
CD38-Induced Metabolic Dysfunction Primes Multiple Myeloma Cells for NAD+-Lowering Agents
 , , , , , add
Show full author list
, , , , , add
Show full author list
Abstract
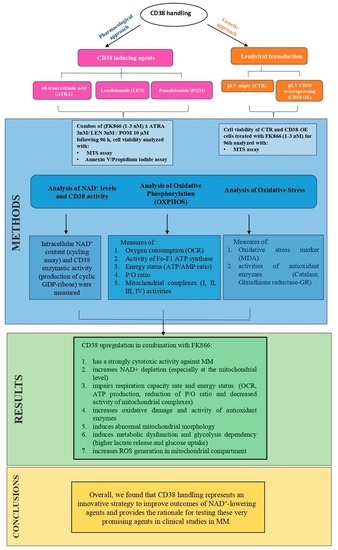
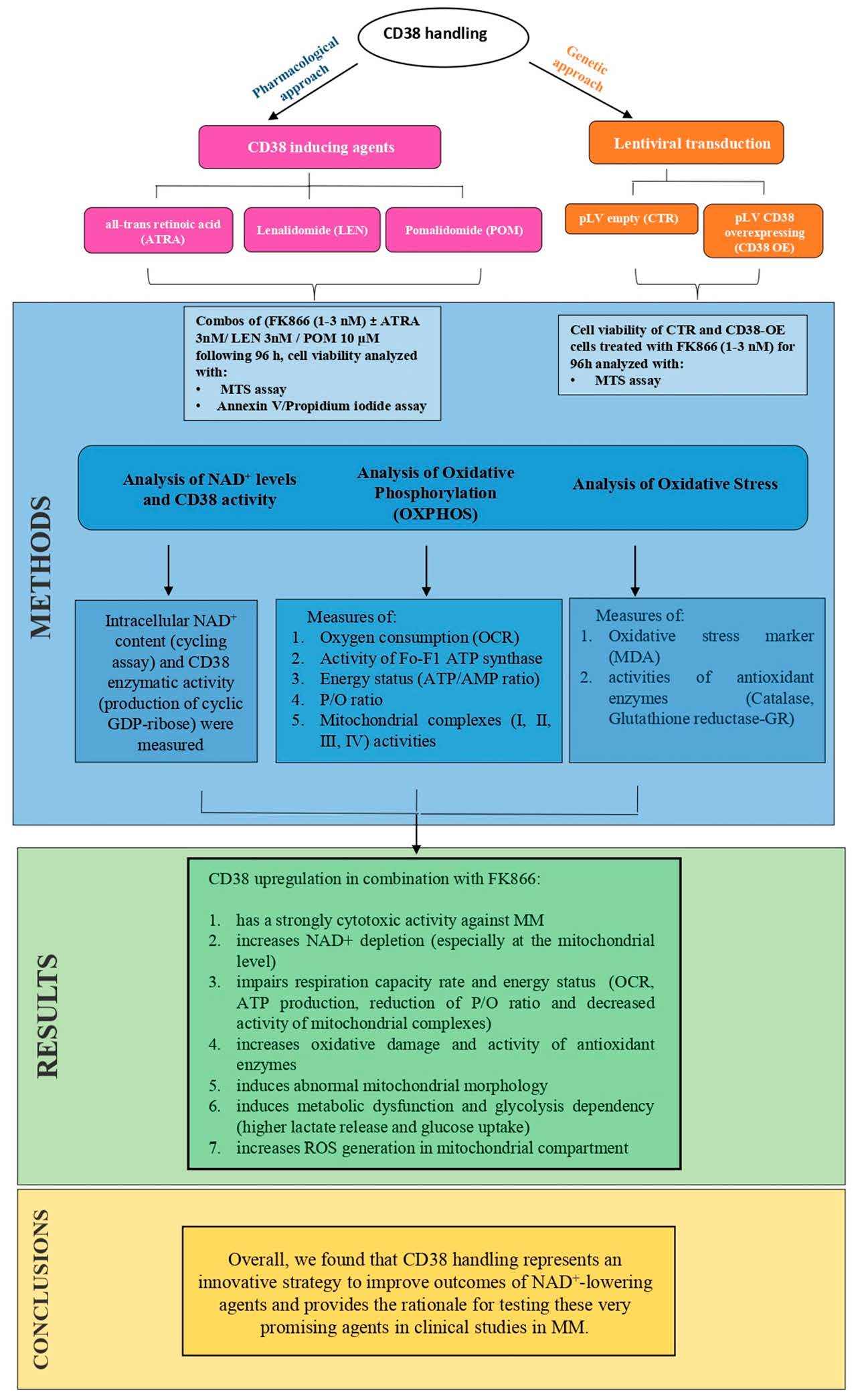
:
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Lines and Culture
2.3. Primary Cells
2.4. Lentiviral Mediated Gene Transfer
2.5. Measurement of CD38 Enzymatic Activity
2.6. Determination of Intracellular NAD+
2.7. Mitochondria Fractionation
2.8. Western Blotting
2.9. Statistical Analyses
3. Results
3.1. CD38 Enzymatic Activity Affects NAD+ Intracellular Level and Influences Anti-MM Activity of NAD+-Depleting Agents
3.2. Synergistic Effects of NAMPT Inhibitor Combined with CD38 Inducing Agents
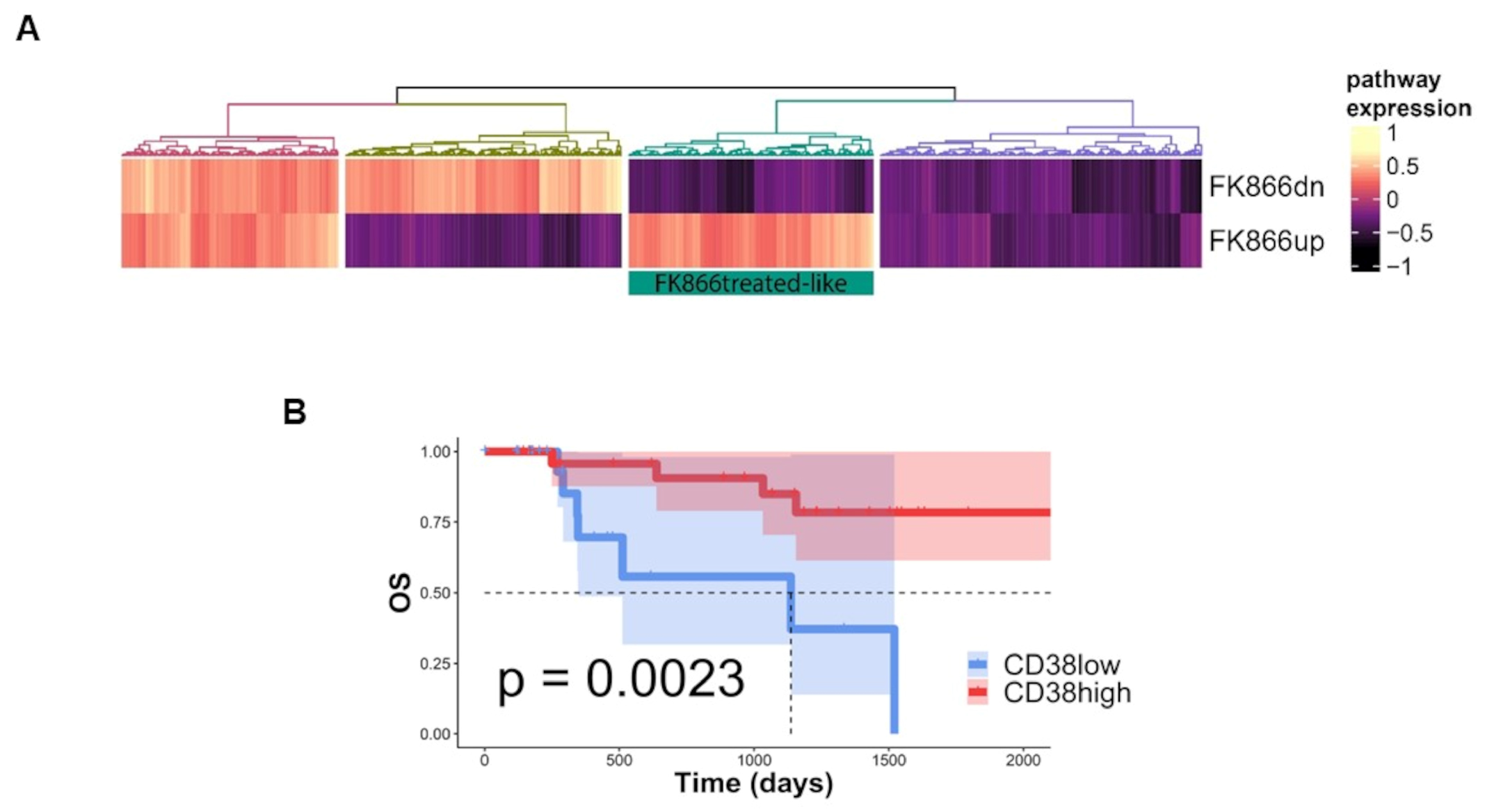
3.3. FK866-Induced Transcriptomic Change among CD38-Overexpressing MM Patients Confers Better Prognosis
3.4. NAD+ Depletion Accounts for the Enhanced Sensibility of CD38-Upregulated MM Cells to Nampt-Inhibitors
3.5. NAD+ and Nicotinic Acid Abolish Activity of Co-Treatment in Myeloma Cells
3.6. Metabolic Reprogramming Elicited by CD38 Overexpression Identifies a Novel Druggable Vulnerability in MM Cells
3.7. Mitochondrial Dynamic Shift Underlies an Organelle-Specific Dysfunction Triggered by CD38 Upregulation
3.8. The Oxidative Stress Triggered by Energetic Depletion Is Crucial for Tested Drug Combinations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kyle, R.A.; Rajkumar, S.V. Multiple Myeloma. N. Engl. J. Med. 2004, 351, 1860–1873. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Anderson, K. Multiple Myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikhael, J.; Ismaila, N.; Cheung, M.C.; Costello, C.; Dhodapkar, M.V.; Kumar, S.; Lacy, M.; Lipe, B.; Little, R.F.; Nikonova, A.; et al. Treatment of Multiple Myeloma: ASCO and CCO Joint Clinical Practice Guideline. J. Clin. Oncol. 2019, 37, 1228–1263. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaku, K.; Okabe, K.; Hikosaka, K.; Nakagawa, T. NAD Metabolism in Cancer Therapeutics. Front. Oncol. 2018, 8, 622. [Google Scholar] [CrossRef] [Green Version]
- Navas, L.E.; Carnero, A. NAD+ metabolism, stemness, the immune response, and cancer. Signal Transduct. Target. Ther. 2021, 6, 2. [Google Scholar] [CrossRef]
- Galli, U.; Travelli, C.; Massarotti, A.; Fakhfouri, G.; Rahimian, R.; Tron, G.C.; Genazzani, A.A. Medicinal Chemistry of Nicotinamide Phosphoribosyltransferase (NAMPT) Inhibitors. J. Med. Chem. 2013, 56, 6279–6296. [Google Scholar] [CrossRef]
- Montecucco, F.; Cea, M.; Cagnetta, A.; Damonte, P.; Nahimana, A.; Ballestrero, A.; Rio, A.; Bruzzone, S.; Nencioni, A. Nicotinamide Phosphoribosyltransferase as a Target in Inflammation- Related Disorders. Curr. Top. Med. Chem. 2013, 13, 2930–2938. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, R.E.; Mayhall, K.; Maxwell, N.M.; Kandil, E.; Coppola, D. Nicotinamide Phosphoribosyltransferase in Malignancy: A Review. Genes Cancer 2013, 4, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Cea, M.; Cagnetta, A.; Fulciniti, M.; Tai, Y.T.; Hideshima, T.; Chauhan, D.; Roccaro, A.; Sacco, A.; Calimeri, T.; Cottini, F.; et al. Targeting NAD+ salvage pathway induces autophagy in multiple myeloma cells via mTORC1 and extracellular signal-regulated kinase (ERK1/2) inhibition. Blood 2012, 120, 3519–3529. [Google Scholar] [CrossRef] [Green Version]
- Cagnetta, A.; Cea, M.; Calimeri, T.; Acharya, C.; Fulciniti, M.; Tai, Y.-T.; Hideshima, T.; Chauhan, D.; Zhong, M.Y.; Patrone, F.; et al. Intracellular NAD+ depletion enhances bortezomib-induced anti-myeloma activity. Blood 2013, 122, 1243–1255. [Google Scholar] [CrossRef] [Green Version]
- Cagnetta, A.; Caffa, I.; Acharya, C.; Soncini, D.; Acharya, P.; Adamia, S.; Pierri, I.; Bergamaschi, M.; Garuti, A.; Fraternali, G.; et al. APO866 Increases Antitumor Activity of Cyclosporin-A by Inducing Mitochondrial and Endoplasmic Reticulum Stress in Leukemia Cells. Clin. Cancer Res. 2015, 21, 3934–3945. [Google Scholar] [CrossRef] [Green Version]
- Cea, M.; Cagnetta, A.; Acharya, C.; Acharya, P.; Tai, Y.-T.; Yang, C.; Lovera, D.; Soncini, D.; Miglino, M.; Fraternali-Orcioni, G.; et al. Dual NAMPT and BTK Targeting Leads to Synergistic Killing of Waldenström Macroglobulinemia Cells Regardless of MYD88 and CXCR4 Somatic Mutation Status. Clin. Cancer Res. 2016, 22, 6099–6109. [Google Scholar] [CrossRef] [Green Version]
- Dimopoulos, M.A.; Oriol, A.; Nahi, H.; San-Miguel, J.; Bahlis, N.J.; Usmani, S.Z.; Rabin, N.; Orlowski, R.Z.; Komarnicki, M.; Suzuki, K.; et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 1319–1331. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V.; et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Facon, T.; Kumar, S.; Plesner, T.; Orlowski, R.Z.; Moreau, P.; Bahlis, N.; Basu, S.; Nahi, H.; Hulin, C.; Quach, H.; et al. Daratumumab plus Lenalidomide and Dexamethasone for Untreated Myeloma. N. Engl. J. Med. 2019, 380, 2104–2115. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Terpos, E.; Boccadoro, M.; Delimpasi, S.; Beksac, M.; Katodritou, E.; Moreau, P.; Baldini, L.; Symeonidis, A.; Bila, J.; et al. Daratumumab plus pomalidomide and dexamethasone versus pomalidomide and dexamethasone alone in previously treated multiple myeloma (APOLLO): An open-label, randomised, phase 3 trial. Lancet Oncol. 2021, 22, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.-V.; Cavo, M.; Blade, J.; Dimopoulos, M.A.; Suzuki, K.; Jakubowiak, A.; Knop, S.; Doyen, C.; Lucio, P.; Nagy, Z.; et al. Overall survival with daratumumab, bortezomib, melphalan, and prednisone in newly diagnosed multiple myeloma (ALCYONE): A randomised, open-label, phase 3 trial. Lancet 2020, 395, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Hulin, C.; Perrot, A.; Arnulf, B.; Belhadj, K.; Benboubker, L.; Béné, M.C.; Zweegman, S.; Caillon, H.; Caillot, D.; et al. Maintenance with daratumumab or observation following treatment with bortezomib, thalidomide, and dexamethasone with or without daratumumab and autologous stem-cell transplant in patients with newly diagnosed multiple myeloma (CASSIOPEIA): An open-label. Lancet Oncol. 2021, 22, 1378–1390. [Google Scholar] [CrossRef] [PubMed]
- Bruzzone, S.; Moreschi, I.; Usai, C.; Guida, L.; Damonte, G.; Salis, A.; Scarfì, S.; Millo, E.; De Flora, A.; Zocchi, E. Abscisic acid is an endogenous cytokine in human granulocytes with cyclic ADP-ribose as second messenger. Proc. Natl. Acad. Sci. USA 2007, 104, 5759–5764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruzzone, S.; Fruscione, F.; Morando, S.; Ferrando, T.; Poggi, A.; Garuti, A.; D’Urso, A.; Selmo, M.; Benvenuto, F.; Cea, M.; et al. Catastrophic NAD+ Depletion in Activated T Lymphocytes through Nampt Inhibition Reduces Demyelination and Disability in EAE. PLoS ONE 2009, 4, e7897. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, R.; Andreyev, A.; Murphy, A.N.; Perkins, G.A.; Ellisman, M.H.; Newmeyer, D.D. Mitochondria frozen with trehalose retain a number of biological functions and preserve outer membrane integrity. Cell Death Differ. 2007, 14, 616–624. [Google Scholar] [CrossRef] [Green Version]
- Cagnetta, A.; Soncini, D.; Orecchioni, S.; Talarico, G.; Minetto, P.; Guolo, F.; Retali, V.; Colombo, N.; Carminati, E.; Clavio, M.; et al. Depletion of SIRT6 enzymatic activity increases acute myeloid leukemia cells’ vulnerability to DNA-damaging agents. Haematologica 2018, 103, 80–90. [Google Scholar] [CrossRef] [Green Version]
- Moreau, P.; Attal, M.; Hulin, C.; Arnulf, B.; Belhadj, K.; Benboubker, L.; Béné, M.C.; Broijl, A.; Caillon, H.; Caillot, D.; et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): A randomised, open-label, phase 3 study. Lancet 2019, 394, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.; Quach, H.; Mateos, M.-V.; Landgren, O.; Leleu, X.; Siegel, D.; Weisel, K.; Yang, H.; Klippel, Z.; Zahlten-Kumeli, A.; et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): Results from a randomised, multicentre, open-label, phase 3 study. Lancet 2020, 396, 186–197. [Google Scholar] [CrossRef]
- Moreau, P.; Dimopoulos, M.-A.; Mikhael, J.; Yong, K.; Capra, M.; Facon, T.; Hajek, R.; Špička, I.; Baker, R.; Kim, K.; et al. Isatuximab, carfilzomib, and dexamethasone in relapsed multiple myeloma (IKEMA): A multicentre, open-label, randomised phase 3 trial. Lancet 2021, 397, 2361–2371. [Google Scholar] [CrossRef] [PubMed]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and Function of the ADP Ribosyl Cyclase/CD38 Gene Family in Physiology and Pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nijhof, I.S.; Groen, R.W.J.; Lokhorst, H.M.; van Kessel, B.; Bloem, A.C.; van Velzen, J.; de Jong-Korlaar, R.; Yuan, H.; Noort, W.A.; Klein, S.K.; et al. Upregulation of CD38 expression on multiple myeloma cells by all-trans retinoic acid improves the efficacy of daratumumab. Leukemia 2015, 29, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- García-Guerrero, E.; Gogishvili, T.; Danhof, S.; Schreder, M.; Pallaud, C.; Pérez-Simón, J.A.; Einsele, H.; Hudecek, M. Panobinostat induces CD38 upregulation and augments the antimyeloma efficacy of daratumumab. Blood 2017, 129, 3386–3388. [Google Scholar] [CrossRef] [PubMed]
- Choudhry, P.; Mariano, M.C.; Geng, H.; Martin, T.G.; Wolf, J.L.; Wong, S.W.; Shah, N.; Wiita, A.P. DNA methyltransferase inhibitors upregulate CD38 protein expression and enhance daratumumab efficacy in multiple myeloma. Leukemia 2020, 34, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Wang, S.; Liu, J.; Yu, T.; Chen, H.; Wen, K.; Li, Y.; Lin, L.; Hsieh, P.A.; Cho, S.-F.; et al. BCMA-Specific ADC MEDI2228 and Daratumumab Induce Synergistic Myeloma Cytotoxicity via IFN-Driven Immune Responses and Enhanced CD38 Expression. Clin. Cancer Res. 2021, 27, 5376–5388. [Google Scholar] [CrossRef] [PubMed]
- Fedele, P.L.; Willis, S.N.; Liao, Y.; Low, M.S.; Rautela, J.; Segal, D.H.; Gong, J.-N.; Huntington, N.D.; Shi, W.; Huang, D.C.S.; et al. IMiDs prime myeloma cells for daratumumab-mediated cytotoxicity through loss of Ikaros and Aiolos. Blood 2018, 132, 2166–2178. [Google Scholar] [CrossRef] [Green Version]
- Ogiya, D.; Liu, J.; Ohguchi, H.; Kurata, K.; Samur, M.K.; Tai, Y.-T.; Adamia, S.; Ando, K.; Hideshima, T.; Anderson, K.C. The JAK-STAT pathway regulates CD38 on myeloma cells in the bone marrow microenvironment: Therapeutic implications. Blood 2020, 136, 2334–2345. [Google Scholar] [CrossRef]
- García-Guerrero, E.; Götz, R.; Doose, S.; Sauer, M.; Rodríguez-Gil, A.; Nerreter, T.; Kortüm, K.M.; Pérez-Simón, J.A.; Einsele, H.; Hudecek, M.; et al. Upregulation of CD38 expression on multiple myeloma cells by novel HDAC6 inhibitors is a class effect and augments the efficacy of daratumumab. Leukemia 2021, 35, 201–214. [Google Scholar] [CrossRef]
- Thongon, N.; Zucal, C.; D’Agostino, V.G.; Tebaldi, T.; Ravera, S.; Zamporlini, F.; Piacente, F.; Moschoi, R.; Raffaelli, N.; Quattrone, A.; et al. Cancer cell metabolic plasticity allows resistance to NAMPT inhibition but invariably induces dependence on LDHA. Cancer Metab. 2018, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Chakrabarty, R.P.; Chandel, N.S. Mitochondria as Signaling Organelles Control Mammalian Stem Cell Fate. Cell Stem Cell 2021, 28, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Stein, L.R.; Imai, S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol. Metab. 2012, 23, 420–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucal, C.; D’Agostino, V.G.; Casini, A.; Mantelli, B.; Thongon, N.; Soncini, D.; Caffa, I.; Cea, M.; Ballestrero, A.; Quattrone, A.; et al. EIF2A-dependent translational arrest protects leukemia cells from the energetic stress induced by NAMPT inhibition. BMC Cancer 2015, 15, 855. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Mitochondrial Dynamics in Regulating the Unique Phenotypes of Cancer and Stem Cells. Cell Metab. 2017, 26, 39–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial Fission, Fusion, and Stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; McCaffery, J.M.; Chan, D.C. Mitochondrial Fusion Protects against Neurodegeneration in the Cerebellum. Cell 2007, 130, 548–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Gammella, E.; Recalcati, S.; Cairo, G. Dual Role of ROS as Signal and Stress Agents: Iron Tips the Balance in favor of Toxic Effects. Oxid. Med. Cell. Longev. 2016, 2016, 8629024. [Google Scholar] [CrossRef] [Green Version]
- Ježek, J.; Cooper, K.; Strich, R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants 2018, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Cloux, A.-J.; Aubry, D.; Heulot, M.; Widmann, C.; ElMokh, O.; Piacente, F.; Cea, M.; Nencioni, A.; Bellotti, A.; Bouzourène, K.; et al. Reactive oxygen/nitrogen species contribute substantially to the antileukemia effect of APO866, a NAD lowering agent. Oncotarget 2019, 10, 6723–6738. [Google Scholar] [CrossRef] [Green Version]
- Cea, M.; Cagnetta, A.; Adamia, S.; Acharya, C.; Tai, Y.-T.T.; Fulciniti, M.; Ohguchi, H.; Munshi, A.; Acharya, P.; Bhasin, M.K.; et al. Evidence for a role of the histone deacetylase SIRT6 in DNA damage response of multiple myeloma cells. Blood 2016, 127, 1138–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, L.; Perez, C.; Zabaleta, A.; Manrique, I.; Alignani, D.; Ajona, D.; Blanco, L.; Lasa, M.; Maiso, P.; Rodriguez, I.; et al. The Mechanism of Action of the Anti-CD38 Monoclonal Antibody Isatuximab in Multiple Myeloma. Clin. Cancer Res. 2019, 25, 3176–3187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deckert, J.; Wetzel, M.-C.; Bartle, L.M.; Skaletskaya, A.; Goldmacher, V.S.; Vallée, F.; Zhou-Liu, Q.; Ferrari, P.; Pouzieux, S.; Lahoute, C.; et al. SAR650984, A Novel Humanized CD38-Targeting Antibody, Demonstrates Potent Antitumor Activity in Models of Multiple Myeloma and Other CD38+ Hematologic Malignancies. Clin. Cancer Res. 2014, 20, 4574–4583. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, J.P.; Bowlby, S.C.; Wheeler, F.B.; Shi, L.; Sui, G.; Davis, A.L.; Howard, T.D.; D’Agostino, R.B.; Miller, L.D.; Sirintrapun, S.J.; et al. CD38 Inhibits Prostate Cancer Metabolism and Proliferation by Reducing Cellular NAD+ Pools. Mol. Cancer Res. 2018, 16, 1687–1700. [Google Scholar] [CrossRef] [Green Version]
- Chini, C.C.S.; Guerrico, A.M.G.; Nin, V.; Camacho-Pereira, J.; Escande, C.; Barbosa, M.T.; Chini, E.N. Targeting of NAD Metabolism in Pancreatic Cancer Cells: Potential Novel Therapy for Pancreatic Tumors. Clin. Cancer Res. 2014, 20, 120–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.; Cantley, L.; Thompson, C. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghanem, M.S.; Monacelli, F.; Nencioni, A. Advances in NAD-Lowering Agents for Cancer Treatment. Nutrients 2021, 13, 1665. [Google Scholar] [CrossRef] [PubMed]
- Camacho-Pereira, J.; Tarragó, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.-H.; Lu, M.; Lee, B.-Y.; Ugurbil, K.; Chen, W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc. Natl. Acad. Sci. USA 2015, 112, 2876–2881. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.P.; Price, N.L.; Ling, A.J.Y.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD+ Induces a Pseudohypoxic State Disrupting Nuclear-Mitochondrial Communication during Aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [Green Version]
- Scheibye-Knudsen, M.; Mitchell, S.J.; Fang, E.F.; Iyama, T.; Ward, T.; Wang, J.; Dunn, C.A.; Singh, N.; Veith, S.; Hasan-Olive, M.M.; et al. A High-Fat Diet and NAD + Activate Sirt1 to Rescue Premature Aging in Cockayne Syndrome. Cell Metab. 2014, 20, 840–855. [Google Scholar] [CrossRef] [Green Version]
- Abeliovich, H.; Zarei, M.; Rigbolt, K.T.G.; Youle, R.J.; Dengjel, J. Involvement of mitochondrial dynamics in the segregation of mitochondrial matrix proteins during stationary phase mitophagy. Nat. Commun. 2013, 4, 2789. [Google Scholar] [CrossRef] [Green Version]
- Soncini, D.; Orecchioni, S.; Ruberti, S.; Minetto, P.; Martinuzzi, C.; Agnelli, L.; Todoerti, K.; Cagnetta, A.; Miglino, M.; Clavio, M.; et al. The new small tyrosine kinase inhibitor ARQ531 targets acute myeloid leukemia cells by disrupting multiple tumor-addicted programs. Haematologica 2020, 105, 2420–2431. [Google Scholar] [CrossRef]
- Soncini, D.; Minetto, P.; Martinuzzi, C.; Becherini, P.; Fenu, V.; Guolo, F.; Todoerti, K.; Calice, G.; Contini, P.; Miglino, M.; et al. Amino acid depletion triggered by ʟ-asparaginase sensitizes MM cells to carfilzomib by inducing mitochondria ROS-mediated cell death. Blood Adv. 2020, 4, 4312–4326. [Google Scholar] [CrossRef]
- Marini, C.; Ravera, S.; Buschiazzo, A.; Bianchi, G.; Orengo, A.M.; Bruno, S.; Bottoni, G.; Emionite, L.; Pastorino, F.; Monteverde, E.; et al. Discovery of a novel glucose metabolism in cancer: The role of endoplasmic reticulum beyond glycolysis and pentose phosphate shunt. Sci. Rep. 2016, 6, 2509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravera, S.; Aluigi, M.G.; Calzia, D.; Ramoino, P.; Morelli, A.; Panfoli, I. Evidence for ectopic aerobic ATP production on C6 glioma cell plasma membrane. Cell. Mol. Neurobiol. 2011, 31, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Bartolucci, M.; Ravera, S.; Garbarino, G.; Ramoino, P.; Ferrando, S.; Calzia, D.; Candiani, S.; Morelli, A.; Panfoli, I. Functional Expression of Electron Transport Chain and FoF1-ATP Synthase in Optic Nerve Myelin Sheath. Neurochem. Res. 2015, 40, 2230–2241. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; White, K.; Turnbull, D.M. Mitochondrial morphology, topology, and membrane interactions in skeletal muscle: A quantitative three-dimensional electron microscopy study. J. Appl. Physiol. 2013, 114, 161–171. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients | AGE | SEX | ISOTYPE | ISS | R-ISS | Disease Status | FISH | * Immunophenotypic Analysis on Bone Marrow | BMPCs (%) | Biochemical Parameters | Therapy | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD138 | CD38 | CD56 | CD19 | CD20 | CD45 | Crea (mg/dL) | Hb (mg/dL) | PLT (×103) | WBC (×106) | Beta2 Micro (mg/dL) | LDH (U/L) | Crea (mg/dL) | ||||||||||
| MM#1 | 77 | M | IgGk | II | II | RRMM | SR | + | + | +/− | − | +/− | +/− | 24 | 0.8 | 10.6 | 232 | 2.95 | 3.3 | 198 | DVMP | 0.8 |
| MM#2 | 48 | F | IgGk | I | I | NDMM | t(4;14) | + | + | − | + | − | + | 30 | 0.6 | 13.5 | 318 | 13.42 | 1.3 | 185 | KRD | 0.6 |
| MM#3 | 63 | M | IgAk | III | III | NDMM | SR | + | + | + | − | − | +/− | 18 | 1.4 | 11.5 | 170 | 7.22 | 7 | 180 | KRD | 1.4 |
| MM#4 | 77 | M | IgG-l | II | III | RRMM | t(4;14) | + | + | + | − | − | − | 19 | 1.05 | 9.8 | 338 | 12.5 | 3.78 | 216 | MPV | 1.05 |
| MM#5 | 66 | M | IgGk | ND | III | RRMM | t(11;14) | + | + | + | − | − | + | 45 | 1 | 10 | 348 | 9.69 | 3 | 340 | DVTD | 1 |
| MM#6 | 66 | F | Micromolecular | III | III | NDMM | SR | + | ++ | + | − | − | +/− | 25 | 0.6 | 8.7 | 273 | 4.87 | 2.2 | 178 | DVTD | 0.6 |
| MM#7 | 85 | F | IgGk | II | II | NDMM | del(17p) | ++ | ++ | + | − | − | +/− | 30 | 0.8 | 10.4 | 237 | 5.99 | 3.2 | 182 | MPV | 0.8 |
| GENE | SIGNATURE | GENE | SIGNATURE |
|---|---|---|---|
| FLJ32224 | FK866dn | SLC30A1 | FK866up |
| LOC100128239 | FK866dn | CHD6 | FK866up |
| C4orf47 | FK866dn | KIAA1024L | FK866up |
| PTAFR | FK866dn | PHLDA2 | FK866up |
| CCR5 | FK866dn | FLCN | FK866up |
| LINC00469 | FK866dn | LRRC55 | FK866up |
| PON1 | FK866dn | PTPN14 | FK866up |
| GLB1L3 | FK866dn | VPS13B | FK866up |
| TMEM132E | FK866dn | POLR2M | FK866up |
| NPAS2 | FK866dn | CSF2 | FK866up |
| DIS3L2 | FK866dn | BTBD9 | FK866up |
| MIRLET7BHG | FK866dn | NOM1 | FK866up |
| CFTR | FK866up | LOC647107 | FK866up |
| ZC3HAV1 | FK866up | KLHL11 | FK866up |
| NPHS2 | FK866up | ZC3H12D | FK866up |
| MYLIP | FK866up | RASD1 | FK866up |
| P2RX6 | FK866up | RGS1 | FK866up |
| FAM22G | FK866up | TAGAP | FK866up |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becherini, P.; Soncini, D.; Ravera, S.; Gelli, E.; Martinuzzi, C.; Giorgetti, G.; Cagnetta, A.; Guolo, F.; Ivaldi, F.; Miglino, M.; et al. CD38-Induced Metabolic Dysfunction Primes Multiple Myeloma Cells for NAD+-Lowering Agents. Antioxidants 2023, 12, 494. https://doi.org/10.3390/antiox12020494
Becherini P, Soncini D, Ravera S, Gelli E, Martinuzzi C, Giorgetti G, Cagnetta A, Guolo F, Ivaldi F, Miglino M, et al. CD38-Induced Metabolic Dysfunction Primes Multiple Myeloma Cells for NAD+-Lowering Agents. Antioxidants. 2023; 12(2):494. https://doi.org/10.3390/antiox12020494
Chicago/Turabian StyleBecherini, Pamela, Debora Soncini, Silvia Ravera, Elisa Gelli, Claudia Martinuzzi, Giulia Giorgetti, Antonia Cagnetta, Fabio Guolo, Federico Ivaldi, Maurizio Miglino, and et al. 2023. "CD38-Induced Metabolic Dysfunction Primes Multiple Myeloma Cells for NAD+-Lowering Agents" Antioxidants 12, no. 2: 494. https://doi.org/10.3390/antiox12020494