRef] [Green Version]
- Brusselle, G. Dysregulated fibulin-5 expression and elastogenesis in COPD lungs: Pyromaniac or fire fighter? Thorax 2015, 70, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Brandsma, C.A.; van den Berge, M.; Postma, D.S.; Jonker, M.R.; Brouwer, S.; Pare, P.D.; Sin, D.D.; Bosse, Y.; Laviolette, M.; Karjalainen, J.; et al. A large lung gene expression study identifying fibulin-5 as a novel player in tissue repair in COPD. Thorax 2015, 70, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Yanagisawa, H.; Schluterman, M.K.; Brekken, R.A. Fibulin-5, an integrin-binding matricellular protein: Its function in development and disease. J. Cell Commun. Signal. 2009, 3, 337–347. [Google Scholar] [CrossRef] [Green Version]
- Molet, S.; Belleguic, C.; Lena, H.; Germain, N.; Bertrand, C.P.; Shapiro, S.D.; Planquois, J.M.; Delaval, P.; Lagente, V. Increase in macrophage elastase (MMP-12) in lungs from patients with chronic obstructive pulmonary disease. Inflamm. Res. 2005, 54, 31–36. [Google Scholar] [CrossRef]
- Hautamaki, R.D.; Kobayashi, D.K.; Senior, R.M.; Shapiro, S.D. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science 1997, 277, 2002–2004. [Google Scholar] [CrossRef]
- Reilly, J.J., Jr.; Chen, P.; Sailor, L.Z.; Wilcox, D.; Mason, R.W.; Chapman, H.A., Jr. Cigarette smoking induces an elastolytic cysteine proteinase in macrophages distinct from cathepsin L. Am. J. Physiol. 1991, 261, L41–L48. [Google Scholar] [CrossRef]
- Atkinson, J.J.; Lutey, B.A.; Suzuki, Y.; Toennies, H.M.; Kelley, D.G.; Kobayashi, D.K.; Ijem, W.G.; Deslee, G.; Moore, C.H.; Jacobs, M.E.; et al. The role of matrix metalloproteinase-9 in cigarette smoke-induced emphysema. Am. J. Respir. Crit. Care Med. 2011, 183, 876–884. [Google Scholar] [CrossRef] [Green Version]
- Abboud, R.T.; Vimalanathan, S. Pathogenesis of COPD. Part I. The role of protease-antiprotease imbalance in emphysema. Int. J. Tuberc. Lung Dis. 2008, 12, 361–367. [Google Scholar]
- Hunninghake, G.W.; Davidson, J.M.; Rennard, S.; Szapiel, S.; Gadek, J.E.; Crystal, R.G. Elastin fragments attract macrophage precursors to diseased sites in pulmonary emphysema. Science 1981, 212, 925–927. [Google Scholar] [CrossRef]
- Adair-Kirk, T.L.; Atkinson, J.J.; Broekelmann, T.J.; Doi, M.; Tryggvason, K.; Miner, J.H.; Mecham, R.P.; Senior, R.M. A site on laminin alpha 5, AQARSAASKVKVSMKF, induces inflammatory cell production of matrix metalloproteinase-9 and chemotaxis. J. Immunol. 2003, 171, 398–406. [Google Scholar] [CrossRef] [Green Version]
- Churg, A.; Zhou, S.; Wright, J.L. Series “matrix metalloproteinases in lung health and disease”: Matrix metalloproteinases in COPD. Eur. Respir. J. 2012, 39, 197–209. [Google Scholar] [CrossRef]
- Rahman, I.; MacNee, W. Role of oxidants/antioxidants in smoking-induced lung diseases. Free Radic. Biol. Med. 1996, 21, 669–681. [Google Scholar] [CrossRef]
- Yang, S.R.; Chida, A.S.; Bauter, M.R.; Shafiq, N.; Seweryniak, K.; Maggirwar, S.B.; Kilty, I.; Rahman, I. Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L46–L57. [Google Scholar] [CrossRef] [Green Version]
- Zuo, L.; He, F.; Sergakis, G.G.; Koozehchian, M.S.; Stimpfl, J.N.; Rong, Y.; Diaz, P.T.; Best, T.M. Interrelated role of cigarette smoking, oxidative stress, and immune response in COPD and corresponding treatments. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L205–L218. [Google Scholar] [CrossRef] [Green Version]
- Yuan, C.; Chang; Lu, G.; Deng, X. Genetic polymorphism and chronic obstructive pulmonary disease. Int. J. Chron. Obstruct. Pulmon. Dis. 2017, 12, 1385–1393. [Google Scholar] [CrossRef] [Green Version]
- Song, Q.; Chen, P.; Liu, X.M. The role of cigarette smoke-induced pulmonary vascular endothelial cell apoptosis in COPD. Respir. Res. 2021, 22, 39. [Google Scholar] [CrossRef]
- Barnes, P.J. Mediators of chronic obstructive pulmonary disease. Pharmacol. Rev. 2004, 56, 515–548. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef] [Green Version]
- Seimetz, M.; Parajuli, N.; Pichl, A.; Veit, F.; Kwapiszewska, G.; Weisel, F.C.; Milger, K.; Egemnazarov, B.; Turowska, A.; Fuchs, B.; et al. Inducible NOS inhibition reverses tobacco-smoke-induced emphysema and pulmonary hypertension in mice. Cell 2011, 147, 293–305. [Google Scholar] [CrossRef] [Green Version]
- Osoata, G.O.; Hanazawa, T.; Brindicci, C.; Ito, M.; Barnes, P.J.; Kharitonov, S.; Ito, K. Peroxynitrite elevation in exhaled breath condensate of COPD and its inhibition by fudosteine. Chest 2009, 135, 1513–1520. [Google Scholar] [CrossRef]
- Rahman, I.; van Schadewijk, A.A.; Crowther, A.J.; Hiemstra, P.S.; Stolk, J.; MacNee, W.; De Boer, W.I. 4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2002, 166, 490–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arja, C.; Surapaneni, K.M.; Raya, P.; Adimoolam, C.; Balisetty, B.; Kanala, K.R. Oxidative stress and antioxidant enzyme activity in South Indian male smokers with chronic obstructive pulmonary disease. Respirology 2013, 18, 1069–1075. [Google Scholar] [CrossRef]
- Ahmad, A.; Shameem, M.; Husain, Q. Altered oxidant-antioxidant levels in the disease prognosis of chronic obstructive pulmonary disease. Int. J. Tuberc. Lung Dis. 2013, 17, 1104–1109. [Google Scholar] [CrossRef]
- Barnes, P.J. Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clin. Chest Med. 2014, 35, 71–86. [Google Scholar] [CrossRef]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- Malhotra, D.; Thimmulappa, R.; Navas-Acien, A.; Sandford, A.; Elliott, M.; Singh, A.; Chen, L.; Zhuang, X.; Hogg, J.; Pare, P.; et al. Decline in NRF2-regulated antioxidants in chronic obstructive pulmonary disease lungs due to loss of its positive regulator, DJ-1. Am. J. Respir. Crit. Care Med. 2008, 178, 592–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutten, A.; Goven, D.; Artaud-Macari, E.; Boczkowski, J.; Bonay, M. NRF2 targeting: A promising therapeutic strategy in chronic obstructive pulmonary disease. Trends Mol. Med. 2011, 17, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Sussan, T.E.; Rangasamy, T.; Blake, D.J.; Malhotra, D.; El-Haddad, H.; Bedja, D.; Yates, M.S.; Kombairaju, P.; Yamamoto, M.; Liby, K.T.; et al. Targeting Nrf2 with the triterpenoid CDDO-imidazolide attenuates cigarette smoke-induced emphysema and cardiac dysfunction in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 250–255. [Google Scholar] [CrossRef] [Green Version]
- Paul, T.; Blanco, I.; Aguilar, D.; Tura-Ceide, O.; Bonjoch, C.; Smolders, V.F.; Peinado, V.I.; Barbera, J.A. Therapeutic effects of soluble guanylate cyclase stimulation on pulmonary hemodynamics and emphysema development in guinea pigs chronically exposed to cigarette smoke. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L222–L234. [Google Scholar] [CrossRef] [PubMed]
- Brunnquell, C.R.; Vieira, N.A.; Sabio, L.R.; Sczepanski, F.; Cecchini, A.L.; Cecchini, R.; Guarnier, F.A. Oxidative and proteolysis-related parameters of skeletal muscle from hamsters with experimental pulmonary emphysema: A comparison between papain and elastase induction. Int. J. Exp. Pathol. 2015, 96, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Baila, B.; Ohno, Y.; Nagamoto, H.; Kotosai, K.; Yabuuchi, Y.; Funaguchi, N.; Ito, F.; Endo, J.; Mori, H.; Takemura, G.; et al. Tetomilast attenuates elastase-induced pulmonary emphysema through inhibition of oxidative stress in rabbits. Biol. Pharm. Bull. 2012, 35, 494–502. [Google Scholar] [CrossRef] [Green Version]
- Otsuki, Y.; Go, T.; Kato, A.; Yokota, N.; Fujiwara, A.; Matsuura, N.; Chang, S.S.; Misaki, N.; Yokomise, H. Regeneration of emphysematous lungs using gelatin sheets that release basic fibroblast growth factor. Surg. Today 2022, 52, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, N.; Kitabatake, M.; Ouji-Sageshima, N.; Ibaraki, T.; Kumamoto, M.; Fujita, Y.; Hontsu, S.; Yamauchi, M.; Yoshikawa, M.; Muro, S.; et al. Human Adipose-Derived Mesenchymal Stem Cells Ameliorate Elastase-Induced Emphysema in Mice by Mesenchymal-Epithelial Transition. Int. J. Chron. Obstruct. Pulmon. Dis. 2021, 16, 2783–2793. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.N.; Mazzoli-Rocha, F.; Casquilho, N.V.; Maron-Gutierrez, T.; Ortenzi, V.H.; Morales, M.M.; Fortunato, R.S.; Zin, W.A. Bone Marrow-Derived Mononuclear Cell Therapy in Papain-Induced Experimental Pulmonary Emphysema. Front. Physiol. 2018, 9, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Aaron, C.P.; Madrigano, J.; Hoffman, E.A.; Angelini, E.; Yang, J.; Laine, A.; Vetterli, T.M.; Kinney, P.L.; Sampson, P.D.; et al. Association Between Long-term Exposure to Ambient Air Pollution and Change in Quantitatively Assessed Emphysema and Lung Function. JAMA 2019, 322, 546–556. [Google Scholar] [CrossRef]
- Tung, N.T.; Ho, S.C.; Lu, Y.H.; Chen, T.T.; Lee, K.Y.; Chen, K.Y.; Wu, C.D.; Chung, K.F.; Kuo, H.P.; Thao, H.N.X.; et al. Association Between Air Pollution and Lung Lobar Emphysema in COPD. Front. Med. 2021, 8, 705792. [Google Scholar] [CrossRef]
- Hu, G.; Zhou, Y.; Hong, W.; Tian, J.; Hu, J.; Peng, G.; Cui, J.; Li, B.; Ran, P. Development and systematic oxidative stress of a rat model of chronic bronchitis and emphysema induced by biomass smoke. Exp. Lung Res. 2013, 39, 229–240. [Google Scholar] [CrossRef]
- Skurikhin, E.; Pershina, O.; Zhukova, M.; Widera, D.; Pan, E.; Pakhomova, A.; Krupin, V.; Ermakova, N.; Skurikhina, V.; Sandrikina, L.; et al. Spiperone Stimulates Regeneration in Pulmonary Endothelium Damaged by Cigarette Smoke and Lipopolysaccharide. Int. J. Chron. Obstruct. Pulmon. Dis. 2021, 16, 3575–3591. [Google Scholar] [CrossRef]
- Yang, Y.; Di, T.; Zhang, Z.; Liu, J.; Fu, C.; Wu, Y.; Bian, T. Dynamic evolution of emphysema and airway remodeling in two mouse models of COPD. BMC Pulm. Med. 2021, 21, 134. [Google Scholar] [CrossRef] [PubMed]
- Valenca, S.S.; Castro, P.; Pimenta, W.A.; Lanzetti, M.; Silva, S.V.; Barja-Fidalgo, C.; Koatz, V.L.; Porto, L.C. Light cigarette smoke-induced emphysema and NFkappaB activation in mouse lung. Int. J. Exp. Pathol. 2006, 87, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Rashid, K.; Sundar, I.K.; Gerloff, J.; Li, D.; Rahman, I. Lung cellular senescence is independent of aging in a mouse model of COPD/emphysema. Sci. Rep. 2018, 8, 9023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, M.; Koger, F.; Klingelhofer, D.; Muller, R.; Groneberg, D.A. Particulate Matter Emissions of Four Different Cigarette Types of One Popular Brand: Influence of Tobacco Strength and Additives. Int. J. Environ. Res. Public Health 2019, 16, 263. [Google Scholar] [CrossRef] [Green Version]
- Trappenburg, J.C.; Niesink, A.; de Weert-van Oene, G.H.; van der Zeijden, H.; van Snippenburg, R.; Peters, A.; Lammers, J.W.; Schrijvers, A.J. Effects of telemonitoring in patients with chronic obstructive pulmonary disease. Telemed. e-Health 2008, 14, 138–146. [Google Scholar] [CrossRef]
- Mulhall, P.; Criner, G. Non-pharmacological treatments for COPD. Respirology 2016, 21, 791–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, P.J. Oxidative stress-based therapeutics in COPD. Redox Biol. 2020, 33, 101544. [Google Scholar] [CrossRef]
- Van der Toorn, M.; Rezayat, D.; Kauffman, H.F.; Bakker, S.J.; Gans, R.O.; Koeter, G.H.; Choi, A.M.; van Oosterhout, A.J.; Slebos, D.J. Lipid-soluble components in cigarette smoke induce mitochondrial production of reactive oxygen species in lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L109–L114. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.M.; Lin, P.H.; Yao, Q.; Chen, C. Chemical and molecular mechanisms of antioxidants: Experimental approaches and model systems. J. Cell. Mol. Med. 2010, 14, 840–860. [Google Scholar] [CrossRef]
- Hunyadi, A. The mechanism(s) of action of antioxidants: From scavenging reactive oxygen/nitrogen species to redox signaling and the generation of bioactive secondary metabolites. Med. Res. Rev. 2019, 39, 2505–2533. [Google Scholar] [CrossRef] [Green Version]
- Luczaj, W.; Gegotek, A.; Skrzydlewska, E. Antioxidants and HNE in redox homeostasis. Free Radic. Biol. Med. 2017, 111, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Kehm, R.; Baldensperger, T.; Raupbach, J.; Hohn, A. Protein oxidation—Formation mechanisms, detection and relevance as biomarkers in human diseases. Redox Biol. 2021, 42, 101901. [Google Scholar] [CrossRef] [PubMed]
- Ugarte, N.; Petropoulos, I.; Friguet, B. Oxidized mitochondrial protein degradation and repair in aging and oxidative stress. Antioxid. Redox Signal. 2010, 13, 539–549. [Google Scholar] [CrossRef]
- Zheng, J.P.; Wen, F.Q.; Bai, C.X.; Wan, H.Y.; Kang, J.; Chen, P.; Yao, W.Z.; Ma, L.J.; Li, X.; Raiteri, L.; et al. Twice daily N-acetylcysteine 600 mg for exacerbations of chronic obstructive pulmonary disease (PANTHEON): A randomised, double-blind placebo-controlled trial. Lancet Respir. Med. 2014, 2, 187–194. [Google Scholar] [CrossRef]
- Papi, A.; Zheng, J.; Criner, G.J.; Fabbri, L.M.; Calverley, P.M.A. Impact of smoking status and concomitant medications on the effect of high-dose N-acetylcysteine on chronic obstructive pulmonary disease exacerbations: A post-hoc analysis of the PANTHEON study. Respir. Med. 2019, 147, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.S.; Hu, Y.; Li, C.L.; Li, Y.; Wei, Y.R.; Yin, Z.F.; Du, Y.K.; Min, Z.; Weng, D.; Chen, J.M.; et al. N-acetylcysteine attenuates cigaret smoke-induced pulmonary exacerbation in a mouse model of emphysema. Inhal. Toxicol. 2015, 27, 802–809. [Google Scholar] [CrossRef]
- Messier, E.M.; Day, B.J.; Bahmed, K.; Kleeberger, S.R.; Tuder, R.M.; Bowler, R.P.; Chu, H.W.; Mason, R.J.; Kosmider, B. N-acetylcysteine protects murine alveolar type II cells from cigarette smoke injury in a nuclear erythroid 2-related factor-2-independent manner. Am. J. Respir. Cell Mol. Biol. 2013, 48, 559–567. [Google Scholar] [CrossRef] [Green Version]
- Lanzetti, M.; da Costa, C.A.; Nesi, R.T.; Barroso, M.V.; Martins, V.; Victoni, T.; Lagente, V.; Pires, K.M.; e Silva, P.M.; Resende, A.C.; et al. Oxidative stress and nitrosative stress are involved in different stages of proteolytic pulmonary emphysema. Free Radic. Biol. Med. 2012, 53, 1993–2001. [Google Scholar] [CrossRef]
- Yao, H.; Arunachalam, G.; Hwang, J.W.; Chung, S.; Sundar, I.K.; Kinnula, V.L.; Crapo, J.D.; Rahman, I. Extracellular superoxide dismutase protects against pulmonary emphysema by attenuating oxidative fragmentation of ECM. Proc. Natl. Acad. Sci. USA 2010, 107, 15571–15576. [Google Scholar] [CrossRef] [Green Version]
- Duong, C.; Seow, H.J.; Bozinovski, S.; Crack, P.J.; Anderson, G.P.; Vlahos, R. Glutathione peroxidase-1 protects against cigarette smoke-induced lung inflammation in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L425–L433. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, S.S.; Nassar, N.N. Diallyl sulfide protects against N-nitrosodiethylamine-induced liver tumorigenesis: Role of aldose reductase. World J. Gastroenterol. 2008, 14, 6145–6153. [Google Scholar] [CrossRef] [PubMed]
- Kalayarasan, S.; Prabhu, P.N.; Sriram, N.; Manikandan, R.; Arumugam, M.; Sudhandiran, G. Diallyl sulfide enhances antioxidants and inhibits inflammation through the activation of Nrf2 against gentamicin-induced nephrotoxicity in Wistar rats. Eur. J. Pharmacol. 2009, 606, 162–171. [Google Scholar] [CrossRef]
- Davenport, D.M.; Wargovich, M.J. Modulation of cytochrome P450 enzymes by organosulfur compounds from garlic. Food Chem. Toxicol. 2005, 43, 1753–1762. [Google Scholar] [CrossRef]
- Cardoso, A.O.P.; Pecli, E.S.C.; Dos Anjos, F.F.; Quesnot, N.; Valenca, H.D.M.; Cattani-Cavalieri, I.; Brito-Gitirana, L.; Valenca, S.S.; Lanzetti, M. Diallyl disulfide prevents cigarette smoke-induced emphysema in mice. Pulm. Pharmacol. Ther. 2021, 69, 102053. [Google Scholar] [CrossRef]
- Liu, Y.; Li, A.; Feng, X.; Sun, X.; Zhu, X.; Zhao, Z. Pharmacological Investigation of the Anti-Inflammation and Anti-Oxidation Activities of Diallyl Disulfide in a Rat Emphysema Model Induced by Cigarette Smoke Extract. Nutrients 2018, 10, 79. [Google Scholar] [CrossRef] [Green Version]
- Stancu, C.; Sima, A. Statins: Mechanism of action and effects. J. Cell. Mol. Med. 2001, 5, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Pinho-Ribeiro, V.; Melo, A.C.; Kennedy-Feitosa, E.; Graca-Reis, A.; Barroso, M.V.; Cattani-Cavalieri, I.; Carvalho, G.M.C.; Zin, W.A.; Porto, L.C.; Gitirana, L.B.; et al. Atorvastatin and Simvastatin Promoted Mouse Lung Repair After Cigarette Smoke-Induced Emphysema. Inflammation 2017, 40, 965–979. [Google Scholar] [CrossRef]
- Melo, A.C.; Cattani-Cavalieri, I.; Barroso, M.V.; Quesnot, N.; Gitirana, L.B.; Lanzetti, M.; Valenca, S.S. Atorvastatin dose-dependently promotes mouse lung repair after emphysema induced by elastase. Biomed. Pharmacother. 2018, 102, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Rudrapal, M.; Khairnar, S.J.; Khan, J.; Dukhyil, A.B.; Ansari, M.A.; Alomary, M.N.; Alshabrmi, F.M.; Palai, S.; Deb, P.K.; Devi, R. Dietary Polyphenols and Their Role in Oxidative Stress-Induced Human Diseases: Insights Into Protective Effects, Antioxidant Potentials and Mechanism(s) of Action. Front. Pharmacol. 2022, 13, 806470. [Google Scholar] [CrossRef]
- Polverino, F.; Wu, T.D.; Rojas-Quintero, J.; Wang, X.; Mayo, J.; Tomchaney, M.; Tram, J.; Packard, S.; Zhang, D.; Cleveland, K.H.; et al. Metformin: Experimental and Clinical Evidence for a Potential Role in Emphysema Treatment. Am. J. Respir. Crit. Care Med. 2021, 204, 651–666. [Google Scholar] [CrossRef] [PubMed]
- Lanzetti, M.; Lopes, A.A.; Ferreira, T.S.; de Moura, R.S.; Resende, A.C.; Porto, L.C.; Valenca, S.S. Mate tea ameliorates emphysema in cigarette smoke-exposed mice. Exp. Lung Res. 2011, 37, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Kennedy-Feitosa, E.; Cattani-Cavalieri, I.; Barroso, M.V.; Romana-Souza, B.; Brito-Gitirana, L.; Valenca, S.S. Eucalyptol promotes lung repair in mice following cigarette smoke-induced emphysema. Phytomedicine 2019, 55, 70–79. [Google Scholar] [CrossRef]
- Barroso, M.V.; Cattani-Cavalieri, I.; de Brito-Gitirana, L.; Fautrel, A.; Lagente, V.; Schmidt, M.; Porto, L.C.; Romana-Souza, B.; Valenca, S.S.; Lanzetti, M. Propolis reversed cigarette smoke-induced emphysema through macrophage alternative activation independent of Nrf2. Bioorg. Med. Chem. 2017, 25, 5557–5568. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, F.; Lemaur, V.; Cornil, J.; Lazzaroni, R.; Duroux, J.L.; Olivier, Y.; Trouillas, P. Free radical scavenging by natural polyphenols: Atom versus electron transfer. J. Phys. Chem. A 2013, 117, 2082–2092. [Google Scholar] [CrossRef]
- Tanigawa, S.; Fujii, M.; Hou, D.X. Action of Nrf2 and Keap1 in ARE-mediated NQO1 expression by quercetin. Free Radic. Biol. Med. 2007, 42, 1690–1703. [Google Scholar] [CrossRef] [PubMed]
- Araújo, N.P.d.S.; de Matos, N.A.; Oliveira, M.; de Souza, A.B.F.; Castro, T.d.F.; Machado-Júnior, P.A.; de Souza, D.M.S.; Talvani, A.; Cangussú, S.D.; de Menezes, R.C.A.J.A.; et al. Quercetin Improves Pulmonary Function and Prevents Emphysema Caused by Exposure to Cigarette Smoke in Male Mice. Antioxidants 2022, 11, 181. [Google Scholar] [CrossRef]
- Ganesan, S.; Faris, A.N.; Comstock, A.T.; Chattoraj, S.S.; Chattoraj, A.; Burgess, J.R.; Curtis, J.L.; Martinez, F.J.; Zick, S.; Hershenson, M.B.; et al. Quercetin prevents progression of disease in elastase/LPS-exposed mice by negatively regulating MMP expression. Respir. Res. 2010, 11, 131. [Google Scholar] [CrossRef] [Green Version]
- Catalgol, B.; Batirel, S.; Taga, Y.; Ozer, N.K. Resveratrol: French paradox revisited. Front. Pharmacol. 2012, 3, 141. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.B.; Lan, Y.W.; Chen, L.G.; Huang, T.T.; Choo, K.B.; Cheng, W.T.; Lee, H.S.; Chong, K.Y. Mesenchymal stem cell-based HSP70 promoter-driven VEGFA induction by resveratrol alleviates elastase-induced emphysema in a mouse model. Cell Stress Chaperones 2015, 20, 979–989. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.; Ling, C. Curcumin ameliorates chronic obstructive pulmonary disease by modulating autophagy and endoplasmic reticulum stress through regulation of SIRT1 in a rat model. J. Int. Med. Res. 2019, 47, 4764–4774. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Sun, J.; Mohammadtursun, N.; Wu, J.; Dong, J.; Li, L. Curcumin inhibits cigarette smoke-induced inflammation via modulating the PPARgamma-NF-kappaB signaling pathway. Food Funct. 2019, 10, 7983–7994. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Z.; Chang, J.; Li, J.; Nie, D.; Cui, H.; Guo, D. Sulforaphane increases Nrf2 expression and protects alveolar epithelial cells against injury caused by cigarette smoke extract. Mol. Med. Rep. 2017, 16, 1241–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, X.; Liu, X.; Bao, H. Sulforaphane suppresses lipopolysaccharide- and Pam3CysSerLys4-mediated inflammation in chronic obstructive pulmonary disease via toll-like receptors. FEBS Open Bio. 2021, 11, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, X.; Zhang, K.; Deng, S.; Jiao, P.; Qi, M.; Lian, Z.; Yao, Y. Overexpression of Toll-Like Receptor 4 Affects Autophagy, Oxidative Stress, and Inflammatory Responses in Monocytes of Transgenic Sheep. Front. Cell Dev. Biol. 2020, 8, 248. [Google Scholar] [CrossRef]
- Koike, K.; Ishigami, A.; Sato, Y.; Hirai, T.; Yuan, Y.; Kobayashi, E.; Tobino, K.; Sato, T.; Sekiya, M.; Takahashi, K.; et al. Vitamin C prevents cigarette smoke-induced pulmonary emphysema in mice and provides pulmonary restoration. Am. J. Respir. Cell Mol. Biol. 2014, 50, 347–357. [Google Scholar] [CrossRef]
- Peh, H.Y.; Tan, W.S.D.; Chan, T.K.; Pow, C.W.; Foster, P.S.; Wong, W.S.F. Vitamin E isoform gamma-tocotrienol protects against emphysema in cigarette smoke-induced COPD. Free Radic. Biol. Med. 2017, 110, 332–344. [Google Scholar] [CrossRef]
- Zaman, T.; Lee, J.S. Risk factors for the development of idiopathic pulmonary fibrosis: A review. Curr. Pulmonol. Rep. 2018, 7, 118–125. [Google Scholar] [CrossRef]
- Willis, B.C.; Liebler, J.M.; Luby-Phelps, K.; Nicholson, A.G.; Crandall, E.D.; du Bois, R.M.; Borok, Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: Potential role in idiopathic pulmonary fibrosis. Am. J. Pathol. 2005, 166, 1321–1332. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Hirai, S.; Tanaka, Y.; Sumi, T.; Miyajima, M.; Mishina, T.; Yamada, G.; Otsuka, M.; Hasegawa, T.; Kojima, T.; et al. Fibroblastic foci, covered with alveolar epithelia exhibiting epithelial-mesenchymal transition, destroy alveolar septa by disrupting blood flow in idiopathic pulmonary fibrosis. Lab. Investig. 2017, 97, 232–242. [Google Scholar] [CrossRef] [Green Version]
- Ballester, B.; Milara, J.; Cortijo, J. Idiopathic Pulmonary Fibrosis and Lung Cancer: Mechanisms and Molecular Targets. Int. J. Mol. Sci. 2019, 20, 593. [Google Scholar] [CrossRef] [Green Version]
- Chanda, D.; Otoupalova, E.; Smith, S.R.; Volckaert, T.; De Langhe, S.P.; Thannickal, V.J. Developmental pathways in the pathogenesis of lung fibrosis. Mol. Aspects Med. 2019, 65, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Y.; Wu, G.; Xiong, W.; Gu, W.; Wang, C.Y. Macrophages: Friend or foe in idiopathic pulmonary fibrosis? Respir. Res. 2018, 19, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scotton, C.J.; Chambers, R.C. Molecular targets in pulmonary fibrosis: The myofibroblast in focus. Chest 2007, 132, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Rao, L.Z.; Wang, Y.; Zhang, L.; Wu, G.; Zhang, L.; Wang, F.X.; Chen, L.M.; Sun, F.; Jia, S.; Zhang, S.; et al. IL-24 deficiency protects mice against bleomycin-induced pulmonary fibrosis by repressing IL-4-induced M2 program in macrophages. Cell Death Differ. 2021, 28, 2989. [Google Scholar] [CrossRef]
- Nagaoka, I.; Trapnell, B.C.; Crystal, R.G. Upregulation of platelet-derived growth factor-A and -B gene expression in alveolar macrophages of individuals with idiopathic pulmonary fibrosis. J. Clin. Investig. 1990, 85, 2023–2027. [Google Scholar] [CrossRef] [Green Version]
- Nanthakumar, C.B.; Hatley, R.J.; Lemma, S.; Gauldie, J.; Marshall, R.P.; Macdonald, S.J. Dissecting fibrosis: Therapeutic insights from the small-molecule toolbox. Nat. Rev. Drug Discov. 2015, 14, 693–720. [Google Scholar] [CrossRef]
- Kinnula, V.L.; Fattman, C.L.; Tan, R.J.; Oury, T.D. Oxidative stress in pulmonary fibrosis: A possible role for redox modulatory therapy. Am. J. Respir. Crit. Care Med. 2005, 172, 417–422. [Google Scholar] [CrossRef] [Green Version]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar] [CrossRef] [Green Version]
- Amara, N.; Goven, D.; Prost, F.; Muloway, R.; Crestani, B.; Boczkowski, J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts. Thorax 2010, 65, 733–738. [Google Scholar] [CrossRef] [Green Version]
- McKeown, S.; Richter, A.G.; O’Kane, C.; McAuley, D.F.; Thickett, D.R. MMP expression and abnormal lung permeability are important determinants of outcome in IPF. Eur. Respir. J. 2009, 33, 77–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarman, E.R.; Khambata, V.S.; Cope, C.; Jones, P.; Roger, J.; Ye, L.Y.; Duggan, N.; Head, D.; Pearce, A.; Press, N.J.; et al. An inhibitor of NADPH oxidase-4 attenuates established pulmonary fibrosis in a rodent disease model. Am. J. Respir. Cell Mol. Biol. 2014, 50, 158–169. [Google Scholar] [CrossRef]
- Saleh, D.; Barnes, P.J.; Giaid, A. Increased production of the potent oxidant peroxynitrite in the lungs of patients with idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1997, 155, 1763–1769. [Google Scholar] [CrossRef]
- Daniil, Z.D.; Papageorgiou, E.; Koutsokera, A.; Kostikas, K.; Kiropoulos, T.; Papaioannou, A.I.; Gourgoulianis, K.I. Serum levels of oxidative stress as a marker of disease severity in idiopathic pulmonary fibrosis. Pulm. Pharmacol. Ther. 2008, 21, 26–31. [Google Scholar] [CrossRef]
- Malli, F.; Bardaka, F.; Tsilioni, I.; Karetsi, E.; Gourgoulianis, K.I.; Daniil, Z. 8-isoprostane levels in serum and bronchoalveolar lavage in idiopathic pulmonary fibrosis and sarcoidosis. Food Chem. Toxicol. 2013, 61, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, Y.; Kawashima, T.; Kuwabara, R.; Hayakawa, S.; Irie, T.; Yoshida, T.; Rikitake, H.; Wakabayashi, T.; Okada, N.; Kawashima, K.; et al. Change in serum marker of oxidative stress in the progression of idiopathic pulmonary fibrosis. Pulm. Pharmacol. Ther. 2015, 32, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Lenz, A.G.; Costabel, U.; Maier, K.L. Oxidized BAL fluid proteins in patients with interstitial lung diseases. Eur. Respir. J. 1996, 9, 307–312. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Yin, G.; Jiang, H.; Zhang, C. High-dose N-acetylcysteine decreases silica-induced lung fibrosis in the rat. J. Int. Med. Res. 2013, 41, 1179–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansour, H.H.; Omran, M.M.; Hasan, H.F.; El Kiki, S.M. Modulation of bleomycin-induced oxidative stress and pulmonary fibrosis by N-acetylcysteine in rats via AMPK/SIRT1/NF-kappabeta. Clin. Exp. Pharmacol. Physiol. 2020, 47, 1943–1952. [Google Scholar] [CrossRef] [PubMed]
- Poletti, V.; Ravaglia, C.; Tomassetti, S. Pirfenidone for the treatment of idiopathic pulmonary fibrosis. Expert Rev. Respir. Med. 2014, 8, 539–545. [Google Scholar] [CrossRef]
- Fois, A.G.; Posadino, A.M.; Giordo, R.; Cossu, A.; Agouni, A.; Rizk, N.M.; Pirina, P.; Carru, C.; Zinellu, A.; Pintus, G. Antioxidant Activity Mediates Pirfenidone Antifibrotic Effects in Human Pulmonary Vascular Smooth Muscle Cells Exposed to Sera of Idiopathic Pulmonary Fibrosis Patients. Oxid. Med. Cell. Longev. 2018, 2018, 2639081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Lu, F.; Kang, L.; Wang, Z.; Wang, Y. Pirfenidone attenuates bleomycin-induced pulmonary fibrosis in mice by regulating Nrf2/Bach1 equilibrium. BMC Pulm. Med. 2017, 17, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina-Molina, M.; Machahua-Huamani, C.; Vicens-Zygmunt, V.; Llatjos, R.; Escobar, I.; Sala-Llinas, E.; Luburich-Hernaiz, P.; Dorca, J.; Montes-Worboys, A. Anti-fibrotic effects of pirfenidone and rapamycin in primary IPF fibroblasts and human alveolar epithelial cells. BMC Pulm. Med. 2018, 18, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwana, M.; Azuma, A. Nintedanib: New indication for systemic sclerosis-associated interstitial lung disease. Mod. Rheumatol. 2020, 30, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Costabel, U.; Selman, M.; Kim, D.S.; Hansell, D.M.; Nicholson, A.G.; Brown, K.K.; Flaherty, K.R.; Noble, P.W.; Raghu, G.; et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2011, 365, 1079–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, M.; Young, P.J.; Laffey, J.G.; Asfar, P.; Taccone, F.S.; Skrifvars, M.B.; Meyhoff, C.S.; Radermacher, P. Dangers of hyperoxia. Crit. Care 2021, 25, 440. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol. 2007, 8, 722–728. [Google Scholar] [CrossRef]
- Amarelle, L.; Quintela, L.; Hurtado, J.; Malacrida, L. Hyperoxia and Lungs: What We Have Learned From Animal Models. Front. Med. 2021, 8, 606678. [Google Scholar] [CrossRef]
- McAllister, S.; Thorn, L.; Boladuadua, S.; Gil, M.; Audas, R.; Edmonds, T.; Rafai, E.; Hill, P.C.; Howie, S.R.C. Cost analysis and critical success factors of the use of oxygen concentrators versus cylinders in sub-divisional hospitals in Fiji. BMC Health Serv. Res. 2021, 21, 636. [Google Scholar] [CrossRef]
- Brueckl, C.; Kaestle, S.; Kerem, A.; Habazettl, H.; Krombach, F.; Kuppe, H.; Kuebler, W.M. Hyperoxia-induced reactive oxygen species formation in pulmonary capillary endothelial cells in situ. Am. J. Respir. Cell Mol. Biol. 2006, 34, 453–463. [Google Scholar] [CrossRef] [Green Version]
- O’Driscoll, B.R.; Howard, L.S.; Davison, A.G.; British Thoracic, S. BTS guideline for emergency oxygen use in adult patients. Thorax 2008, 63 (Suppl. 6), vi1–vi68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrache, I.; Choi, M.E.; Otterbein, L.E.; Chin, B.Y.; Mantell, L.L.; Horowitz, S.; Choi, A.M. Mitogen-activated protein kinase pathway mediates hyperoxia-induced apoptosis in cultured macrophage cells. Am. J. Physiol. 1999, 277, L589–L595. [Google Scholar] [CrossRef] [PubMed]
- Weibel, E.R. Oxygen effect on lung cells. Arch. Intern. Med. 1971, 128, 54–56. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wang, J.; Li, C.; Mo, L.; Huang, D. Hyperoxia induces alveolar epithelial cell apoptosis by regulating mitochondrial function through small mothers against decapentaplegic 3 (SMAD3) and extracellular signal-regulated kinase 1/2 (ERK1/2). Bioengineered 2022, 13, 242–252. [Google Scholar] [CrossRef]
- Patel, V.S.; Sitapara, R.A.; Gore, A.; Phan, B.; Sharma, L.; Sampat, V.; Li, J.H.; Yang, H.; Chavan, S.S.; Wang, H.; et al. High Mobility Group Box-1 mediates hyperoxia-induced impairment of Pseudomonas aeruginosa clearance and inflammatory lung injury in mice. Am. J. Respir. Cell. Mol. Biol. 2013, 48, 280–287. [Google Scholar] [CrossRef] [Green Version]
- Jones, N.; Agani, F.H. Hyperoxia induces Egr-1 expression through activation of extracellular signal-regulated kinase 1/2 pathway. J. Cell. Physiol. 2003, 196, 326–333. [Google Scholar] [CrossRef]
- Tateda, K.; Deng, J.C.; Moore, T.A.; Newstead, M.W.; Paine, R., 3rd; Kobayashi, N.; Yamaguchi, K.; Standiford, T.J. Hyperoxia mediates acute lung injury and increased lethality in murine Legionella pneumonia: The role of apoptosis. J. Immunol. 2003, 170, 4209–4216. [Google Scholar] [CrossRef] [Green Version]
- Reddy, N.M.; Potteti, H.R.; Mariani, T.J.; Biswal, S.; Reddy, S.P. Conditional deletion of Nrf2 in airway epithelium exacerbates acute lung injury and impairs the resolution of inflammation. Am. J. Respir. Cell Mol. Biol. 2011, 45, 1161–1168. [Google Scholar] [CrossRef] [Green Version]
- Reddy, N.M.; Suryanarayana, V.; Kalvakolanu, D.V.; Yamamoto, M.; Kensler, T.W.; Hassoun, P.M.; Kleeberger, S.R.; Reddy, S.P. Innate immunity against bacterial infection following hyperoxia exposure is impaired in NRF2-deficient mice. J. Immunol. 2009, 183, 4601–4608. [Google Scholar] [CrossRef] [Green Version]
- Barry, B.E.; Crapo, J.D. Patterns of accumulation of platelets and neutrophils in rat lungs during exposure to 100% and 85% oxygen. Am. Rev. Respir. Dis. 1985, 132, 548–555. [Google Scholar] [CrossRef]
- Yee, M.; McDavid, A.N.; Cohen, E.D.; Huyck, H.L.; Poole, C.; Altman, B.J.; Maniscalco, W.M.; Deutsch, G.H.; Pryhuber, G.S.; O’Reilly, M.A. Neonatal Hyperoxia Activates Activating Transcription Factor 4 to Stimulate Folate Metabolism and Alveolar Epithelial Type 2 Cell Proliferation. Am. J. Respir. Cell Mol. Biol. 2022, 66, 402–414. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, N.R.; Brower, R.G.; Hager, D.N.; Thompson, B.T.; Netzer, G.; Shanholtz, C.; Lagakos, A.; Checkley, W.; National Institutes of Health Acute Respiratory Distress Syndrome Network, I. Oxygen Exposure Resulting in Arterial Oxygen Tensions Above the Protocol Goal Was Associated With Worse Clinical Outcomes in Acute Respiratory Distress Syndrome. Crit. Care Med. 2018, 46, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Page, D.; Ablordeppey, E.; Wessman, B.T.; Mohr, N.M.; Trzeciak, S.; Kollef, M.H.; Roberts, B.W.; Fuller, B.M. Emergency department hyperoxia is associated with increased mortality in mechanically ventilated patients: A cohort study. Crit. Care 2018, 22, 9. [Google Scholar] [CrossRef] [Green Version]
- Jankov, R.P.; Johnstone, L.; Luo, X.; Robinson, B.H.; Tanswell, A.K. Macrophages as a major source of oxygen radicals in the hyperoxic newborn rat lung. Free Radic. Biol. Med. 2003, 35, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Berkelhamer, S.K.; Kim, G.A.; Radder, J.E.; Wedgwood, S.; Czech, L.; Steinhorn, R.H.; Schumacker, P.T. Developmental differences in hyperoxia-induced oxidative stress and cellular responses in the murine lung. Free Radic. Biol. Med. 2013, 61, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Beyer, A.M.; Durand, M.; Clough, A.V.; Zhu, D.; Norwood Toro, L.; Terashvili, M.; Ebben, J.D.; Hill, R.B.; Audi, S.H.; et al. Hyperoxia Causes Mitochondrial Fragmentation in Pulmonary Endothelial Cells by Increasing Expression of Pro-Fission Proteins. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 622–635. [Google Scholar] [CrossRef] [Green Version]
- Papaiahgari, S.; Kleeberger, S.R.; Cho, H.Y.; Kalvakolanu, D.V.; Reddy, S.P. NADPH oxidase and ERK signaling regulates hyperoxia-induced Nrf2-ARE transcriptional response in pulmonary epithelial cells. J. Biol. Chem. 2004, 279, 42302–42312. [Google Scholar] [CrossRef] [Green Version]
- Parinandi, N.L.; Kleinberg, M.A.; Usatyuk, P.V.; Cummings, R.J.; Pennathur, A.; Cardounel, A.J.; Zweier, J.L.; Garcia, J.G.; Natarajan, V. Hyperoxia-induced NAD(P)H oxidase activation and regulation by MAP kinases in human lung endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L26–L38. [Google Scholar] [CrossRef] [Green Version]
- D’Autreaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef]
- Carnesecchi, S.; Deffert, C.; Pagano, A.; Garrido-Urbani, S.; Metrailler-Ruchonnet, I.; Schappi, M.; Donati, Y.; Matthay, M.A.; Krause, K.H.; Barazzone Argiroffo, C. NADPH oxidase-1 plays a crucial role in hyperoxia-induced acute lung injury in mice. Am. J. Respir. Crit. Care Med. 2009, 180, 972–981. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.N.; Lee, K.E.; Hong, J.Y.; Heo, W.I.; Kim, K.W.; Kim, K.E.; Sohn, M.H. Involvement of the MAPK and PI3K pathways in chitinase 3-like 1-regulated hyperoxia-induced airway epithelial cell death. Biochem. Biophys. Res. Commun. 2012, 421, 790–796. [Google Scholar] [CrossRef]
- Kim, M.J.; Ryu, J.C.; Kwon, Y.; Lee, S.; Bae, Y.S.; Yoon, J.H.; Ryu, J.H. Dual oxidase 2 in lung epithelia is essential for hyperoxia-induced acute lung injury in mice. Antioxid. Redox Signal. 2014, 21, 1803–1818. [Google Scholar] [CrossRef] [Green Version]
- Perkowski, S.; Scherpereel, A.; Murciano, J.C.; Arguiri, E.; Solomides, C.C.; Albelda, S.M.; Muzykantov, V.; Christofidou-Solomidou, M. Dissociation between alveolar transmigration of neutrophils and lung injury in hyperoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L1050–L1058. [Google Scholar] [CrossRef]
- Wang, Y.; Feinstein, S.I.; Manevich, Y.; Ho, Y.S.; Fisher, A.B. Lung injury and mortality with hyperoxia are increased in peroxiredoxin 6 gene-targeted mice. Free Radic. Biol. Med. 2004, 37, 1736–1743. [Google Scholar] [CrossRef] [PubMed]
- Tsan, M.F.; Tacy, N.J.; Lindau-Shepard, B.A.; White, J.E. Protection of rats against oxygen toxicity by tracheal administration of plasmid DNA: Role of endogenous tumor necrosis factor. Proc. Assoc. Am. Physicians 1997, 109, 409–419. [Google Scholar] [PubMed]
- Li, W.; Chang, L.; Rong, Z.; Zhang, Q.; Wang, H.; Wang, H.; Liu, C.; Liu, W. Mechanism of retinoic acid and mitogen-activated protein kinases regulating hyperoxia lung injury. J. Huazhong Univ. Sci. Technol. Med. Sci. 2006, 26, 178–181. [Google Scholar] [CrossRef]
- Truong, S.V.; Monick, M.M.; Yarovinsky, T.O.; Powers, L.S.; Nyunoya, T.; Hunninghake, G.W. Extracellular signal-regulated kinase activation delays hyperoxia-induced epithelial cell death in conditions of Akt downregulation. Am. J. Respir. Cell Mol. Biol. 2004, 31, 611–618. [Google Scholar] [CrossRef]
- Pagano, A.; Barazzone-Argiroffo, C. Alveolar cell death in hyperoxia-induced lung injury. Ann. N. Y. Acad. Sci. 2003, 1010, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Franek, W.R.; Morrow, D.M.; Zhu, H.; Vancurova, I.; Miskolci, V.; Darley-Usmar, K.; Simms, H.H.; Mantell, L.L. NF-kappaB protects lung epithelium against hyperoxia-induced nonapoptotic cell death-oncosis. Free Radic. Biol. Med. 2004, 37, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Romashko, J., 3rd; Horowitz, S.; Franek, W.R.; Palaia, T.; Miller, E.J.; Lin, A.; Birrer, M.J.; Scott, W.; Mantell, L.L. MAPK pathways mediate hyperoxia-induced oncotic cell death in lung epithelial cells. Free Radic. Biol. Med. 2003, 35, 978–993. [Google Scholar] [CrossRef]
- Nagato, A.C.; Bezerra, F.S.; Talvani, A.; Aarestrup, B.J.; Aarestrup, F.M. Hyperoxia promotes polarization of the immune response in ovalbumin-induced airway inflammation, leading to a TH17 cell phenotype. Immun. Inflamm. Dis. 2015, 3, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Cazals, V.; Nabeyrat, E.; Corroyer, S.; de Keyzer, Y.; Clement, A. Role for NF-kappa B in mediating the effects of hyperoxia on IGF-binding protein 2 promoter activity in lung alveolar epithelial cells. Biochim. Biophys. Acta 1999, 1448, 349–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porzionato, A.; Sfriso, M.M.; Mazzatenta, A.; Macchi, V.; De Caro, R.; Di Giulio, C. Effects of hyperoxic exposure on signal transduction pathways in the lung. Respir. Physiol. Neurobiol. 2015, 209, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Popovic, V.; Gerschman, R.; Gilbert, D.L. Effect of High Oxygen Pressure on Ground Squirrels in Hypothermia and Hibernation. Am. J. Physiol. 1964, 206, 49–50. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Otterbein, S.L.; Ifedigbo, E.; Liu, F.; Morse, D.E.; Fearns, C.; Ulevitch, R.J.; Knickelbein, R.; Flavell, R.A.; Choi, A.M. MKK3 mitogen-activated protein kinase pathway mediates carbon monoxide-induced protection against oxidant-induced lung injury. Am. J. Pathol. 2003, 163, 2555–2563. [Google Scholar] [CrossRef] [Green Version]
- McGrath-Morrow, S.A.; Lauer, T.; Collaco, J.M.; Lopez, A.; Malhotra, D.; Alekseyev, Y.O.; Neptune, E.; Wise, R.; Biswal, S. Transcriptional responses of neonatal mouse lung to hyperoxia by Nrf2 status. Cytokine 2014, 65, 4–9. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.Y.; Kleeberger, S.R. Nrf2 protects against airway disorders. Toxicol. Appl. Pharmacol. 2010, 244, 43–56. [Google Scholar] [CrossRef]
- Bhandari, V.; Maulik, N.; Kresch, M. Hyperoxia causes an increase in antioxidant enzyme activity in adult and fetal rat type II pneumocytes. Lung 2000, 178, 53–60. [Google Scholar] [CrossRef]
- Valenca Sdos, S.; Kloss, M.L.; Bezerra, F.S.; Lanzetti, M.; Silva, F.L.; Porto, L.C. Effects of hyperoxia on Wistar rat lungs. J. Bras. Pneumol. 2007, 33, 655–662. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Gorasiya, S.; Antoine, D.J.; Sitapara, R.A.; Wu, W.; Sharma, L.; Yang, H.; Ashby, C.R., Jr.; Vasudevan, D.; Zur, M.; et al. The compromise of macrophage functions by hyperoxia is attenuated by ethacrynic acid via inhibition of NF-kappaB-mediated release of high-mobility group box-1. Am. J. Respir. Cell Mol. Biol. 2015, 52, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Nagato, A.; Silva, F.L.; Silva, A.R.; Bezerra, F.S.; Oliveira, M.L.; Bello-Klein, A.; Cristovao Porto, L.; Santos Valenca, S. Hyperoxia-induced lung injury is dose dependent in Wistar rats. Exp. Lung Res. 2009, 35, 713–728. [Google Scholar] [CrossRef]
- Nagato, A.C.; Bezerra, F.S.; Lanzetti, M.; Lopes, A.A.; Silva, M.A.; Porto, L.C.; Valenca, S.S. Time course of inflammation, oxidative stress and tissue damage induced by hyperoxia in mouse lungs. Int. J. Exp. Pathol. 2012, 93, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Langley, S.C.; Kelly, F.J. Depletion of pulmonary glutathione using diethylmaleic acid accelerates the development of oxygen-induced lung injury in term and preterm guinea-pig neonates. J Pharm. Pharmacol. 1994, 46, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Kiers, D.; Gerretsen, J.; Janssen, E.; John, A.; Groeneveld, R.; van der Hoeven, J.G.; Scheffer, G.J.; Pickkers, P.; Kox, M. Short-term hyperoxia does not exert immunologic effects during experimental murine and human endotoxemia. Sci. Rep. 2015, 5, 17441. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Guthrie, J.R.; Mabry, S.; Sack, T.M.; Truog, W.E. Mitochondrial aldehyde dehydrogenase attenuates hyperoxia-induced cell death through activation of ERK/MAPK and PI3K-Akt pathways in lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L966–L975. [Google Scholar] [CrossRef] [PubMed]
- Esterbauer, H.; Gebicki, J.; Puhl, H.; Jurgens, G. The role of lipid peroxidation and antioxidants in oxidative modification of LDL. Free Radic. Biol. Med. 1992, 13, 341–390. [Google Scholar] [CrossRef]
- Turanlahti, M.; Pesonen, E.; Lassus, P.; Andersson, S. Nitric oxide and hyperoxia in oxidative lung injury. Acta Paediatr. 2000, 89, 966–970. [Google Scholar] [CrossRef]
- Kannan, S.; Pang, H.; Foster, D.C.; Rao, Z.; Wu, M. Human 8-oxoguanine DNA glycosylase increases resistance to hyperoxic cytotoxicity in lung epithelial cells and involvement with altered MAPK activity. Cell Death Differ. 2006, 13, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Buckley, S.; Driscoll, B.; Barsky, L.; Weinberg, K.; Anderson, K.; Warburton, D. ERK activation protects against DNA damage and apoptosis in hyperoxic rat AEC2. Am. J. Physiol. 1999, 277, L159–L166. [Google Scholar] [CrossRef]
- Bowman, C.M.; Harada, R.N.; Repine, J.E. Hyperoxia stimulates alveolar macrophages to produce and release a factor which increases neutrophil adherence. Inflammation 1983, 7, 331–338. [Google Scholar] [CrossRef]
- Grisafi, D.; Tassone, E.; Dedja, A.; Oselladore, B.; Masola, V.; Guzzardo, V.; Porzionato, A.; Salmaso, R.; Albertin, G.; Artusi, C.; et al. L-citrulline prevents alveolar and vascular derangement in a rat model of moderate hyperoxia-induced lung injury. Lung 2012, 190, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wei, D.; Zhao, J.; Xu, X.; Chen, J. Reduction of hyperoxic acute lung injury in mice by Formononetin. PLoS ONE 2021, 16, e0245050. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Chang, L.; Rong, Z.; Liu, W. Retinoic acid diminished the expression of MMP-2 in hyperoxia-exposed premature rat lung fibroblasts through regulating mitogen-activated protein kinases. J. Huazhong Univ. Sci. Technol. Med. Sci. 2011, 31, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Danel, C.; Erzurum, S.C.; Prayssac, P.; Eissa, N.T.; Crystal, R.G.; Herve, P.; Baudet, B.; Mazmanian, M.; Lemarchand, P. Gene therapy for oxidant injury-related diseases: Adenovirus-mediated transfer of superoxide dismutase and catalase cDNAs protects against hyperoxia but not against ischemia-reperfusion lung injury. Hum. Gene Ther. 1998, 9, 1487–1496. [Google Scholar] [CrossRef]
- Xu, W.; Zhao, Y.; Zhang, B.; Xu, B.; Yang, Y.; Wang, Y.; Liu, C. Resveratrol attenuates hyperoxia-induced oxidative stress, inflammation and fibrosis and suppresses Wnt/beta-catenin signalling in lungs of neonatal rats. Clin. Exp. Pharmacol. Physiol. 2015, 42, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.M.; Tung, Y.T.; Wei, C.H.; Lee, P.Y.; Chen, W. Anti-Inflammatory and Reactive Oxygen Species Suppression through Aspirin Pretreatment to Treat Hyperoxia-Induced Acute Lung Injury in NF-kappaB-Luciferase Inducible Transgenic Mice. Antioxidants 2020, 9, 429. [Google Scholar] [CrossRef]
- Bezerra, F.S.; Ramos, C.O.; Castro, T.F.; Araujo, N.; de Souza, A.B.F.; Bandeira, A.C.B.; Costa, G.P.; Cartelle, C.T.; Talvani, A.; Cangussu, S.D.; et al. Exogenous surfactant prevents hyperoxia-induced lung injury in adult mice. Intensive Care Med. Exp. 2019, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Folz, R.J.; Abushamaa, A.M.; Suliman, H.B. Extracellular superoxide dismutase in the airways of transgenic mice reduces inflammation and attenuates lung toxicity following hyperoxia. J. Clin. Investig. 1999, 103, 1055–1066. [Google Scholar] [CrossRef] [Green Version]
- Ho, Y.S.; Vincent, R.; Dey, M.S.; Slot, J.W.; Crapo, J.D. Transgenic models for the study of lung antioxidant defense: Enhanced manganese-containing superoxide dismutase activity gives partial protection to B6C3 hybrid mice exposed to hyperoxia. Am. J. Respir. Cell Mol. Biol. 1998, 18, 538–547. [Google Scholar] [CrossRef] [Green Version]
- Choi, A.M.; Alam, J. Heme oxygenase-1: Function, regulation, and implication of a novel stress-inducible protein in oxidant-induced lung injury. Am. J. Respir. Cell Mol. Biol. 1996, 15, 9–19. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Kolls, J.K.; Mantell, L.L.; Cook, J.L.; Alam, J.; Choi, A.M. Exogenous administration of heme oxygenase-1 by gene transfer provides protection against hyperoxia-induced lung injury. J. Clin. Investig. 1999, 103, 1047–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, V.; Dial, K.; Wu, J.; Gauthier, A.G.; Wu, W.; Lin, M.; Espey, M.G.; Thomas, D.D.; Ashby, C.R., Jr.; Mantell, L.L. Dietary Antioxidants Significantly Attenuate Hyperoxia-Induced Acute Inflammatory Lung Injury by Enhancing Macrophage Function via Reducing the Accumulation of Airway HMGB1. Int. J. Mol. Sci. 2020, 21, 977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Wang, S.H.; Zheng, X.M.; Tang, Z.M. Calcitonin Gene-Related Peptide Attenuates Hyperoxia-Induced Oxidative Damage in Alveolar Epithelial Type II Cells Through Regulating Viability and Transdifferentiation. Inflammation 2022, 45, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Dang, H.X.; Li, J.; Liu, C.; Fu, Y.; Zhou, F.; Tang, L.; Li, L.; Xu, F. CGRP attenuates hyperoxia-induced oxidative stress-related injury to alveolar epithelial type II cells via the activation of the Sonic hedgehog pathway. Int. J. Mol. Med. 2017, 40, 209–216. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, M.A.; Staversky, R.J.; Stripp, B.R.; Finkelstein, J.N. Exposure to hyperoxia induces p53 expression in mouse lung epithelium. Am. J. Respir. Cell Mol. Biol. 1998, 18, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Shan, P.; Sasidhar, M.; Chupp, G.L.; Flavell, R.A.; Choi, A.M.; Lee, P.J. Reactive oxygen species and extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase mediate hyperoxia-induced cell death in lung epithelium. Am. J. Respir. Cell Mol. Biol. 2003, 28, 305–315. [Google Scholar] [CrossRef]
- Son, Y.; Cheong, Y.K.; Kim, N.H.; Chung, H.T.; Kang, D.G.; Pae, H.O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal Transduct. 2011, 2011, 792639. [Google Scholar] [CrossRef] [Green Version]
- Nyunoya, T.; Monick, M.M.; Powers, L.S.; Yarovinsky, T.O.; Hunninghake, G.W. Macrophages survive hyperoxia via prolonged ERK activation due to phosphatase down-regulation. J. Biol. Chem. 2005, 280, 26295–26302. [Google Scholar] [CrossRef] [Green Version]
- Kondrikov, D.; Caldwell, R.B.; Dong, Z.; Su, Y. Reactive oxygen species-dependent RhoA activation mediates collagen synthesis in hyperoxic lung fibrosis. Free Radic. Biol. Med. 2011, 50, 1689–1698. [Google Scholar] [CrossRef] [Green Version]
- Usatyuk, P.V.; Gorshkova, I.A.; He, D.; Zhao, Y.; Kalari, S.K.; Garcia, J.G.; Natarajan, V. Phospholipase D-mediated activation of IQGAP1 through Rac1 regulates hyperoxia-induced p47phox translocation and reactive oxygen species generation in lung endothelial cells. J. Biol. Chem. 2009, 284, 15339–15352. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Fu, H.; Yang, M.; Fang, F.; Kuang, F.; Xu, F. Neuropeptide substance P attenuates hyperoxia-induced oxidative stress injury in type II alveolar epithelial cells via suppressing the activation of JNK pathway. Lung 2009, 187, 421–426. [Google Scholar] [CrossRef]
- Li, Z.; Choo-Wing, R.; Sun, H.; Sureshbabu, A.; Sakurai, R.; Rehan, V.K.; Bhandari, V. A potential role of the JNK pathway in hyperoxia-induced cell death, myofibroblast transdifferentiation and TGF-beta1-mediated injury in the developing murine lung. BMC Cell Biol. 2011, 12, 54. [Google Scholar] [CrossRef] [Green Version]
- D’Angio, C.T.; LoMonaco, M.B.; Chaudhry, S.A.; Paxhia, A.; Ryan, R.M. Discordant pulmonary proinflammatory cytokine expression during acute hyperoxia in the newborn rabbit. Exp. Lung. Res. 1999, 25, 443–465. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, V.; Elias, J.A. Cytokines in tolerance to hyperoxia-induced injury in the developing and adult lung. Free Radic. Biol. Med. 2006, 41, 4–18. [Google Scholar] [CrossRef] [PubMed]
- White, C.W.; Ghezzi, P.; Dinarello, C.A.; Caldwell, S.A.; McMurtry, I.F.; Repine, J.E. Recombinant tumor necrosis factor/cachectin and interleukin 1 pretreatment decreases lung oxidized glutathione accumulation, lung injury, and mortality in rats exposed to hyperoxia. J. Clin. Investig. 1987, 79, 1868–1873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, N.S.; Waxman, A.B.; Homer, R.J.; Mantell, L.L.; Einarsson, O.; Du, Y.; Elias, J.A. Interleukin-6-induced protection in hyperoxic acute lung injury. Am. J. Respir. Cell Mol. Biol. 2000, 22, 535–542. [Google Scholar] [CrossRef]
- Shea, L.M.; Beehler, C.; Schwartz, M.; Shenkar, R.; Tuder, R.; Abraham, E. Hyperoxia activates NF-kappaB and increases TNF-alpha and IFN-gamma gene expression in mouse pulmonary lymphocytes. J. Immunol. 1996, 157, 3902–3908. [Google Scholar] [CrossRef] [PubMed]
- Burges, A.; Allmeling, A.; Krombach, F. Hyperoxia induces upregulation of CD11b and amplifies LPS-induced TNF-alpha release by alveolar macrophages. Eur. J. Med. Res. 1997, 2, 149–154. [Google Scholar]
- Desmarquest, P.; Chadelat, K.; Corroyer, S.; Cazals, V.; Clement, A. Effect of hyperoxia on human macrophage cytokine response. Respir. Med. 1998, 92, 951–960. [Google Scholar] [CrossRef] [Green Version]
- Jensen, J.C.; Pogrebniak, H.W.; Pass, H.I.; Buresh, C.; Merino, M.J.; Kauffman, D.; Venzon, D.; Langstein, H.N.; Norton, J.A. Role of tumor necrosis factor in oxygen toxicity. J. Appl. Physiol. 1992, 72, 1902–1907. [Google Scholar] [CrossRef]
- Piedboeuf, B.; Horowitz, S.; Johnston, C.J.; Gamache, M.; Belanger, S.; Poubelle, P.E.; Welty, S.E.; Watkins, R.H. Interleukin-1 expression during hyperoxic lung injury in the mouse. Free Radic. Biol. Med. 1998, 24, 1446–1454. [Google Scholar] [CrossRef] [PubMed]
- Johnston, C.J.; Wright, T.W.; Reed, C.K.; Finkelstein, J.N. Comparison of adult and newborn pulmonary cytokine mRNA expression after hyperoxia. Exp. Lung Res. 1997, 23, 537–552. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, G.; Lin, S.; Wang, C.; Zha, J. Loss of interleukin-6 enhances the inflammatory response associated with hyperoxia-induced lung injury in neonatal mice. Exp. Ther. Med. 2019, 17, 3101–3107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bateman, E.D.; Hurd, S.S.; Barnes, P.J.; Bousquet, J.; Drazen, J.M.; FitzGerald, J.M.; Gibson, P.; Ohta, K.; O’Byrne, P.; Pedersen, S.E.; et al. Global strategy for asthma management and prevention: GINA executive summary. Eur. Respir. J. 2008, 31, 143–178. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, F.; Romagnani, C.; Romagnani, S. The 3 major types of innate and adaptive cell-mediated effector immunity. J. Allergy Clin. Immunol. 2015, 135, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.T.; Chou, H.C.; Chen, C.M. Inhibition of FABP4 attenuates hyperoxia-induced lung injury and fibrosis via inhibiting TGF-beta signaling in neonatal rats. J. Cell. Physiol. 2022, 237, 1509–1520. [Google Scholar] [CrossRef]
- Ano, S.; Morishima, Y.; Ishii, Y.; Yoh, K.; Yageta, Y.; Ohtsuka, S.; Matsuyama, M.; Kawaguchi, M.; Takahashi, S.; Hizawa, N. Transcription factors GATA-3 and RORgammat are important for determining the phenotype of allergic airway inflammation in a murine model of asthma. J. Immunol. 2013, 190, 1056–1065. [Google Scholar] [CrossRef] [Green Version]
- Finotto, S. T-cell regulation in asthmatic diseases. Chem. Immunol. Allergy 2008, 94, 83–92. [Google Scholar] [CrossRef]
- Tanaka, S.; Yoshimoto, T.; Naka, T.; Nakae, S.; Iwakura, Y.; Cua, D.; Kubo, M. Natural occurring IL-17 producing T cells regulate the initial phase of neutrophil mediated airway responses. J. Immunol. 2009, 183, 7523–7530. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Nishio, K.; Takeshita, K.; Takeuchi, O.; Watanabe, K.; Sato, N.; Naoki, K.; Kudo, H.; Aoki, T.; Yamaguchi, K. Effect of steroid on hyperoxia-induced ICAM-1 expression in pulmonary endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 278, L245–L252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attaye, I.; Smulders, Y.M.; de Waard, M.C.; Oudemans-van Straaten, H.M.; Smit, B.; Van Wijhe, M.H.; Musters, R.J.; Koolwijk, P.; Spoelstra-de Man, A.M.E. The effects of hyperoxia on microvascular endothelial cell proliferation and production of vaso-active substances. Intensive Care Med. Exp. 2017, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Auten, R.L.; Whorton, M.H.; Nicholas Mason, S. Blocking neutrophil influx reduces DNA damage in hyperoxia-exposed newborn rat lung. Am. J. Respir. Cell Mol. Biol. 2002, 26, 391–397. [Google Scholar] [CrossRef] [Green Version]
- Nerurkar, L.S.; Zeligs, B.J.; Bellanti, J.A. Changes in superoxide dismutase, catalase and glucose-6-phosphate dehydrogenase activities of rabbit alveolar macrophages: Induced by postnatal maturation and/or in vitro hyperoxia. Photochem. Photobiol. 1978, 28, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Rister, M.; Baehner, R.L. Effect of hyperoxia on superoxide anion and hydrogen peroxide production of polymorphonuclear leucocytes and alveolar macrophages. Br. J. Haematol. 1977, 36, 241–248. [Google Scholar] [CrossRef]
- Yamada, M.; Kubo, H.; Kobayashi, S.; Ishizawa, K.; Sasaki, H. Interferon-gamma: A key contributor to hyperoxia-induced lung injury in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L1042–L1047. [Google Scholar] [CrossRef]
- Sporn, P.H.; Murphy, T.M.; Peters-Golden, M. Complex effects of in vitro hyperoxia on alveolar macrophage arachidonic acid metabolism. Am. J. Respir. Cell Mol. Biol. 1990, 2, 81–90. [Google Scholar] [CrossRef]
- Entezari, M.; Javdan, M.; Antoine, D.J.; Morrow, D.M.; Sitapara, R.A.; Patel, V.; Wang, M.; Sharma, L.; Gorasiya, S.; Zur, M.; et al. Inhibition of extracellular HMGB1 attenuates hyperoxia-induced inflammatory acute lung injury. Redox Biol. 2014, 2, 314–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffith, D.E.; Garcia, J.G.; James, H.L.; Callahan, K.S.; Iriana, S.; Holiday, D. Hyperoxic exposure in humans. Effects of 50 percent oxygen on alveolar macrophage leukotriene B4 synthesis. Chest 1992, 101, 392–397. [Google Scholar] [CrossRef]
- Maus, U.; von Grote, K.; Kuziel, W.A.; Mack, M.; Miller, E.J.; Cihak, J.; Stangassinger, M.; Maus, R.; Schlondorff, D.; Seeger, W.; et al. The role of CC chemokine receptor 2 in alveolar monocyte and neutrophil immigration in intact mice. Am. J. Respir. Crit. Care Med. 2002, 166, 268–273. [Google Scholar] [CrossRef]
- Hirani, D.; Alvira, C.M.; Danopoulos, S.; Milla, C.; Donato, M.; Tian, L.; Mohr, J.; Dinger, K.; Vohlen, C.; Selle, J.; et al. Macrophage-derived IL-6 trans-signalling as a novel target in the pathogenesis of bronchopulmonary dysplasia. Eur. Respir. J. 2022, 59, 2002248. [Google Scholar] [CrossRef]
- Kolliputi, N.; Shaik, R.S.; Waxman, A.B. The inflammasome mediates hyperoxia-induced alveolar cell permeability. J. Immunol. 2010, 184, 5819–5826. [Google Scholar] [CrossRef] [Green Version]
- Bhandari, V.; Choo-Wing, R.; Lee, C.G.; Zhu, Z.; Nedrelow, J.H.; Chupp, G.L.; Zhang, X.; Matthay, M.A.; Ware, L.B.; Homer, R.J.; et al. Hyperoxia causes angiopoietin 2-mediated acute lung injury and necrotic cell death. Nat. Med. 2006, 12, 1286–1293. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.; Zambrano, R.; Duncan, M.R.; Chen, S.; Luo, S.; Yuan, H.; Chen, P.; Benny, M.; Schmidt, A.; Young, K.; et al. Hyperoxia-activated circulating extracellular vesicles induce lung and brain injury in neonatal rats. Sci. Rep. 2021, 11, 8791. [Google Scholar] [CrossRef] [PubMed]
- Tiboldi, A.; Fuhrer, J.; Schaubmayr, W.; Hunyadi-Gulyas, E.; Zach, M.L.; Hochreiter, B.; Spittler, A.; Ullrich, R.; Markstaller, K.; Altmann, F.; et al. Oxygen-Dependent Changes in the N-Glycome of Murine Pulmonary Endothelial Cells. Antioxidants 2021, 10, 1947. [Google Scholar] [CrossRef]
- Barazzone, C.; Tacchini-Cottier, F.; Vesin, C.; Rochat, A.F.; Piguet, P.F. Hyperoxia induces platelet activation and lung sequestration: An event dependent on tumor necrosis factor-alpha and CD11a. Am. J. Respir. Cell Mol. Biol. 1996, 15, 107–114. [Google Scholar] [CrossRef]
- Tipping, P.G.; Campbell, D.A.; Boyce, N.W.; Holdsworth, S.R. Alveolar macrophage procoagulant activity is increased in acute hyperoxic lung injury. Am. J. Pathol. 1988, 131, 206–212. [Google Scholar] [PubMed]
- Elias, J.A.; Zheng, T.; Whiting, N.L.; Trow, T.K.; Merrill, W.W.; Zitnik, R.; Ray, P.; Alderman, E.M. IL-1 and transforming growth factor-beta regulation of fibroblast-derived IL-11. J. Immunol. 1994, 152, 2421–2429. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Du, H.; Yu, X.; Zhu, J. Fucoidan attenuates hyperoxia-induced lung injury in newborn rats by mediating lung fibroblasts differentiate into myofibroblasts. Ann. Transl. Med. 2020, 8, 1501. [Google Scholar] [CrossRef] [PubMed]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef]
- Giusto, K.; Wanczyk, H.; Jensen, T.; Finck, C. Hyperoxia-induced bronchopulmonary dysplasia: Better models for better therapies. Dis. Model. Mech. 2021, 14, dmm047753. [Google Scholar] [CrossRef]
- Li, X.G.; Song, X.; Wang, J.Y.; Sun, C.H.; Li, Z.Q.; Meng, L.L.; Chi, S.H. Fibroblast growth factor 18 alleviates hyperoxia-induced lung injury in mice by adjusting oxidative stress and inflammation. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.Y.; Kim, M.N.; Kim, E.G.; Lee, J.W.; Kim, H.R.; Kim, S.Y.; Lee, S.M.; Kim, Y.H.; Kim, K.W.; Sohn, M.H. Clusterin Deficiency Exacerbates Hyperoxia-Induced Acute Lung Injury. Cells 2021, 10, 944. [Google Scholar] [CrossRef]
- Liang, Z.; Zhang, X.; Liu, Y.; Wu, Q.; You, C. SEMA3A protects against hyperoxia-induced lung injury in a bronchopulmonary dysplasia model of newborn rat by inhibiting ERK pathway. Allergol. Immunopathol. 2021, 49, 8–15. [Google Scholar] [CrossRef]
- Ozdemir, R.; Gokce, I.K.; Taslidere, A.C.; Tanbek, K.; Gul, C.C.; Sandal, S.; Turgut, H.; Kaya, H.; Aslan, M. Does Chrysin prevent severe lung damage in Hyperoxia-Induced lung injury Model? Int. Immunopharmacol. 2021, 99, 108033. [Google Scholar] [CrossRef]
- Jia, L.; Hao, H.; Wang, C.; Wei, J. Etomidate attenuates hyperoxia-induced acute lung injury in mice by modulating the Nrf2/HO-1 signaling pathway. Exp. Ther. Med. 2021, 22, 785. [Google Scholar] [CrossRef] [PubMed]
- Qing, C.; Ziyun, L.; Xuefei, Y.; Xinyi, Z.; Xindong, X.; Jianhua, F. Protective Effects of 18beta-Glycyrrhetinic Acid on Neonatal Rats with Hyperoxia Exposure. Inflammation 2022, 45, 1224–1238. [Google Scholar] [CrossRef]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990-2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paoli, C.J.; Reynolds, M.A.; Sinha, M.; Gitlin, M.; Crouser, E. Epidemiology and Costs of Sepsis in the United States-An Analysis Based on Timing of Diagnosis and Severity Level. Crit. Care Med. 2018, 46, 1889–1897. [Google Scholar] [CrossRef]
- Vincent, J.L.; Rello, J.; Marshall, J.; Silva, E.; Anzueto, A.; Martin, C.D.; Moreno, R.; Lipman, J.; Gomersall, C.; Sakr, Y.; et al. International study of the prevalence and outcomes of infection in intensive care units. JAMA 2009, 302, 2323–2329. [Google Scholar] [CrossRef] [Green Version]
- Beale, R.; Reinhart, K.; Brunkhorst, F.M.; Dobb, G.; Levy, M.; Martin, G.; Martin, C.; Ramsey, G.; Silva, E.; Vallet, B.; et al. Promoting Global Research Excellence in Severe Sepsis (PROGRESS): Lessons from an international sepsis registry. Infection 2009, 37, 222–232. [Google Scholar] [CrossRef]
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, Patterns of Care, and Mortality for Patients With Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016, 315, 788–800. [Google Scholar] [CrossRef]
- Varisco, B.M. The pharmacology of acute lung injury in sepsis. Adv. Pharmacol. Sci. 2011, 2011, 254619. [Google Scholar] [CrossRef]
- Bhandary, Y.P.; Shetty, S.K.; Marudamuthu, A.S.; Gyetko, M.R.; Idell, S.; Gharaee-Kermani, M.; Shetty, R.S.; Starcher, B.C.; Shetty, S. Regulation of alveolar epithelial cell apoptosis and pulmonary fibrosis by coordinate expression of components of the fibrinolytic system. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L463–L473. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Kuba, K.; Neely, G.G.; Yaghubian-Malhami, R.; Perkmann, T.; van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.H.; Wang, H.; et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef]
- Patel, B.V.; Wilson, M.R.; O’Dea, K.P.; Takata, M. TNF-induced death signaling triggers alveolar epithelial dysfunction in acute lung injury. J. Immunol. 2013, 190, 4274–4282. [Google Scholar] [CrossRef] [Green Version]
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute respiratory distress syndrome. Nat. Rev. Dis. Primers 2019, 5, 18. [Google Scholar] [CrossRef]
- Peters, M.J.; Dixon, G.; Kotowicz, K.T.; Hatch, D.J.; Heyderman, R.S.; Klein, N.J. Circulating platelet-neutrophil complexes represent a subpopulation of activated neutrophils primed for adhesion, phagocytosis and intracellular killing. Br. J. Haematol. 1999, 106, 391–399. [Google Scholar] [CrossRef]
- Grommes, J.; Soehnlein, O. Contribution of neutrophils to acute lung injury. Mol. Med. 2011, 17, 293–307. [Google Scholar] [CrossRef]
- Fan, E.K.Y.; Fan, J. Regulation of alveolar macrophage death in acute lung inflammation. Respir. Res. 2018, 19, 50. [Google Scholar] [CrossRef] [Green Version]
- Johnston, L.K.; Rims, C.R.; Gill, S.E.; McGuire, J.K.; Manicone, A.M. Pulmonary macrophage subpopulations in the induction and resolution of acute lung injury. Am. J. Respir. Cell Mol. Biol. 2012, 47, 417–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, K.P.; Milberg, J.A.; Martin, T.R.; Maunder, R.J.; Cockrill, B.A.; Hudson, L.D. Evolution of bronchoalveolar cell populations in the adult respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1994, 150, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, G.; Szyndralewiez, C.; Molango, S.; Carnesecchi, S.; Heitz, F.; Wiesel, P.; Wood, J.M. Therapeutic potential of NADPH oxidase 1/4 inhibitors. Br. J. Pharmacol. 2017, 174, 1647–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.C.; Tsai, Y.F.; Pan, Y.L.; Hwang, T.L. Understanding the role of neutrophils in acute respiratory distress syndrome. Biomed. J. 2021, 44, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Arnhold, J.; Furtmuller, P.G.; Regelsberger, G.; Obinger, C. Redox properties of the couple compound I/native enzyme of myeloperoxidase and eosinophil peroxidase. Eur. J. Biochem. 2001, 268, 5142–5148. [Google Scholar] [CrossRef]
- Park, J.S.; Park, Y.J.; Kim, H.R.; Chung, K.H. Polyhexamethylene guanidine phosphate-induced ROS-mediated DNA damage caused cell cycle arrest and apoptosis in lung epithelial cells. J. Toxicol. Sci. 2019, 44, 415–424. [Google Scholar] [CrossRef] [Green Version]
- Fukuhara, K.; Nakashima, T.; Abe, M.; Masuda, T.; Hamada, H.; Iwamoto, H.; Fujitaka, K.; Kohno, N.; Hattori, N. Suplatast tosilate protects the lung against hyperoxic lung injury by scavenging hydroxyl radicals. Free Radic. Biol. Med. 2017, 106, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Dias-Freitas, F.; Metelo-Coimbra, C.; Roncon-Albuquerque, R., Jr. Molecular mechanisms underlying hyperoxia acute lung injury. Respir. Med. 2016, 119, 23–28. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.J.; Fantone, J.C., 3rd; Kaplan, J.; Ward, P.A. In vivo damage of rat lungs by oxygen metabolites. J. Clin. Investig. 1981, 67, 983–993. [Google Scholar] [CrossRef] [Green Version]
- Reddy, S.P.; Hassoun, P.M.; Brower, R. Redox imbalance and ventilator-induced lung injury. Antioxid. Redox Signal. 2007, 9, 2003–2012. [Google Scholar] [CrossRef]
- Metnitz, P.G.; Bartens, C.; Fischer, M.; Fridrich, P.; Steltzer, H.; Druml, W. Antioxidant status in patients with acute respiratory distress syndrome. Intensive Care Med. 1999, 25, 180–185. [Google Scholar] [CrossRef]
- Buras, J.A.; Holzmann, B.; Sitkovsky, M. Animal models of sepsis: Setting the stage. Nat. Rev. Drug Discov. 2005, 4, 854–865. [Google Scholar] [CrossRef]
- Ohsawa, I.; Ishikawa, M.; Takahashi, K.; Watanabe, M.; Nishimaki, K.; Yamagata, K.; Katsura, K.; Katayama, Y.; Asoh, S.; Ohta, S. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat. Med. 2007, 13, 688–694. [Google Scholar] [CrossRef]
- Qiu, P.; Liu, Y.; Chen, K.; Dong, Y.; Liu, S.; Zhang, J. Hydrogen-rich saline regulates the polarization and apoptosis of alveolar macrophages and attenuates lung injury via suppression of autophagy in septic rats. Ann. Transl. Med. 2021, 9, 974. [Google Scholar] [CrossRef]
- Lei, L.; Guo, Y.; Lin, J.; Lin, X.; He, S.; Qin, Z.; Lin, Q. Inhibition of endotoxin-induced acute lung injury in rats by bone marrow-derived mesenchymal stem cells: Role of Nrf2/HO-1 signal axis in inhibition of NLRP3 activation. Biochem. Biophys. Res. Commun. 2021, 551, 7–13. [Google Scholar] [CrossRef]
- Lee, I.; Dodia, C.; Chatterjee, S.; Feinstein, S.I.; Fisher, A.B. Protection against LPS-induced acute lung injury by a mechanism-based inhibitor of NADPH oxidase (type 2). Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L635–L644. [Google Scholar] [CrossRef] [Green Version]
- Fisher, A.B.; Dodia, C.; Chatterjee, S.; Feinstein, S.I. A Peptide Inhibitor of NADPH Oxidase (NOX2) Activation Markedly Decreases Mouse Lung Injury and Mortality Following Administration of Lipopolysaccharide (LPS). Int. J. Mol. Sci. 2020, 20, 2395. [Google Scholar] [CrossRef] [Green Version]
- Holford, P.; Carr, A.C.; Jovic, T.H.; Ali, S.R.; Whitaker, I.S.; Marik, P.E.; Smith, A.D. Vitamin C-An Adjunctive Therapy for Respiratory Infection, Sepsis and COVID-19. Nutrients 2020, 12, 3760. [Google Scholar] [CrossRef]
- Hemila, H.; Chalker, E. Vitamin C Can Shorten the Length of Stay in the ICU: A Meta-Analysis. Nutrients 2019, 11, 708. [Google Scholar] [CrossRef] [Green Version]
- Hemila, H.; Chalker, E. Vitamin C may reduce the duration of mechanical ventilation in critically ill patients: A meta-regression analysis. J. Intensive Care 2020, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Fujii, T.; Deane, A.M.; Nair, P. Metabolic support in sepsis: Corticosteroids and vitamins: The why, the when, the how. Curr. Opin. Crit. Care 2020, 26, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Slutsky, A.S.; Ranieri, V.M. Ventilator-induced lung injury. N. Engl. J. Med. 2013, 369, 2126–2136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cândido, L.d.S.; de Matos, N.A.; Castro, T.d.F.; de Paula, L.C.; Dos Santos, A.M.; Costa, G.d.P.; Talvani, A.; Cangussú, S.D.; Zin, W.A.; Bezerra, F.S.J.O.M.; et al. Different Tidal Volumes May Jeopardize Pulmonary Redox and Inflammatory Status in Healthy Rats Undergoing Mechanical Ventilation. Oxidative Med. Cell. Longev. 2021, 2021, 5196896. [Google Scholar] [CrossRef]
- Curley, G.F.; Laffey, J.G.; Zhang, H.; Slutsky, A.S. Biotrauma and Ventilator-Induced Lung Injury: Clinical Implications. Chest 2016, 150, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, L.N.; Slutsky, A.S. Ventilator-induced lung injury: From the bench to the bedside. Intensive Care Med. 2006, 32, 24–33. [Google Scholar] [CrossRef]
- Santos, R.S.; Maia, L.A.; Oliveira, M.V.; Santos, C.L.; Moraes, L.; Pinto, E.F.; Samary, C.D.S.; Machado, J.A.; Carvalho, A.C.; Fernandes, M.V.S.; et al. Biologic Impact of Mechanical Power at High and Low Tidal Volumes in Experimental Mild Acute Respiratory Distress Syndrome. Anesthesiology 2018, 128, 1193–1206. [Google Scholar] [CrossRef]
- Katira, B.H.; Osada, K.; Engelberts, D.; Bastia, L.; Damiani, L.F.; Li, X.; Chen, H.; Yoshida, T.; Amato, M.B.P.; Ferguson, N.D.; et al. Positive End-Expiratory Pressure, Pleural Pressure, and Regional Compliance during Pronation: An Experimental Study. Am. J. Respir. Crit. Care Med. 2021, 203, 1266–1274. [Google Scholar] [CrossRef]
- Hernandez, L.A.; Peevy, K.J.; Moise, A.A.; Parker, J.C. Chest wall restriction limits high airway pressure-induced lung injury in young rabbits. J. Appl. Physiol. 1989, 66, 2364–2368. [Google Scholar] [CrossRef]
- Amato, M.B.; Meade, M.O.; Slutsky, A.S.; Brochard, L.; Costa, E.L.; Schoenfeld, D.A.; Stewart, T.E.; Briel, M.; Talmor, D.; Mercat, A.; et al. Driving pressure and survival in the acute respiratory distress syndrome. N. Engl. J. Med. 2015, 372, 747–755. [Google Scholar] [CrossRef] [Green Version]
- Almeida, M.R.; Horta, J.G.A.; de Matos, N.A.; de Souza, A.B.F.; Castro, T.F.; Candido, L.D.S.; Andrade, M.C.; Cangussu, S.D.; Costa, G.P.; Talvani, A.; et al. The effects of different ventilatory modes in female adult rats submitted to mechanical ventilation. Respir. Physiol. Neurobiol. 2021, 284, 103583. [Google Scholar] [CrossRef]
- Andrade, M.C.; Souza, A.B.F.; Horta, J.G.; Paula Costa, G.; Talvani, A.; Cangussu, S.D.; Menezes, R.C.A.; Bezerra, F.S. Applying Positive End-Expiratory Pressure During Mechanical Ventilation Causes Pulmonary Redox Imbalance and Inflammation in Rats. Shock 2018, 50, 572–578. [Google Scholar] [CrossRef]
- Joelsson, J.P.; Ingthorsson, S.; Kricker, J.; Gudjonsson, T.; Karason, S. Ventilator-induced lung-injury in mouse models: Is there a trap? Lab. Anim. Res. 2021, 37, 30. [Google Scholar] [CrossRef]
- Grazioli, S.; Dunn-Siegrist, I.; Pauchard, L.A.; Blot, M.; Charles, P.E.; Pugin, J. Mitochondrial alarmins are tissue mediators of ventilator-induced lung injury and ARDS. PLoS ONE 2019, 14, e0225468. [Google Scholar] [CrossRef] [Green Version]
- Spieth, P.M.; Bluth, T.; Gama De Abreu, M.; Bacelis, A.; Goetz, A.E.; Kiefmann, R. Mechanotransduction in the lungs. Minerva Anestesiol. 2014, 80, 933–941. [Google Scholar]
- Ko, Y.A.; Yang, M.C.; Huang, H.T.; Hsu, C.M.; Chen, L.W. NF-kappaB activation in myeloid cells mediates ventilator-induced lung injury. Respir. Res. 2013, 14, 69. [Google Scholar] [CrossRef] [Green Version]
- Kuipers, M.T.; Vogl, T.; Aslami, H.; Jongsma, G.; van den Berg, E.; Vlaar, A.P.; Roelofs, J.J.; Juffermans, N.P.; Schultz, M.J.; van der Poll, T.; et al. High levels of S100A8/A9 proteins aggravate ventilator-induced lung injury via TLR4 signaling. PLoS ONE 2013, 8, e68694. [Google Scholar] [CrossRef] [Green Version]
- Nickles, H.T.; Sumkauskaite, M.; Wang, X.; Wegner, I.; Puderbach, M.; Kuebler, W.M. Mechanical ventilation causes airway distension with proinflammatory sequelae in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L27–L37. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.H.; Lin, J.C.; Wu, S.Y.; Huang, K.L.; Jung, F.; Ma, M.C.; Wang Hsu, G.S.; Jow, G.M. Membrane translocation of IL-33 receptor in ventilator induced lung injury. PLoS ONE 2015, 10, e0121391. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Xie, W.; Wang, Y.; Chen, S.; Han, J.; Wang, L.; Gui, P.; Wu, Q. JAK2/STAT1-mediated HMGB1 translocation increases inflammation and cell death in a ventilator-induced lung injury model. Lab. Investig. 2019, 99, 1810–1821. [Google Scholar] [CrossRef]
- De Souza, A.B.F.; Chirico, M.T.T.; Cartelle, C.T.; de Paula Costa, G.; Talvani, A.; Cangussu, S.D.; de Menezes, R.C.A.; Bezerra, F.S. High-Fat Diet Increases HMGB1 Expression and Promotes Lung Inflammation in Mice Subjected to Mechanical Ventilation. Oxid. Med. Cell. Longev. 2018, 2018, 7457054. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, E.N.; Ishizaka, A.; Tasaka, S.; Koh, H.; Ueno, H.; Amaya, F.; Ebina, M.; Yamada, S.; Funakoshi, Y.; Soejima, J.; et al. Contribution of high-mobility group box-1 to the development of ventilator-induced lung injury. Am. J. Respir. Crit. Care Med. 2006, 174, 400–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhang, R.; Tong, Y.; Ding, X.; Jin, S.; Zhao, X.; Zong, J.; Chen, Z.; Billiar, T.R.; Li, Q. High-mobility group box 1 protein is involved in the protective effect of Saquinavir on ventilation-induced lung injury in mice. Acta Biochim. et Biophys. Sin. 2017, 49, 907–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, G.; Dull, R.O.; Schwartz, D.E.; Hu, G. Autophagy in pulmonary macrophages mediates lung inflammatory injury via NLRP3 inflammasome activation during mechanical ventilation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L173–L185. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, A.C.L.; de Matos, N.A.; de Souza, A.B.F.; Castro, T.F.; Candido, L.D.S.; Oliveira, M.; Costa, G.P.; Talvani, A.; Cangussu, S.D.; Bezerra, F.S. Sigh maneuver protects healthy lungs during mechanical ventilation in adult Wistar rats. Exp. Biol. Med. 2020, 245, 1404–1413. [Google Scholar] [CrossRef]
- Arason, A.J.; Joelsson, J.P.; Valdimarsdottir, B.; Sigurdsson, S.; Gudjonsson, A.; Halldorsson, S.; Johannsson, F.; Rolfsson, O.; Lehmann, F.; Ingthorsson, S.; et al. Azithromycin induces epidermal differentiation and multivesicular bodies in airway epithelia. Respir. Res. 2019, 20, 129. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Pan, P.; Su, X.; Liu, S.; Zhang, L.; Wu, D.; Li, H.; Dai, M.; Li, Y.; Hu, C.; et al. Neutrophil Extracellular Traps Are Pathogenic in Ventilator-Induced Lung Injury and Partially Dependent on TLR4. Biomed. Res. Int. 2017, 2017, 8272504. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Frank, J.A.; Pittet, J.F.; Lee, H.; Godzich, M.; Matthay, M.A. High tidal volume ventilation induces NOS2 and impairs cAMP- dependent air space fluid clearance. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L791–L798. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; An, X.; Wang, X.; Bao, C.; Li, J.; Yang, D.; Bai, C. Curcumin ameliorated ventilator-induced lung injury in rats. Biomed. Pharmacother. 2018, 98, 754–761. [Google Scholar] [CrossRef]
- Carvalho, G.C.; de Camargo, B.A.F.; de Araújo, J.T.C.; Chorilli, M. Lycopene: From tomato to its nutraceutical use and its association with nanotechnology. Trends Food Sci. Technol. 2021, 118, 447–458. [Google Scholar] [CrossRef]
- Da Silva Araujo, N.P.; de Matos, N.A.; Leticia Antunes Mota, S.; Farias de Souza, A.B.; Dantas Cangussu, S.; Cunha Alvim de Menezes, R.; Silva Bezerra, F. Quercetin Attenuates Acute Lung Injury Caused by Cigarette Smoke Both In Vitro and In Vivo. COPD 2020, 17, 205–214. [Google Scholar] [CrossRef] [PubMed]
- De Souza, A.B.F.; de Matos, N.A.; Castro, T.F.; Costa, G.P.; Oliveira, L.A.M.; Nogueira, K.; Ribeiro, I.M.L.; Talvani, A.; Cangussu, S.D.; de Menezes, R.C.A.; et al. Effects in vitro and in vivo of hesperidin administration in an experimental model of acute lung inflammation. Free Radic. Biol. Med. 2022, 180, 253–262. [Google Scholar] [CrossRef] [PubMed]








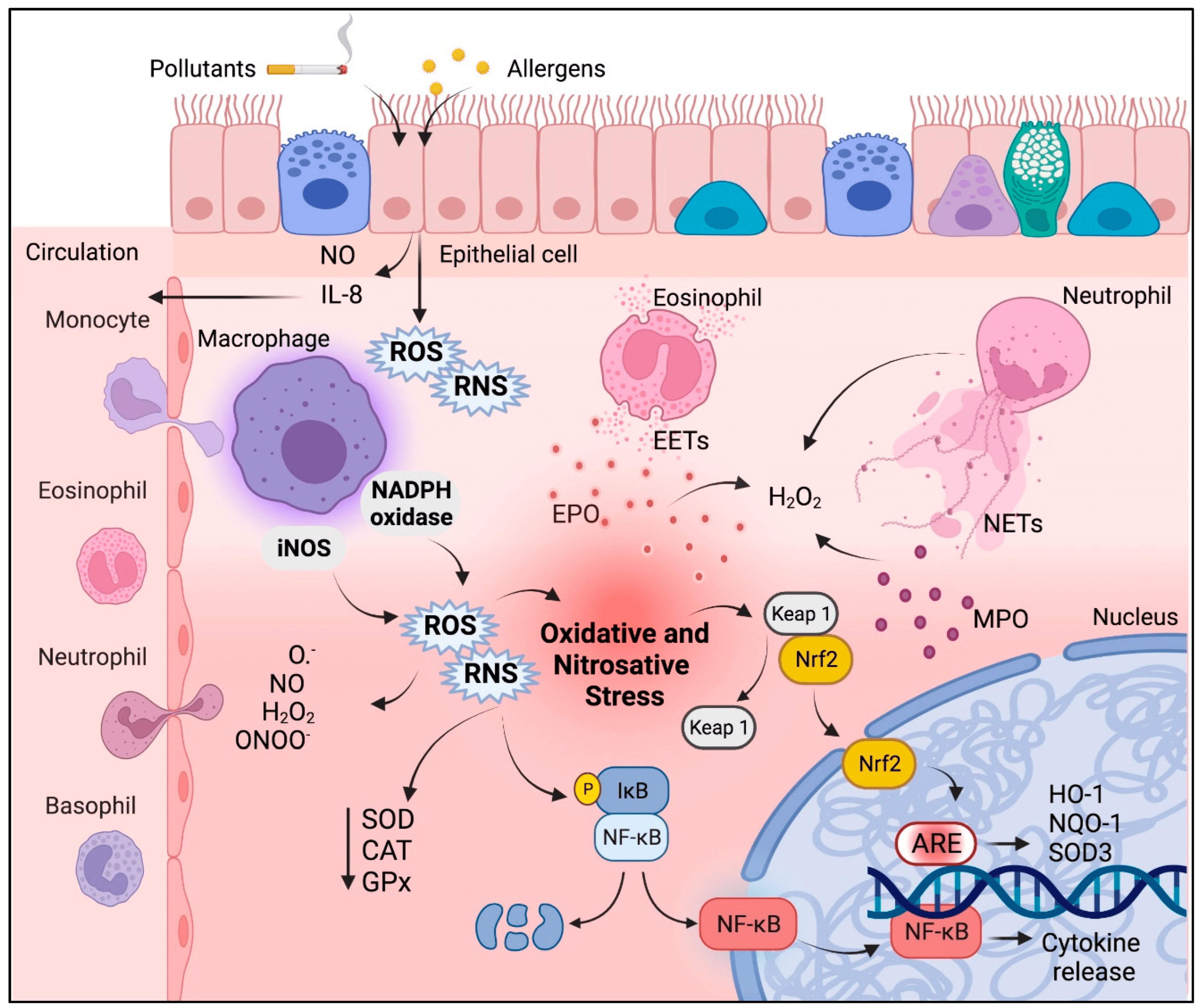{kind=link}
{kind=link}
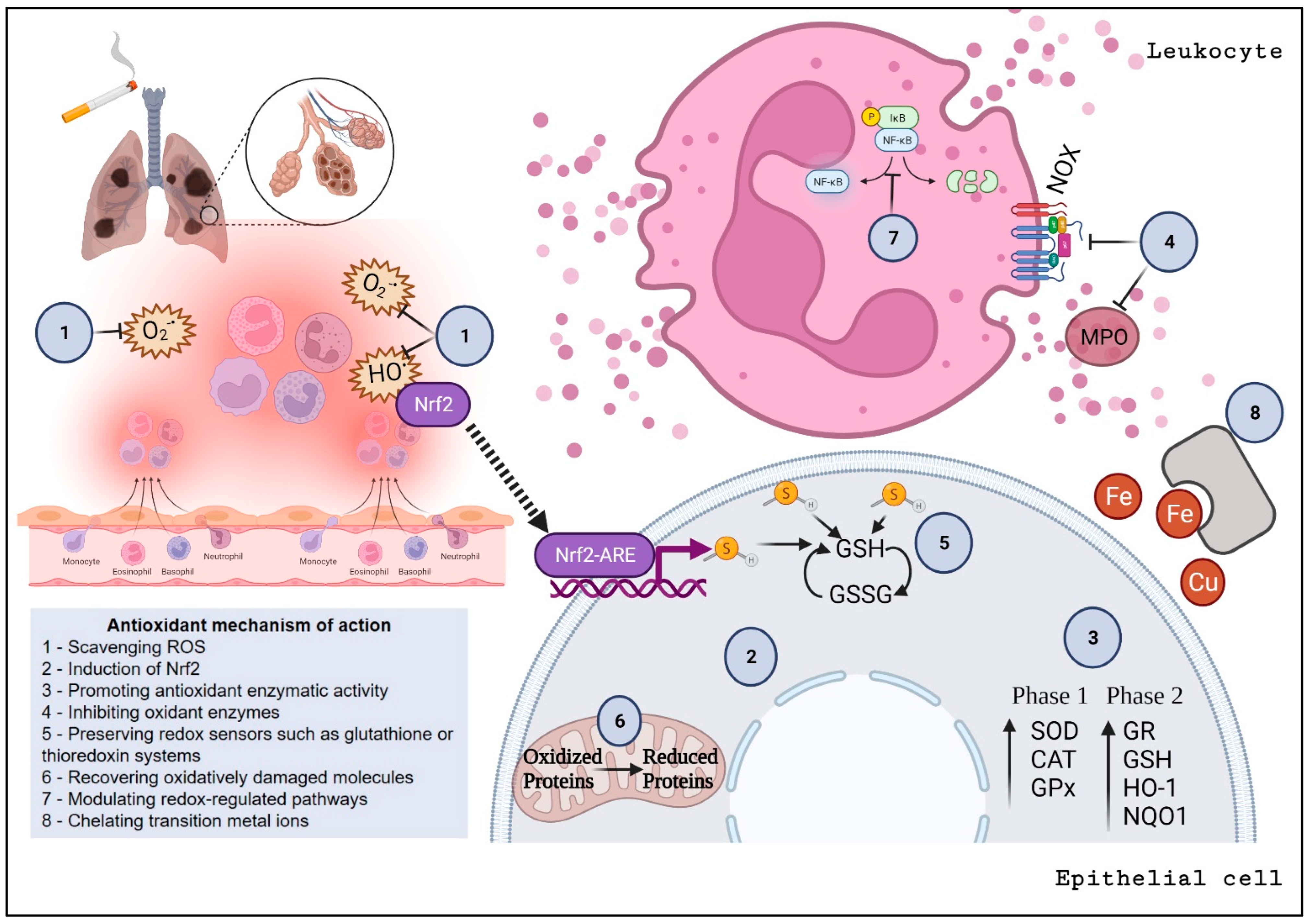{kind=link}
{kind=link}
{kind=link}
{kind=link}
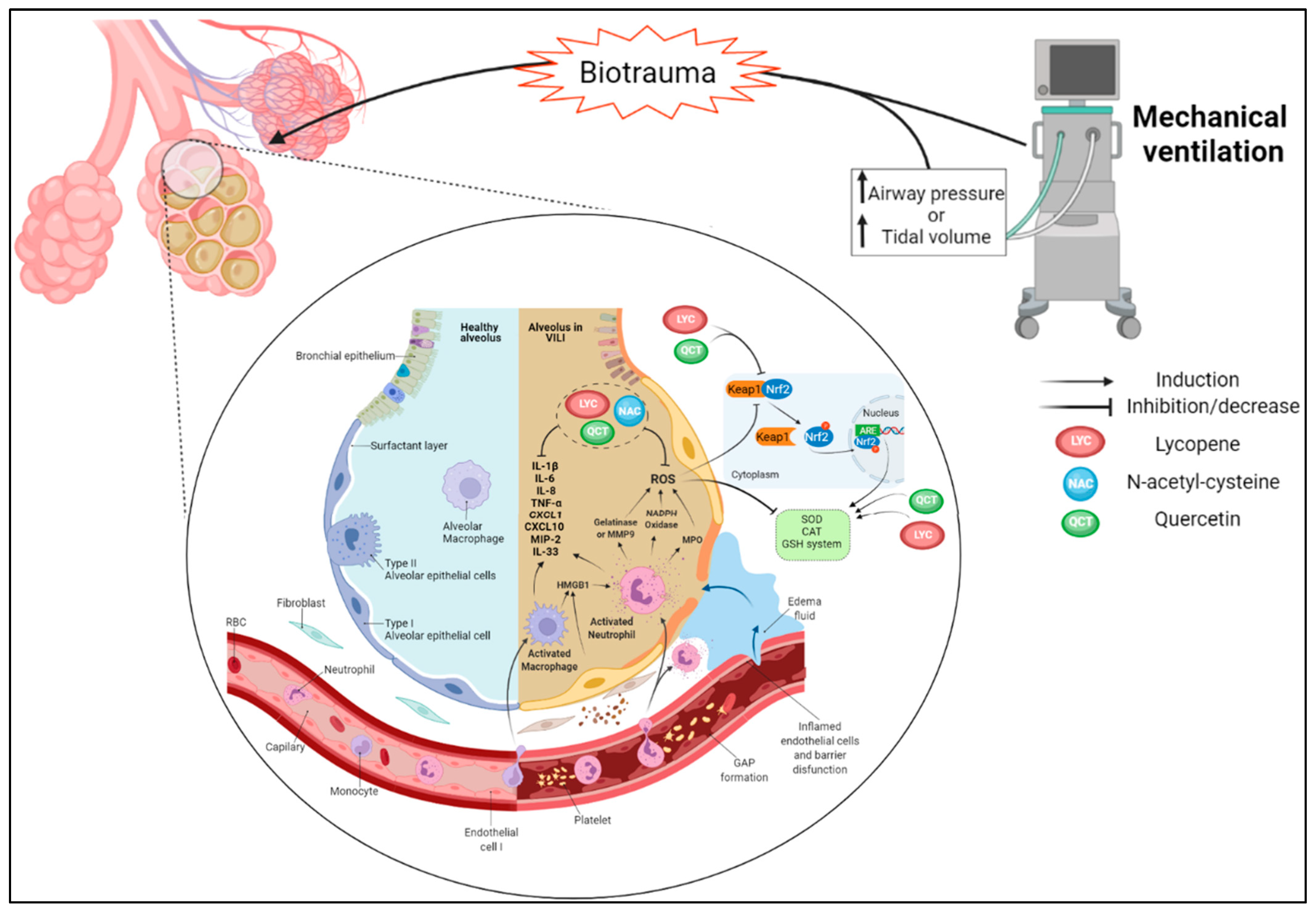{kind=link}
| Synthetic AO against Emphysema | Outcome Observed | References |
|---|---|---|
| N-acetylcysteine | Preserves GSH/GSSG, anti-inflammatory, reduces exacerbations | [186,187,188,189] |
| Aminoguanidine | Reduces peroxynitrite and tissue damage | [190] |
| MnTE-2-PyP | SOD3 mimetic reduces extracellular matrix damage | [191] |
| Diallyl disulfide | Prevents tissue and oxidative damage, inhibits cytokine production, and potentiates antioxidant enzymes. | [196,197] |
| Statins | Reduces ROS, inhibiting tissue as well as oxidative damage, and activates Nrf2 and AO enzymes. | [199,200,202] |
| Natural AO against Emphysema | Outcome Observed | Reference |
|---|---|---|
| Mate tea (chlorogenic acid main polyphenol) | Reduces MPO activity; prevents oxidative and tissue damage; increases CAT activity and GSH/GSSG | [203] |
| Eucalyptol | Reduces oxidative and tissue damage; increases SOD activity | [204] |
| Propolis | Induces pro-resolutive macrophages; reduces tissue damage | [205] |
| Quercetin | Reduces oxidative damage; suppresses iNOS, cytokines, MMPs; increases AO enzyme activity | [208,209] |
| Resveratrol | Ameliorates lung function; activates Nrf2, SOD, Sirt1; reduces oxidative damage and cytokines | [88,211] |
| Curcumin | Induces autophagy; reduces ER stress by Sirt1 activation; reduces cytokines; ameliorates lung function | [212,213] |
| Sulforaphane | Upregulates Nrf2; reduces ROS, oxidative damage, and inflammatory pathways involving TLR2, TLR4 and Myd88 * | [214,215] |
| Vitamin C | Reduces oxidative and tissue damage; increases collagen synthesis and vascular endothelial growth factor levels | [217] |
| Vitamin E | Reduces oxidative and tissue damage as well as inflammation; upregulates Nrf2 and AO enzyme activity | [218] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bezerra, F.S.; Lanzetti, M.; Nesi, R.T.; Nagato, A.C.; Silva, C.P.e.; Kennedy-Feitosa, E.; Melo, A.C.; Cattani-Cavalieri, I.; Porto, L.C.; Valenca, S.S. Oxidative Stress and Inflammation in Acute and Chronic Lung Injuries. Antioxidants 2023, 12, 548. https://doi.org/10.3390/antiox12030548
Bezerra FS, Lanzetti M, Nesi RT, Nagato AC, Silva CPe, Kennedy-Feitosa E, Melo AC, Cattani-Cavalieri I, Porto LC, Valenca SS. Oxidative Stress and Inflammation in Acute and Chronic Lung Injuries. Antioxidants. 2023; 12(3):548. https://doi.org/10.3390/antiox12030548
Chicago/Turabian StyleBezerra, Frank Silva, Manuella Lanzetti, Renata Tiscoski Nesi, Akinori Cardozo Nagato, Cyntia Pecli e Silva, Emanuel Kennedy-Feitosa, Adriana Correa Melo, Isabella Cattani-Cavalieri, Luís Cristóvão Porto, and Samuel Santos Valenca. 2023. "Oxidative Stress and Inflammation in Acute and Chronic Lung Injuries" Antioxidants 12, no. 3: 548. https://doi.org/10.3390/antiox12030548