
Aurora Kinase A Regulation by Cysteine Oxidative Modification
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Redox-Based Cell Cycle Regulation
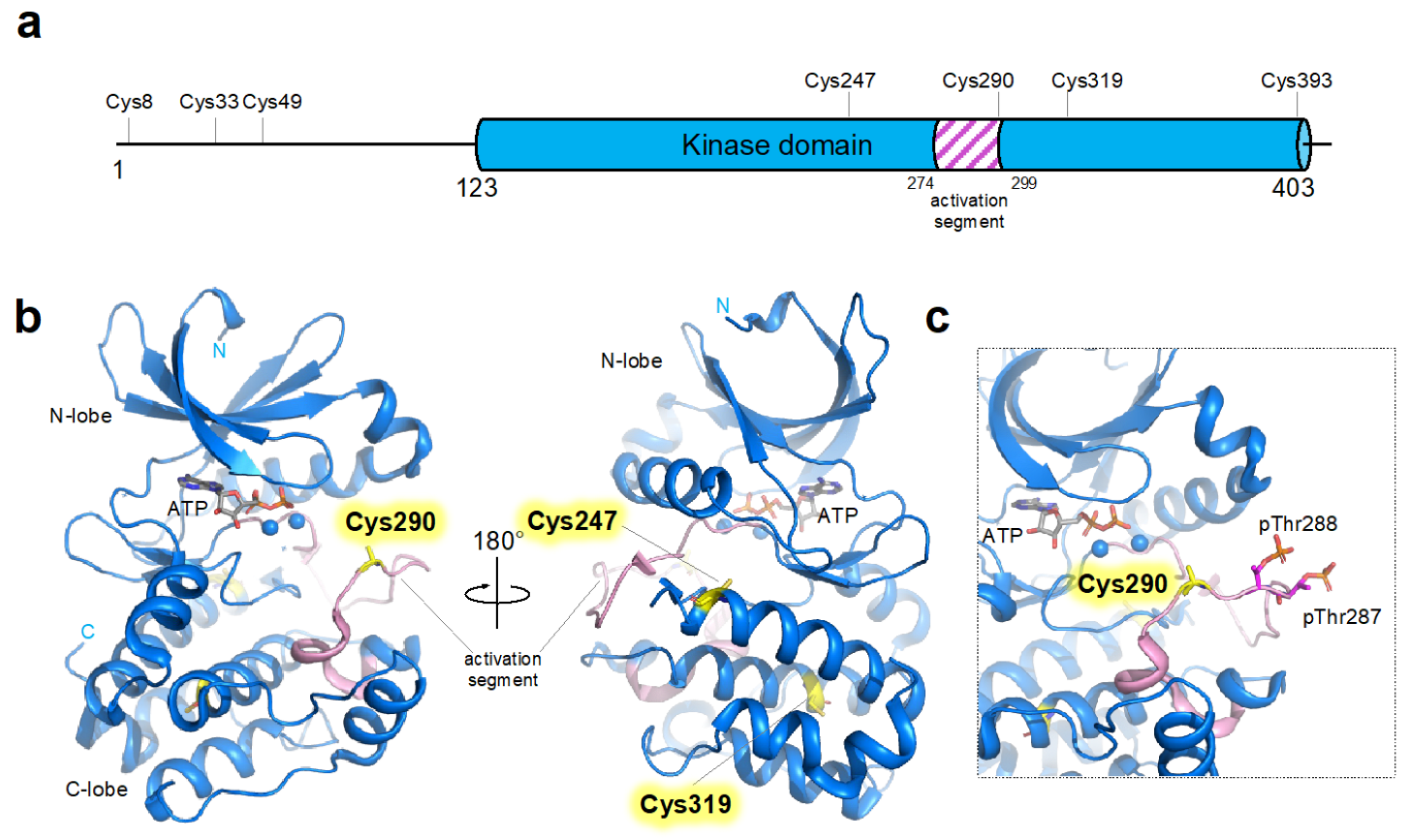
3. Cysteine Residues in AURKA
4. Functional and Structural Consequences of Cysteine Modification in AURKA
4.1. Two Distinct Pathways of AURKA Activation
4.2. H2O2 Induced Oxidative Modification of Cysteine
4.3. Structural Transitions Induced upon Covalent Modification of Cysteine in AURKA with Coenzyme A
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carmena, M.; Earnshaw, W.C. The cellular geography of aurora kinases. Nat. Rev. Mol. Cell Biol. 2003, 4, 842–854. [Google Scholar] [CrossRef] [PubMed]
- Vader, G.; Lens, S.M. The Aurora kinase family in cell division and cancer. Biochim. Biophys. Acta -Rev. Cancer 2008, 1786, 60–72. [Google Scholar] [CrossRef]
- Adhikari, B.; Bozilovic, J.; Diebold, M.; Schwarz, J.D.; Hofstetter, J.; Schröder, M.; Wanior, M.; Narain, A.; Vogt, M.; Dudvarski Stankovic, N. PROTAC-mediated degradation reveals a non-catalytic function of AURORA-A kinase. Nat. Chem. Biol. 2020, 16, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Mahankali, M.; Henkels, K.M.; Speranza, F.; Gomez-Cambronero, J. A non-mitotic role for Aurora kinase A as a direct activator of cell migration upon interaction with PLD, FAK and Src. J. Cell Sci. 2015, 128, 516–526. [Google Scholar] [CrossRef]
- Guarino Almeida, E.; Renaudin, X.; Venkitaraman, A.R. A kinase-independent function for AURORA-A in replisome assembly during DNA replication initiation. Nucleic Acids Res. 2020, 48, 7844–7855. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.S.; Botstein, D. Isolation and characterization of chromosome-gain and increase-in-ploidy mutants in yeast. Genetics 1993, 135, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.C.Y.; Frolov, A.; Li, R.; Ayala, G.; Greenberg, N.M. Targeting Aurora kinases for the treatment of prostate cancer. Cancer Res. 2006, 66, 4996–5002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, R.; Huang, C.; Liu, K.; Li, X.; Dong, Z. Targeting AURKA in Cancer: Molecular mechanisms and opportunities for Cancer therapy. Mol. Cancer 2021, 20, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Kanai, N.; Shiwaku, H.; Soga, N.; Uehara, A.; Horii, A. AURKA is one of the downstream targets of MAPK1/ERK2 in pancreatic cancer. Oncogene 2006, 25, 4831–4839. [Google Scholar] [CrossRef] [Green Version]
- Cox, D.G.; Hankinson, S.E.; Hunter, D.J. Polymorphisms of the AURKA (STK15/Aurora Kinase) gene and breast cancer risk (United States). Cancer Causes Control 2006, 17, 81–83. [Google Scholar] [CrossRef]
- Goos, J.A.; Coupe, V.M.; Diosdado, B.; Diemen, D.-V.; Karga, C.; Belien, J.A.; Carvalho, B.; van den Tol, M.P.; Verheul, H.M.; Geldof, A.A. Aurora kinase A (AURKA) expression in colorectal cancer liver metastasis is associated with poor prognosis. Br. J. Cancer 2013, 109, 2445–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, A.; Gao, K.; Chu, L.; Zhang, R.; Yang, J.; Zheng, J. Aurora kinases: Novel therapy targets in cancers. Oncotarget 2017, 8, 23937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bavetsias, V.; Linardopoulos, S. Aurora kinase inhibitors: Current status and outlook. Front. Oncol. 2015, 5, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keen, N.; Taylor, S. Aurora-kinase inhibitors as anticancer agents. Nat. Rev. Cancer 2004, 4, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Borisa, A.C.; Bhatt, H.G. A comprehensive review on Aurora kinase: Small molecule inhibitors and clinical trial studies. Eur. J. Med. Chem. 2017, 140, 1–19. [Google Scholar] [CrossRef]
- Falchook, G.S.; Bastida, C.C.; Kurzrock, R. Aurora kinase inhibitors in oncology clinical trials: Current state of the progress. Semin. Oncol. 2015, 42, 832–848. [Google Scholar] [CrossRef]
- O’connor, O.A.; Ozcan, M.; Jacobsen, E.D.; Roncero, J.M.; Trotman, J.; Demeter, J.; Masszi, T.; Pereira, J.; Ramchandren, R.; Beaven, A. Randomized phase III study of alisertib or investigator’s choice (selected single agent) in patients with relapsed or refractory peripheral T-cell lymphoma. J. Clin. Oncol. 2019, 37, 613–623. [Google Scholar] [CrossRef]
- Chu, Q.S.-C.; Bouganim, N.; Fortier, C.; Zaknoen, S.; Stille, J.R.; Kremer, J.D.; Yuen, E.; Hui, Y.-H.; de la Peña, A.; Lithio, A. Aurora kinase A inhibitor, LY3295668 erbumine: A phase 1 monotherapy safety study in patients with locally advanced or metastatic solid tumors. Investig. New Drugs 2021, 39, 1001–1010. [Google Scholar] [CrossRef]
- Rosenthal, A.; Kumar, S.; Hofmeister, C.; Laubach, J.; Vij, R.; Dueck, A.; Gano, K.; Stewart, A.K. A Phase Ib Study of the combination of Aurora Kinase Inhibitor alisertib (MLN8237) and bortezomib in Relapsed or Refractory Multiple Myeloma. Br. J. Haematol. 2016, 174, 323. [Google Scholar] [CrossRef] [Green Version]
- Macarulla, T.; Cervantes, A.; Elez, E.; Rodríguez-Braun, E.; Baselga, J.; Roselló, S.; Sala, G.; Blasco, I.; Danaee, H.; Lee, Y. Phase I Study of the Selective Aurora A Kinase Inhibitor MLN8054 in Patients with Advanced Solid Tumors: Safety, Pharmacokinetics, and Pharmacodynamics MLN8054: Outcomes of a Phase I Study. Mol. Cancer Ther. 2010, 9, 2844–2852. [Google Scholar] [CrossRef] [Green Version]
- Joukov, V.; De Nicolo, A. Aurora-PLK1 cascades as key signaling modules in the regulation of mitosis. Sci. Signal. 2018, 11, eaar4195. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Ferreria, M.A.; Rath, U.; Buster, D.W.; Chanda, S.K.; Caldwell, J.S.; Rines, D.R.; Sharp, D.J. Human Cep192 is required for mitotic centrosome and spindle assembly. Curr. Biol. 2007, 17, 1960–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joukov, V.; De Nicolo, A.; Rodriguez, A.; Walter, J.C.; Livingston, D.M. Centrosomal protein of 192 kDa (Cep192) promotes centrosome-driven spindle assembly by engaging in organelle-specific Aurora A activation. Proc. Natl. Acad. Sci. USA 2010, 107, 21022–21027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joukov, V.; Walter, J.C.; De Nicolo, A. The Cep192-organized aurora A-Plk1 cascade is essential for centrosome cycle and bipolar spindle assembly. Mol. Cell 2014, 55, 578–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karthigeyan, D.; Prasad, S.B.B.; Shandilya, J.; Agrawal, S.; Kundu, T.K. Biology of Aurora A kinase: Implications in cancer manifestation and therapy. Med. Res. Rev. 2011, 31, 757–793. [Google Scholar] [CrossRef]
- Carmena, M.; Ruchaud, S.; Earnshaw, W.C. Making the Auroras glow: Regulation of Aurora A and B kinase function by interacting proteins. Curr. Opin. Cell Biol. 2009, 21, 796–805. [Google Scholar] [CrossRef] [Green Version]
- Hutterer, A.; Berdnik, D.; Wirtz-Peitz, F.; Žigman, M.; Schleiffer, A.; Knoblich, J.A. Mitotic activation of the kinase Aurora-A requires its binding partner Bora. Dev. Cell 2006, 11, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Zang, J.; Chen, Y.; Liu, C.; Lin, S. Probing the Role of Aurora Kinase A Threonylation with Site-Specific Lysine Threonylation. ACS Chem. Biol. 2022. [Google Scholar] [CrossRef]
- Byrne, D.P.; Shrestha, S.; Galler, M.; Cao, M.; Daly, L.A.; Campbell, A.E.; Eyers, C.E.; Veal, E.A.; Kannan, N.; Eyers, P.A. Aurora A regulation by reversible cysteine oxidation reveals evolutionarily conserved redox control of Ser/Thr protein kinase activity. Sci. Signal. 2020, 13, eaax2713. [Google Scholar] [CrossRef]
- Lim, D.C.; Joukov, V.; Rettenmaier, T.J.; Kumagai, A.; Dunphy, W.G.; Wells, J.A.; Yaffe, M.B. Redox priming promotes Aurora A activation during mitosis. Sci. Signal. 2020, 13, eabb6707. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Byrne, D.P.; Burgess, S.G.; Bormann, J.; Baković, J.; Huang, Y.; Zhyvoloup, A.; Yu, B.Y.K.; Peak-Chew, S.; Tran, T. Covalent Aurora A regulation by the metabolic integrator coenzyme A. Redox Biol. 2020, 28, 101318. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.-F.; Dong, Q.; Bai, Y.; Yuan, J.; Xu, Q.; Cao, C.; Liu, X. Oxidative stress induces mitotic arrest by inhibiting Aurora A-involved mitotic spindle formation. Free. Radic. Biol. Med. 2017, 103, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Janeček, M.; Rossmann, M.; Sharma, P.; Emery, A.; Huggins, D.J.; Stockwell, S.R.; Stokes, J.E.; Tan, Y.S.; Almeida, E.G.; Hardwick, B. Allosteric modulation of AURKA kinase activity by a small-molecule inhibitor of its protein-protein interaction with TPX2. Sci. Rep. 2016, 6, 28528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntyre, P.J.; Collins, P.M.; Vrzal, L.s.; Birchall, K.; Arnold, L.H.; Mpamhanga, C.; Coombs, P.J.; Burgess, S.G.; Richards, M.W.; Winter, A. Characterization of three druggable hot-spots in the Aurora-A/TPX2 interaction using biochemical, biophysical, and fragment-based approaches. ACS Chem. Biol. 2017, 12, 2906–2914. [Google Scholar] [CrossRef] [Green Version]
- Asteriti, I.A.; Daidone, F.; Colotti, G.; Rinaldo, S.; Lavia, P.; Guarguaglini, G.; Paiardini, A. Identification of small molecule inhibitors of the Aurora-A/TPX2 complex. Oncotarget 2017, 8, 32117. [Google Scholar] [CrossRef] [Green Version]
- Cole, D.J.; Janecek, M.; Stokes, J.E.; Rossmann, M.; Faver, J.C.; McKenzie, G.J.; Venkitaraman, A.R.; Hyvönen, M.; Spring, D.R.; Huggins, D.J. Computationally-guided optimization of small-molecule inhibitors of the Aurora A kinase–TPX2 protein–protein interaction. Chem. Commun. 2017, 53, 9372–9375. [Google Scholar] [CrossRef] [Green Version]
- Bayliss, R.; Sardon, T.; Vernos, I.; Conti, E. Structural basis of Aurora-A activation by TPX2 at the mitotic spindle. Mol. Cell 2003, 12, 851–862. [Google Scholar] [CrossRef]
- Kufer, T.A.; Silljé, H.H.; Körner, R.; Gruss, O.J.; Meraldi, P.; Nigg, E.A. Human TPX2 is required for targeting Aurora-A kinase to the spindle. J. Cell Biol. 2002, 158, 617–623. [Google Scholar] [CrossRef] [Green Version]
- Wittmann, T.; Wilm, M.; Karsenti, E.; Vernos, I. TPX2, A novel xenopus MAP involved in spindle pole organization. J. Cell Biol. 2000, 149, 1405–1418. [Google Scholar] [CrossRef]
- Zeng, K.; Bastos, R.N.; Barr, F.A.; Gruneberg, U. Protein phosphatase 6 regulates mitotic spindle formation by controlling the T-loop phosphorylation state of Aurora A bound to its activator TPX2. J. Cell Biol. 2010, 191, 1315–1332. [Google Scholar] [CrossRef] [Green Version]
- Dauch, D.; Rudalska, R.; Cossa, G.; Nault, J.-C.; Kang, T.-W.; Wuestefeld, T.; Hohmeyer, A.; Imbeaud, S.; Yevsa, T.; Hoenicke, L. A MYC–aurora kinase A protein complex represents an actionable drug target in p53-altered liver cancer. Nat. Med. 2016, 22, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Eilers, M.; Eisenman, R.N. Myc’s broad reach. Genes Dev. 2008, 22, 2755–2766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef]
- Richards, M.W.; Burgess, S.G.; Poon, E.; Carstensen, A.; Eilers, M.; Chesler, L.; Bayliss, R. Structural basis of N-Myc binding by Aurora-A and its destabilization by kinase inhibitors. Proc. Natl. Acad. Sci. USA 2016, 113, 13726–13731. [Google Scholar] [CrossRef] [Green Version]
- Otto, T.; Horn, S.; Brockmann, M.; Eilers, U.; Schüttrumpf, L.; Popov, N.; Kenney, A.M.; Schulte, J.H.; Beijersbergen, R.; Christiansen, H. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell 2009, 15, 67–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brockmann, M.; Poon, E.; Berry, T.; Carstensen, A.; Deubzer, H.E.; Rycak, L.; Jamin, Y.; Thway, K.; Robinson, S.P.; Roels, F. Small molecule inhibitors of aurora-a induce proteasomal degradation of N-myc in childhood neuroblastoma. Cancer Cell 2013, 24, 75–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faisal, A.; Vaughan, L.; Bavetsias, V.; Sun, C.; Atrash, B.; Avery, S.; Jamin, Y.; Robinson, S.P.; Workman, P.; Blagg, J. The aurora kinase inhibitor CCT137690 downregulates MYCN and sensitizes MYCN-amplified neuroblastoma in vivo. Mol. Cancer Ther. 2011, 10, 2115–2123. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, W.C.; Meyerowitz, J.G.; Nekritz, E.A.; Chen, J.; Benes, C.; Charron, E.; Simonds, E.F.; Seeger, R.; Matthay, K.K.; Hertz, N.T. Drugging MYCN through an allosteric transition in Aurora kinase A. Cancer Cell 2014, 26, 414–427. [Google Scholar] [CrossRef] [Green Version]
- Bellany, F.; Tsuchiya, Y.; Tran, T.M.; Chan, A.E.; Allan, H.; Gout, I.; Tabor, A.B. Design and synthesis of Coenzyme A analogues as Aurora kinase A inhibitors: An exploration of the roles of the pyrophosphate and pantetheine moieties. Bioorganic Med. Chem. 2020, 28, 115740. [Google Scholar] [CrossRef]
- Dupré-Crochet, S.; Erard, M.; Nü, O. ROS production in phagocytes: Why, when, and where? J. Leukoc. Biol. 2013, 94, 657–670. [Google Scholar] [CrossRef]
- Lambert, A.J.; Brand, M.D. Reactive oxygen species production by mitochondria. Mitochondrial DNA 2009, 2009, 165–181. [Google Scholar]
- Del Río, L.A.; López-Huertas, E. ROS generation in peroxisomes and its role in cell signaling. Plant Cell Physiol. 2016, 57, 1364–1376. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Mittler, R.; Vanderauwera, S.; Suzuki, N.; Miller, G.; Tognetti, V.B.; Vandepoele, K.; Gollery, M.; Shulaev, V.; Van Breusegem, F. ROS signaling: The new wave? Trends Plant Sci. 2011, 16, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-mediated cellular signaling. Oxidative Med. Cell. Longev. 2016, 2016, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Veiga Moreira, J.; Peres, S.; Steyaert, J.-M.; Bigan, E.; Paulevé, L.; Nogueira, M.L.; Schwartz, L. Cell cycle progression is regulated by intertwined redox oscillators. Theor. Biol. Med. Model. 2015, 12, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, B.C.; Choi, Y.-D.; Oh, I.-J.; Kim, J.H.; Park, J.-I.; Lee, S.-w. GPx3-mediated redox signaling arrests the cell cycle and acts as a tumor suppressor in lung cancer cell lines. PLoS ONE 2018, 13, e0204170. [Google Scholar] [CrossRef] [Green Version]
- Mailand, N.; Podtelejnikov, A.V.; Groth, A.; Mann, M.; Bartek, J.; Lukas, J. Regulation of G2/M events by Cdc25A through phosphorylation-dependent modulation of its stability. EMBO J. 2002, 21, 5911–5920. [Google Scholar] [CrossRef]
- Rudolph, J. Redox regulation of the Cdc25 phosphatases. Antioxid. Redox Signal. 2005, 7, 761–767. [Google Scholar] [CrossRef]
- Nilsson, I.; Hoffmann, I. Cell cycle regulation by the Cdc25 phosphatase family. Prog. Cell Cycle Res. 2000, 4, 107–114. [Google Scholar]
- Boutros, R.; Dozier, C.; Ducommun, B. The when and wheres of CDC25 phosphatases. Curr. Opin. Cell Biol. 2006, 18, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Savitsky, P.A.; Finkel, T. Redox regulation of Cdc25C. J. Biol. Chem. 2002, 277, 20535–20540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virshup, D.M. Protein phosphatase 2A: A panoply of enzymes. Curr. Opin. Cell Biol. 2000, 12, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Hunt, T. On the regulation of protein phosphatase 2A and its role in controlling entry into and exit from mitosis. Adv. Biol. Regul. 2013, 53, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Walter, A.O.; Seghezzi, W.; Korver, W.; Sheung, J.; Lees, E. The mitotic serine/threonine kinase Aurora2/AIK is regulated by phosphorylation and degradation. Oncogene 2000, 19, 4906–4916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forester, C.M.; Maddox, J.; Louis, J.V.; Goris, J.; Virshup, D.M. Control of mitotic exit by PP2A regulation of Cdc25C and Cdk1. Proc. Natl. Acad. Sci. USA 2007, 104, 19867–19872. [Google Scholar] [CrossRef] [Green Version]
- Margolis, S.S.; Perry, J.A.; Forester, C.M.; Nutt, L.K.; Guo, Y.; Jardim, M.J.; Thomenius, M.J.; Freel, C.D.; Darbandi, R.; Ahn, J.-H. Role for the PP2A/B56δ phosphatase in regulating 14-3-3 release from Cdc25 to control mitosis. Cell 2006, 127, 759–773. [Google Scholar] [CrossRef] [Green Version]
- Low, I.C.C.; Loh, T.; Huang, Y.; Virshup, D.M.; Pervaiz, S. Ser70 phosphorylation of Bcl-2 by selective tyrosine nitration of PP2A-B56δ stabilizes its antiapoptotic activity. Blood 2014, 124, 2223–2234. [Google Scholar] [CrossRef]
- Chen, L.; Liu, L.; Yin, J.; Luo, Y.; Huang, S. Hydrogen peroxide-induced neuronal apoptosis is associated with inhibition of protein phosphatase 2A and 5, leading to activation of MAPK pathway. Int. J. Biochem. Cell Biol. 2009, 41, 1284–1295. [Google Scholar] [CrossRef]
- Gu, Y.; Barzegar, M.; Chen, X.; Wu, Y.; Shang, C.; Mahdavian, E.; Salvatore, B.A.; Jiang, S.; Huang, S. Fusarochromanone-induced reactive oxygen species results in activation of JNK cascade and cell death by inhibiting protein phosphatases 2A and 5. Oncotarget 2015, 6, 42322. [Google Scholar] [CrossRef] [Green Version]
- Foley, T.D.; Petro, L.A.; Stredny, C.M.; Coppa, T.M. Oxidative inhibition of protein phosphatase 2A activity: Role of catalytic subunit disulfides. Neurochem. Res. 2007, 32, 1957–1964. [Google Scholar] [CrossRef] [PubMed]
- Sommer, D.; Coleman, S.; Swanson, S.A.; Stemmer, P.M. Differential susceptibilities of serine/threonine phosphatases to oxidative and nitrosative stress. Arch. Biochem. Biophys. 2002, 404, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Janssens, V.; Longin, S.; Goris, J. PP2A holoenzyme assembly: In cauda venenum (the sting is in the tail). Trends Biochem. Sci. 2008, 33, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.B.; Nelson, K.J. Discovering mechanisms of signaling-mediated cysteine oxidation. Curr. Opin. Chem. Biol. 2008, 12, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Reddie, K.G.; Carroll, K.S. Expanding the functional diversity of proteins through cysteine oxidation. Curr. Opin. Chem. Biol. 2008, 12, 746–754. [Google Scholar] [CrossRef]
- Bak, D.W.; Bechtel, T.J.; Falco, J.A.; Weerapana, E. Cysteine reactivity across the subcellular universe. Curr. Opin. Chem. Biol. 2019, 48, 96–105. [Google Scholar] [CrossRef]
- Netto, L.E.S.; de Oliveira, M.A.; Monteiro, G.; Demasi, A.P.D.; Cussiol, J.R.R.; Discola, K.F.; Demasi, M.; Silva, G.M.; Alves, S.V.; Faria, V.G. Reactive cysteine in proteins: Protein folding, antioxidant defense, redox signaling and more. Comp. Biochem. Physiol. Part C: Toxicol. Pharmacol. 2007, 146, 180–193. [Google Scholar] [CrossRef]
- Kallis, G.-B.; Holmgren, A. Differential reactivity of the functional sulfhydryl groups of cysteine-32 and cysteine-35 present in the reduced form of thioredoxin from Escherichia coli. J. Biol. Chem. 1980, 255, 10261–10265. [Google Scholar] [CrossRef]
- Dyson, H.J.; Tennant, L.L.; Holmgren, A. Proton-transfer effects in the active-site region of Escherichia coli thioredoxin using two-dimensional proton NMR. Biochemistry 1991, 30, 4262–4268. [Google Scholar] [CrossRef]
- Nelson, K.J.; Day, A.E.; Zeng, B.-B.; King, S.B.; Poole, L.B. Isotope-coded, iodoacetamide-based reagent to determine individual cysteine pKa values by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Anal. Biochem. 2008, 375, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Sundaramoorthy, E.; Maiti, S.; Brahmachari, S.K.; Sengupta, S. Predicting protein homocysteinylation targets based on dihedral strain energy and pKa of cysteines. Proteins: Struct. Funct. Bioinform. 2008, 71, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Madzelan, P.; Labunska, T.; Wilson, M.A. Influence of peptide dipoles and hydrogen bonds on reactive cysteine pK a values in fission yeast DJ-1. FEBS J. 2012, 279, 4111–4120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino, S.M.; Gladyshev, V.N. Analysis and functional prediction of reactive cysteine residues. J. Biol. Chem. 2012, 287, 4419–4425. [Google Scholar] [CrossRef] [Green Version]
- Soylu, İ.; Marino, S.M. Cy-preds: An algorithm and a web service for the analysis and prediction of cysteine reactivity. Proteins Struct. Funct. Bioinform. 2016, 84, 278–291. [Google Scholar] [CrossRef] [PubMed]
- Hirota, T.; Kunitoku, N.; Sasayama, T.; Marumoto, T.; Zhang, D.; Nitta, M.; Hatakeyama, K.; Saya, H. Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell 2003, 114, 585–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, M.; Ni, J.; Wu, J.; Wang, B.; Shen, S.; Yu, L. A novel mechanism for activation of Aurora-A kinase by Ajuba. Gene 2014, 543, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Cheetham, G.M.; Knegtel, R.M.; Coll, J.T.; Renwick, S.B.; Swenson, L.; Weber, P.; Lippke, J.A.; Austen, D.A. Crystal structure of aurora-2, an oncogenic serine/threonine kinase. J. Biol. Chem. 2002, 277, 42419–42422. [Google Scholar] [CrossRef] [Green Version]
- Nolen, B.; Taylor, S.; Ghosh, G. Regulation of protein kinases: Controlling activity through activation segment conformation. Mol. Cell 2004, 15, 661–675. [Google Scholar] [CrossRef]
- Levinson, N.M. The multifaceted allosteric regulation of Aurora kinase A. Biochem. J. 2018, 475, 2025–2042. [Google Scholar] [CrossRef] [Green Version]
- Zorba, A.; Buosi, V.; Kutter, S.; Kern, N.; Pontiggia, F.; Cho, Y.-J.; Kern, D. Molecular mechanism of Aurora A kinase autophosphorylation and its allosteric activation by TPX2. eLife 2014, 3, e02667. [Google Scholar] [CrossRef]
- Eyers, P.A.; Erikson, E.; Chen, L.G.; Maller, J.L. A novel mechanism for activation of the protein kinase Aurora A. Curr. Biol. 2003, 13, 691–697. [Google Scholar] [CrossRef]
- Ruff, E.F.; Muretta, J.M.; Thompson, A.R.; Lake, E.W.; Cyphers, S.; Albanese, S.K.; Hanson, S.M.; Behr, J.M.; Thomas, D.D.; Chodera, J.D. A dynamic mechanism for allosteric activation of Aurora kinase A by activation loop phosphorylation. eLife 2018, 7, e32766. [Google Scholar] [CrossRef]
- Cyphers, S.; Ruff, E.F.; Behr, J.M.; Chodera, J.D.; Levinson, N.M. A water-mediated allosteric network governs activation of Aurora kinase A. Nat. Chem. Biol. 2017, 13, 402–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolin, G.; Sizaire, F.; Herbomel, G.; Reboutier, D.; Prigent, C.; Tramier, M. A FRET biosensor reveals spatiotemporal activation and functions of aurora kinase A in living cells. Nat. Commun. 2016, 7, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lake, E.W.; Muretta, J.M.; Thompson, A.R.; Rasmussen, D.M.; Majumdar, A.; Faber, E.B.; Ruff, E.F.; Thomas, D.D.; Levinson, N.M. Quantitative conformational profiling of kinase inhibitors reveals origins of selectivity for Aurora kinase activation states. Proc. Natl. Acad. Sci. USA 2018, 115, E11894–E11903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodson, C.A.; Bayliss, R. Activation of Aurora-A kinase by protein partner binding and phosphorylation are independent and synergistic. J. Biol. Chem. 2012, 287, 1150–1157. [Google Scholar] [CrossRef] [Green Version]
- Hammond, D.; Zeng, K.; Espert, A.; Bastos, R.N.; Baron, R.D.; Gruneberg, U.; Barr, F.A. Melanoma-associated mutations in protein phosphatase 6 cause chromosome instability and DNA damage owing to dysregulated Aurora-A. J. Cell Sci. 2013, 126, 3429–3440. [Google Scholar]
- Toya, M.; Terasawa, M.; Nagata, K.; Iida, Y.; Sugimoto, A. A kinase-independent role for Aurora A in the assembly of mitotic spindle microtubules in Caenorhabditis elegans embryos. Nat. Cell Biol. 2011, 13, 708–714. [Google Scholar] [CrossRef]
- Dutertre, S.; Cazales, M.; Quaranta, M.; Froment, C.; Trabut, V.; Dozier, C.; Mirey, G.; Bouché, J.-P.; Theis-Febvre, N.; Schmitt, E. Phosphorylation of CDC25B by Aurora-A at the centrosome contributes to the G2–M transition. J. Cell Sci. 2004, 117, 2523–2531. [Google Scholar] [CrossRef] [Green Version]
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol. 2007, 8, 722–728. [Google Scholar] [CrossRef]
- Veal, E.; Day, A. Hydrogen peroxide as a signaling molecule. Antioxid. Redox Signal. 2011, 15, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, N.; Chisci, E.; Giovannoni, R. The role of hydrogen peroxide in redox-dependent signaling: Homeostatic and pathological responses in mammalian cells. Cells 2018, 7, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.M.; Lee, K.S.; Woo, H.A.; Kang, D.; Rhee, S.G. Control of the pericentrosomal H2O2 level by peroxiredoxin I is critical for mitotic progression. J. Cell Biol. 2015, 210, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Rhee, S.G. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Lee, S.-R.; Yang, K.-S.; Ahn, Y.; Kim, Y.J.; Stadtman, E.R.; Rhee, S.G. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc. Natl. Acad. Sci. USA 2004, 101, 16419–16424. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, N.N.; Sorescu, D.; Seshiah, P.; Ushio-Fukai, M.; Akers, M.; Yin, Q.; Griendling, K.K. Mechanism of hydrogen peroxide-induced cell cycle arrest in vascular smooth muscle. Antioxid. Redox Signal. 2002, 4, 845–854. [Google Scholar] [CrossRef]
- Havens, C.G.; Ho, A.; Yoshioka, N.; Dowdy, S.F. Regulation of late G1/S phase transition and APCCdh1 by reactive oxygen species. Mol. Cell. Biol. 2006, 26, 4701–4711. [Google Scholar] [CrossRef] [Green Version]
- Goswami, P.C.; Sheren, J.; Albee, L.D.; Parsian, A.; Sim, J.E.; Ridnour, L.A.; Higashikubo, R.; Gius, D.; Hunt, C.R.; Spitz, D.R. Cell cycle-coupled variation in topoisomerase IIα mRNA is regulated by the 3′-untranslated region: Possible role of redox-sensitive protein binding in mRNA accumulation. J. Biol. Chem. 2000, 275, 38384–38392. [Google Scholar] [CrossRef] [Green Version]
- Yamaura, M.; Mitsushita, J.; Furuta, S.; Kiniwa, Y.; Ashida, A.; Goto, Y.; Shang, W.H.; Kubodera, M.; Kato, M.; Takata, M. NADPH oxidase 4 contributes to transformation phenotype of melanoma cells by regulating G2-M cell cycle progression. Cancer Res. 2009, 69, 2647–2654. [Google Scholar] [CrossRef] [Green Version]
- Hochegger, H.; Hégarat, N.; Pereira-Leal, J. Aurora at the pole and equator: Overlapping functions of Aurora kinases in the mitotic spindle. Open Biol. 2013, 3, 120185. [Google Scholar] [CrossRef] [Green Version]
- Marumoto, T.; Zhang, D.; Saya, H. Aurora-A—A guardian of poles. Nat. Rev. Cancer 2005, 5, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Gout, I. Coenzyme A, protein CoAlation and redox regulation in mammalian cells. Biochem. Soc. Trans. 2018, 46, 721–728. [Google Scholar] [CrossRef] [Green Version]
- Leonardi, R.; Zhang, Y.-M.; Rock, C.O.; Jackowski, S. Coenzyme A: Back in action. Prog. Lipid Res. 2005, 44, 125–153. [Google Scholar] [CrossRef]
- Baković, J.; Yu, B.Y.K.; Silva, D.; Chew, S.P.; Kim, S.; Ahn, S.-H.; Palmer, L.; Aloum, L.; Stanzani, G.; Malanchuk, O. A key metabolic integrator, coenzyme A, modulates the activity of peroxiredoxin 5 via covalent modification. Mol. Cell. Biochem. 2019, 461, 91–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchiya, Y.; Zhyvoloup, A.; Baković, J.; Thomas, N.; Yu, B.Y.K.; Das, S.; Orengo, C.; Newell, C.; Ward, J.; Saladino, G. Protein CoAlation and antioxidant function of coenzyme A in prokaryotic cells. Biochem. J. 2018, 475, 1909–1937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy, F.; Darbandi, R.; Lee, H.C.; Bharatham, K.; Moldoveanu, T.; Grace, C.R.; Dodd, K.; Lin, W.; Chen, S.-I.; Tangallapally, R.P. Metabolic activation of CaMKII by coenzyme A. Mol. Cell 2013, 52, 325–339. [Google Scholar] [CrossRef] [Green Version]
- Ford, D.A.; Horner, C.C.; Gross, R.W. Protein kinase C acylation by palmitoyl coenzyme A facilitates its translocation to membranes. Biochemistry 1998, 37, 11953–11961. [Google Scholar] [CrossRef]
- Guimarães, C.R.; Rai, B.K.; Munchhof, M.J.; Liu, S.; Wang, J.; Bhattacharya, S.K.; Buckbinder, L. Understanding the impact of the P-loop conformation on kinase selectivity. J. Chem. Inf. Model. 2011, 51, 1199–1204. [Google Scholar] [CrossRef]
- La Sala, G.; Riccardi, L.; Gaspari, R.; Cavalli, A.; Hantschel, O.; De Vivo, M. HRD motif as the central hub of the signaling network for activation loop autophosphorylation in Abl kinase. J. Chem. Theory Comput. 2016, 12, 5563–5574. [Google Scholar] [CrossRef] [Green Version]
- Abdeldayem, A.; Raouf, Y.S.; Constantinescu, S.N.; Moriggl, R.; Gunning, P.T. Advances in covalent kinase inhibitors. Chem. Soc. Rev. 2020, 49, 2617–2687. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, I.-G.; Lee, B.-J. Aurora Kinase A Regulation by Cysteine Oxidative Modification. Antioxidants 2023, 12, 531. https://doi.org/10.3390/antiox12020531
Lee I-G, Lee B-J. Aurora Kinase A Regulation by Cysteine Oxidative Modification. Antioxidants. 2023; 12(2):531. https://doi.org/10.3390/antiox12020531
Chicago/Turabian StyleLee, In-Gyun, and Bong-Jin Lee. 2023. "Aurora Kinase A Regulation by Cysteine Oxidative Modification" Antioxidants 12, no. 2: 531. https://doi.org/10.3390/antiox12020531