
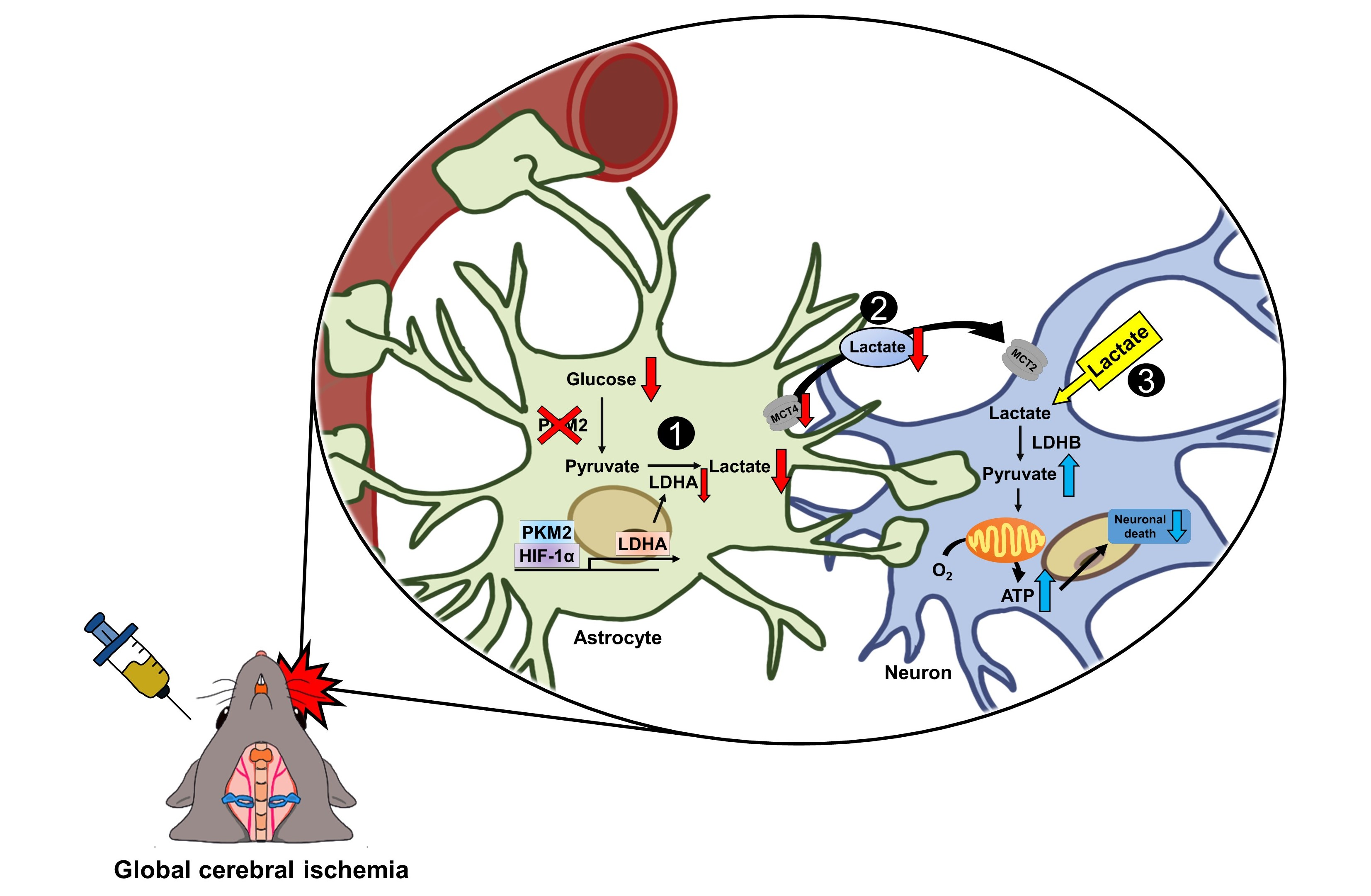
Effects of Pyruvate Kinase M2 (PKM2) Gene Deletion on Astrocyte-Specific Glycolysis and Global Cerebral Ischemia-Induced Neuronal Death
 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animal Care Management and Ethics Statement
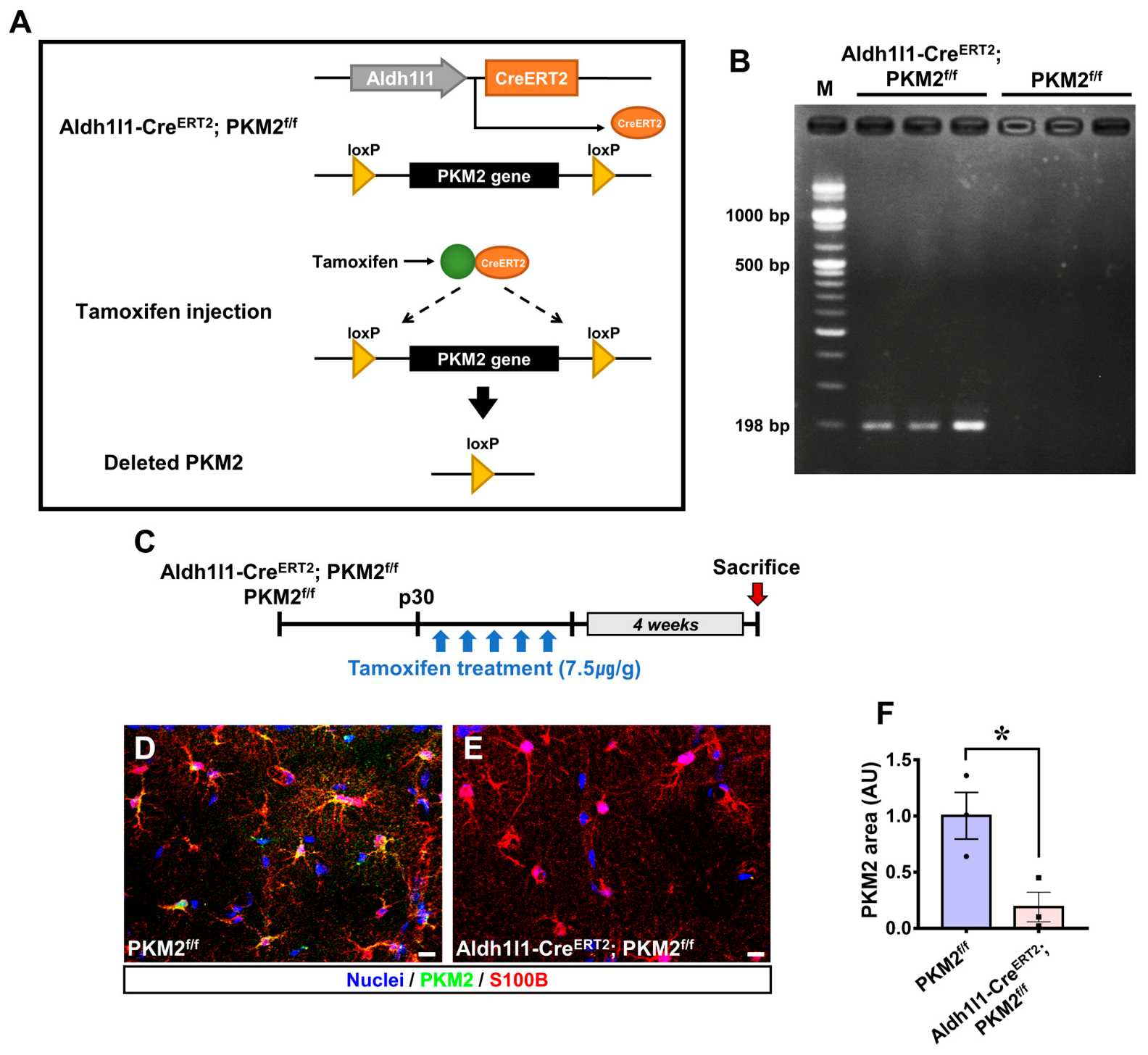
2.2. PCR Genotyping
2.3. Tamoxifen and Lactate Administration
2.4. Mouse Global Cerebral Ischemia
2.5. Preparing Brain Tissue Samples
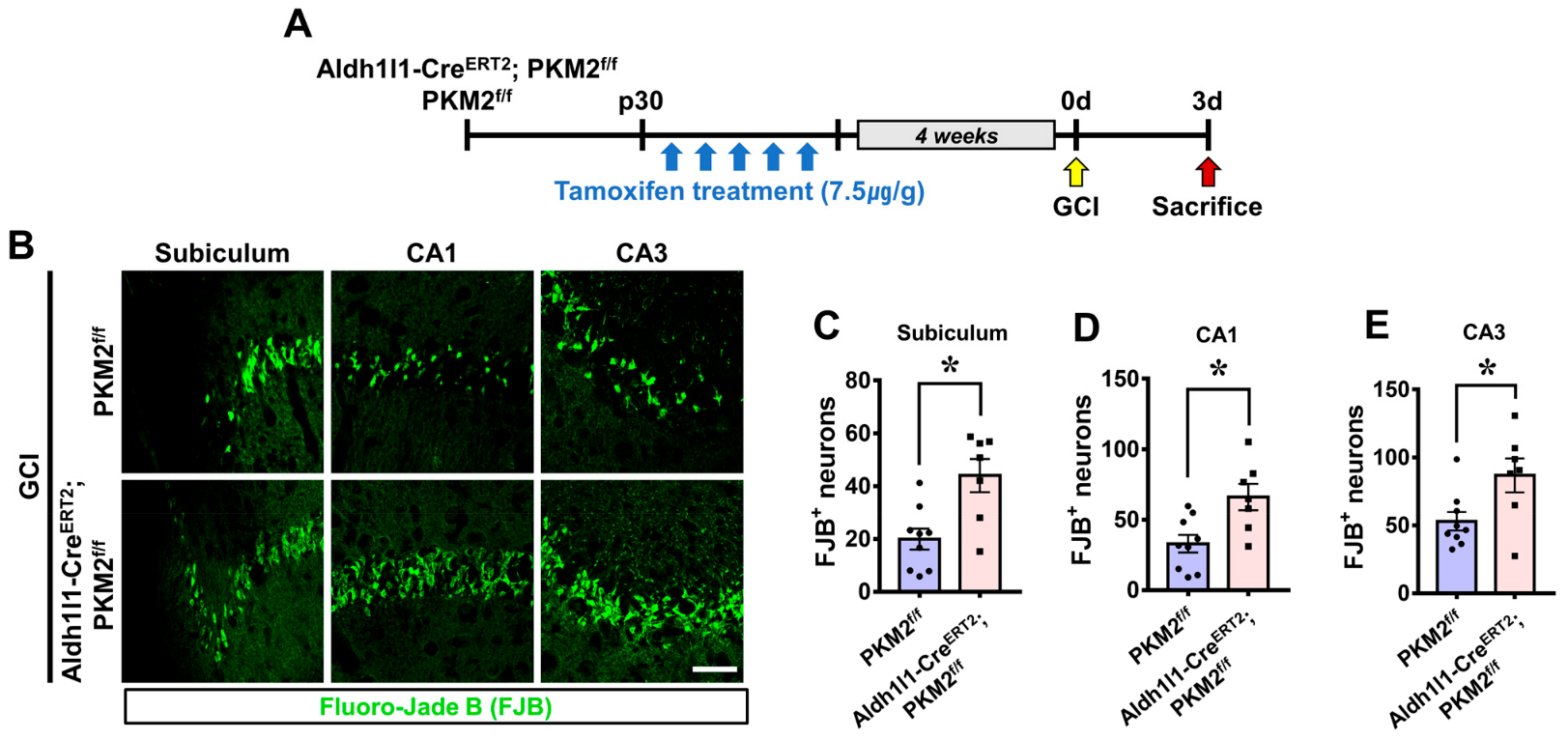
2.6. Confirmation of Neuronal Death
2.7. Immunofluorescence Analysis
2.8. Immunohistochemistry
2.9. Behavioral Test
2.9.1. Neurological Severity Score (NSS) Test
2.9.2. Morris Water Maze Test
2.10. Statistical Analysis
3. Results
3.1. PKM2 Gene Deletion Reduces the Expression of PKM2 in Astrocytes
3.2. PKM2 Gene Deletion Exacerbates Hippocampal Neuron Death after GCI
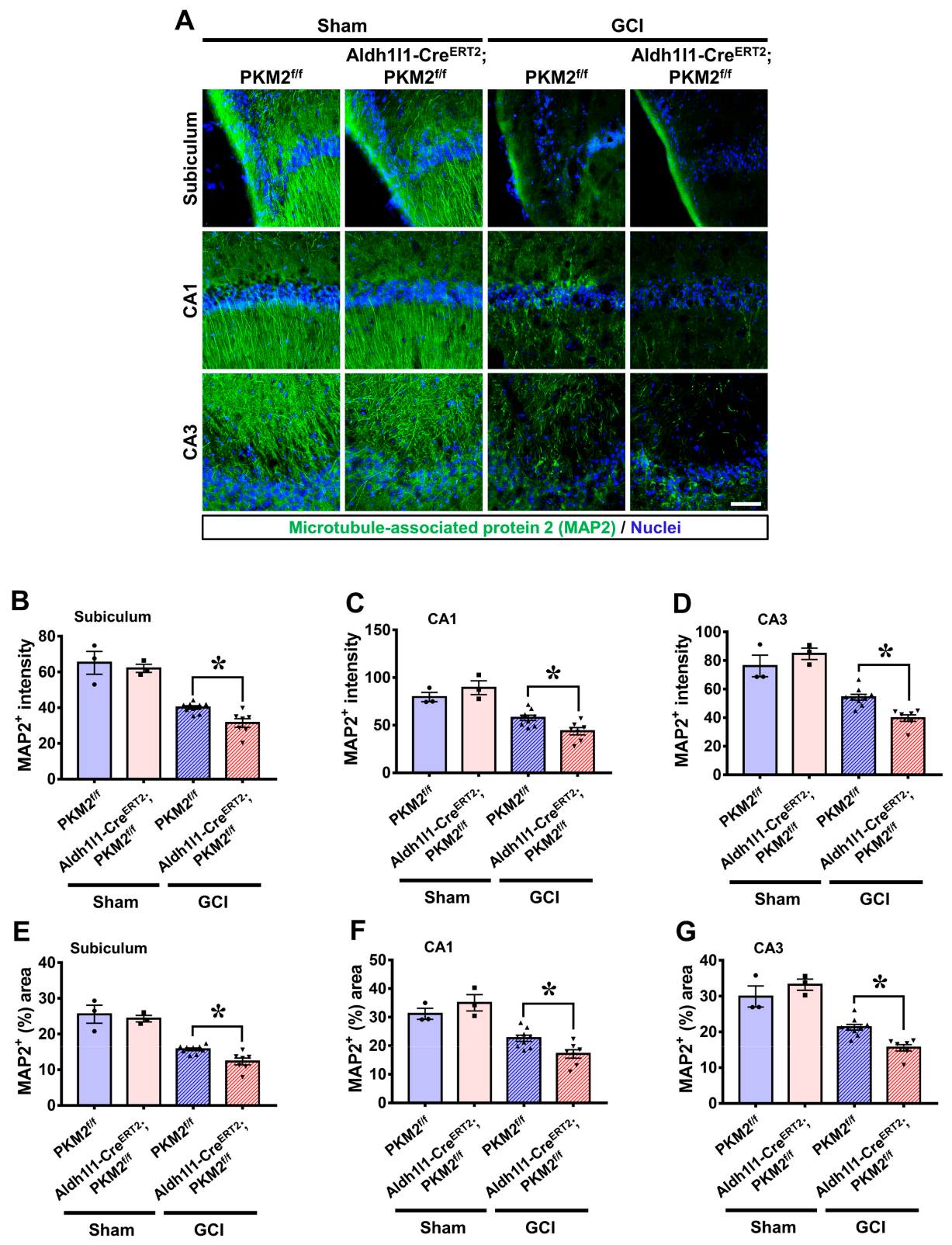
3.3. PKM2 Gene Deletion Increases Disturbance of Microtubule Structure after GCI
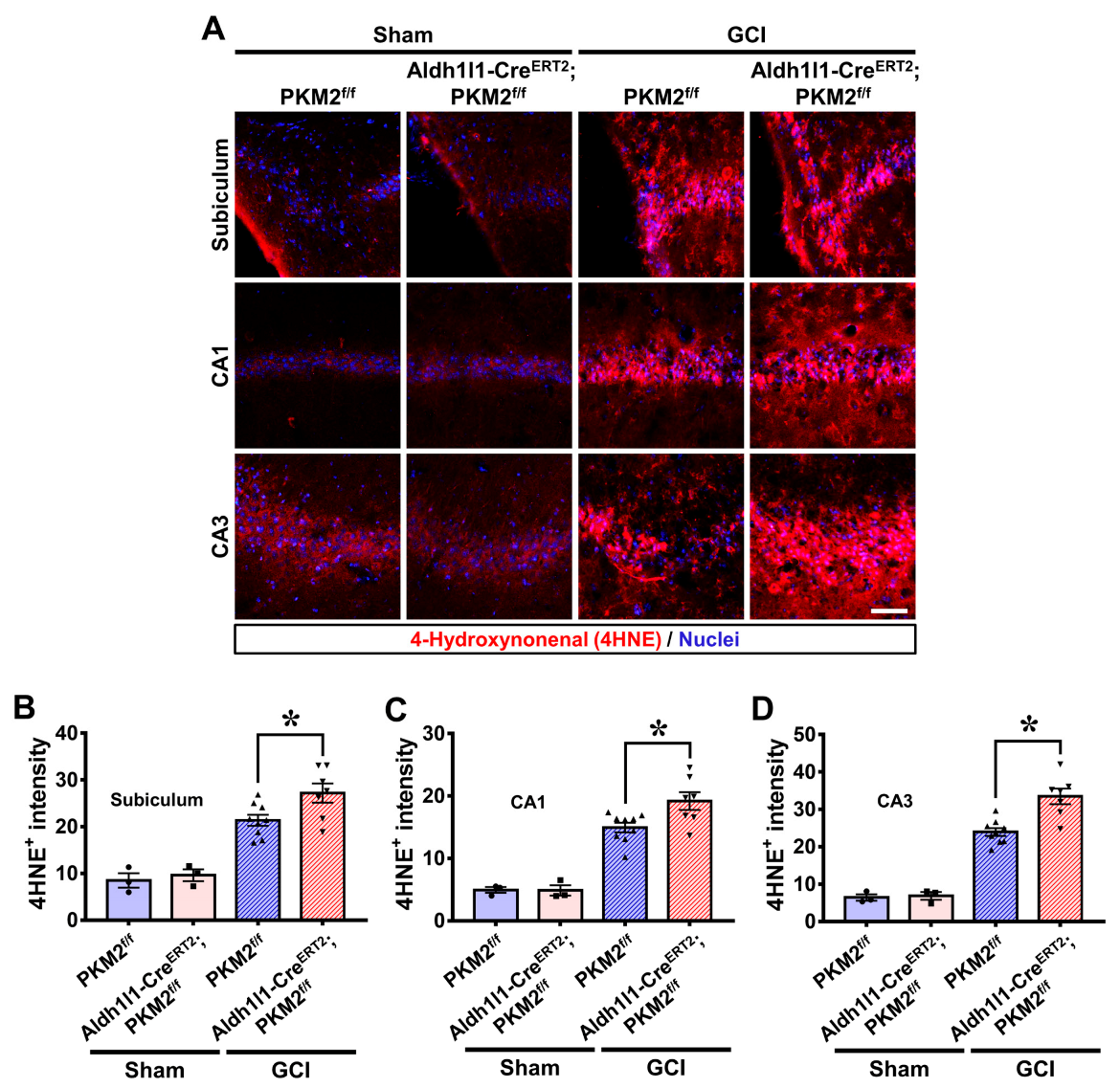
3.4. PKM2 Gene Deletion Increases Oxidative Damage after GCI
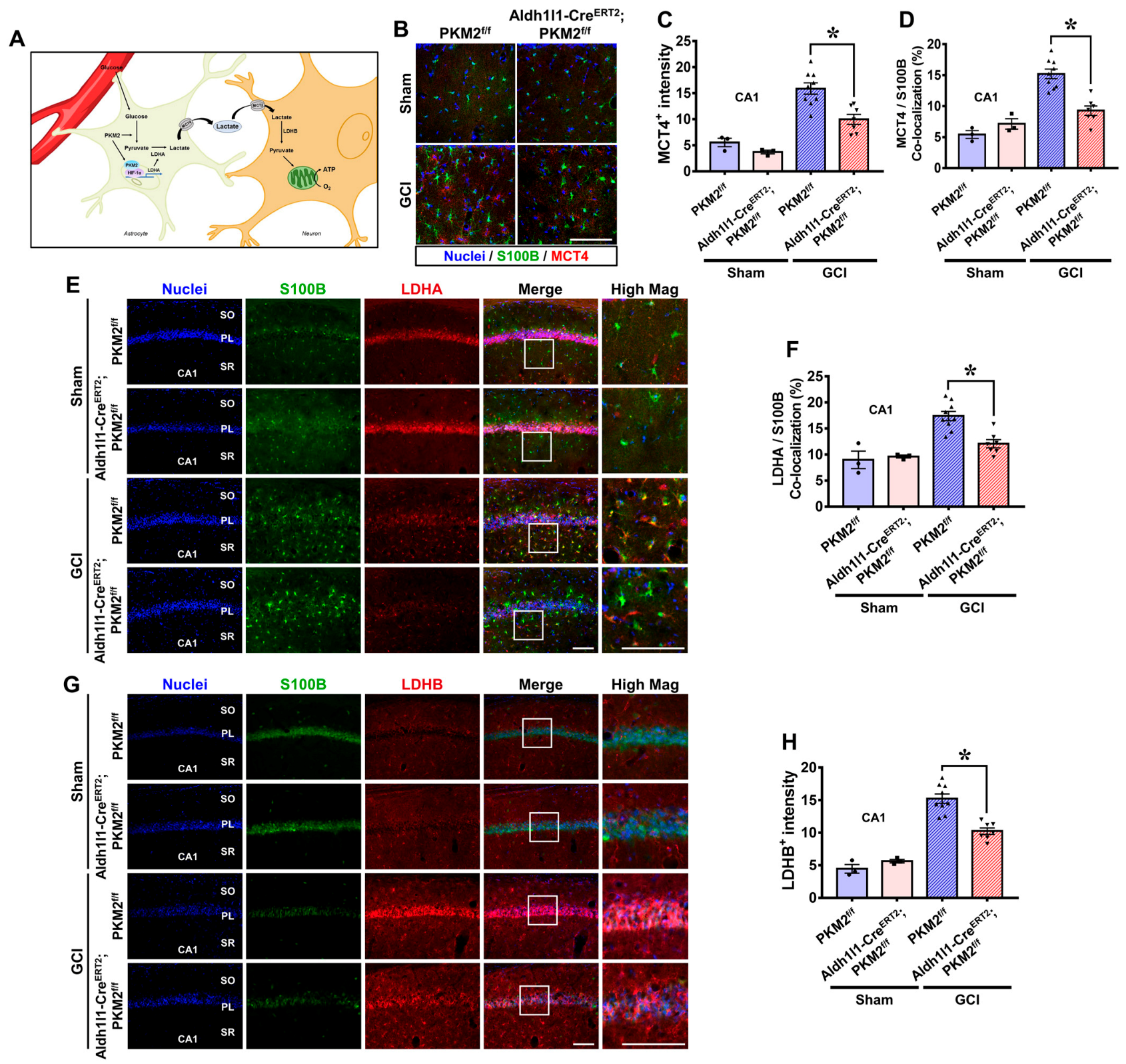
3.5. PKM2 Gene Deletion Reduces the Expression of MCT4, LDHA, and LDHB in the Hippocampus after GCI
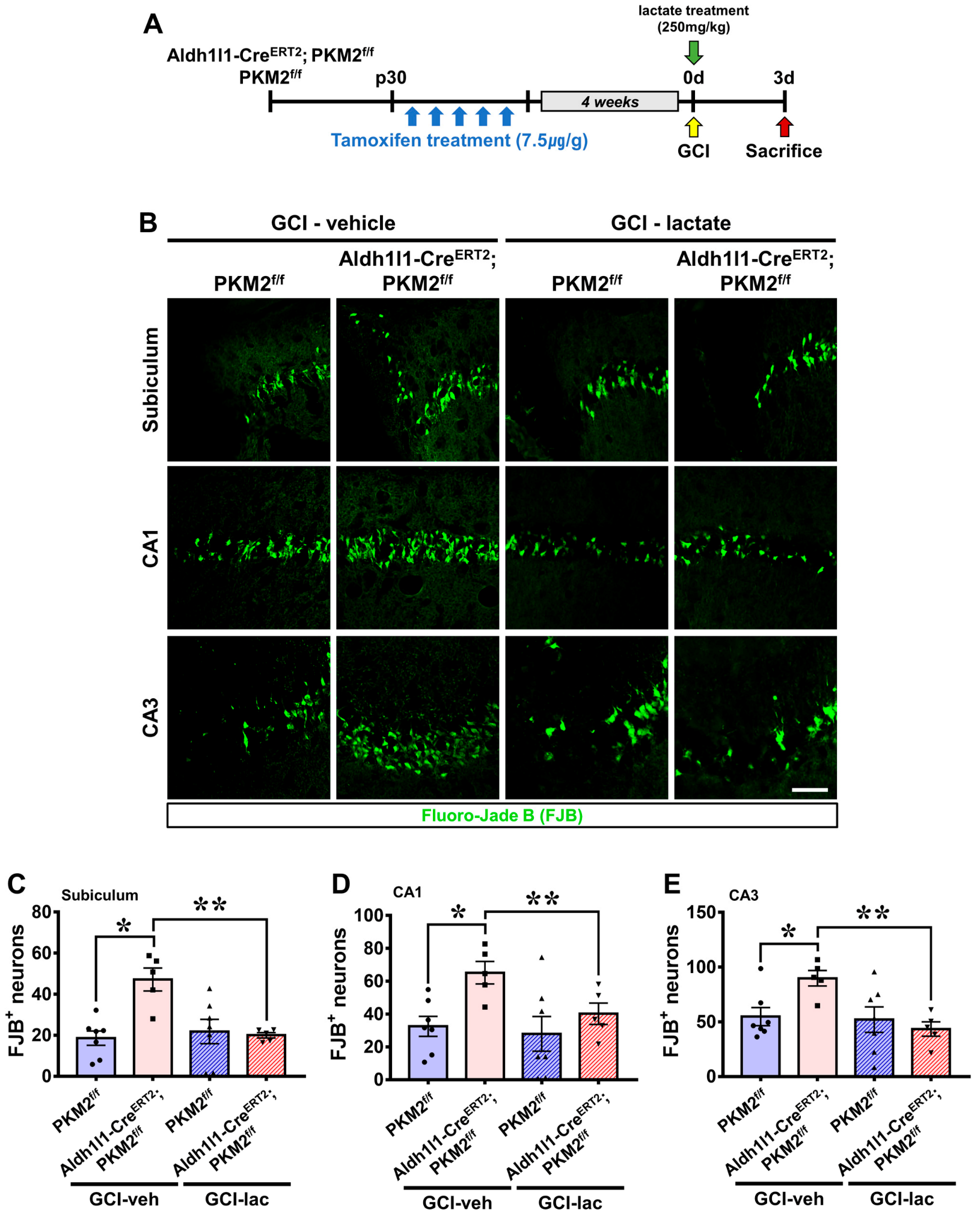
3.6. Exacerbation of Neuronal Damage by PKM2 Gene Deletion Is Restored by Lactate Supplementation after GCI
3.7. PKM2 Gene Deletion Allows Recovery of Live Neurons and Neurological and Cognitive Function by Lactate Supplementation after GCI
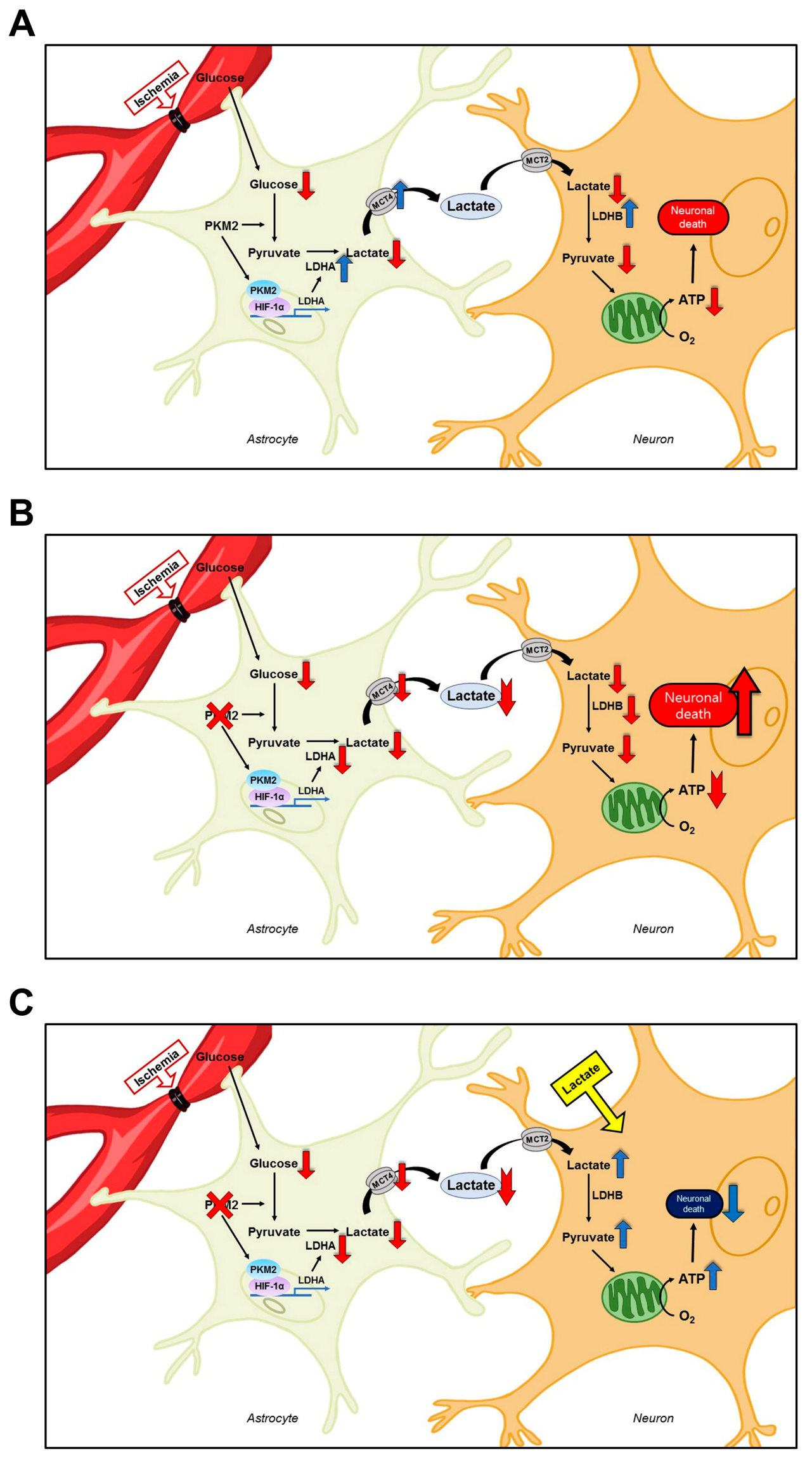
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yamagata, K. Lactate Supply from Astrocytes to Neurons and its Role in Ischemic Stroke-induced Neurodegeneration. Neuroscience 2022, 481, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Quintard, H.; Patet, C.; Zerlauth, J.B.; Suys, T.; Bouzat, P.; Pellerin, L.; Meuli, R.; Magistretti, P.J.; Oddo, M. Improvement of Neuroenergetics by Hypertonic Lactate Therapy in Patients with Traumatic Brain Injury Is Dependent on Baseline Cerebral Lactate/Pyruvate Ratio. J. Neurotrauma 2016, 33, 681–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, D.K.; Kho, A.R.; Lee, S.H.; Jeong, J.H.; Kang, B.S.; Kang, D.H.; Park, M.K.; Park, K.H.; Lim, M.S.; Choi, B.Y.; et al. Transient Receptor Potential Melastatin 2 (TRPM2) Inhibition by Antioxidant, N-Acetyl-l-Cysteine, Reduces Global Cerebral Ischemia-Induced Neuronal Death. Int. J. Mol. Sci. 2020, 21, 6026. [Google Scholar] [CrossRef]
- Mehta, S.L.; Pandi, G.; Vemuganti, R. Circular RNA Expression Profiles Alter Significantly in Mouse Brain After Transient Focal Ischemia. Stroke 2017, 48, 2541–2548. [Google Scholar] [CrossRef]
- Zhou, L.; Lin, Q.; Wang, P.; Yao, L.; Leong, K.; Tan, Z.; Huang, Z. Enhanced neuroprotective efficacy of bone marrow mesenchymal stem cells co-overexpressing BDNF and VEGF in a rat model of cardiac arrest-induced global cerebral ischemia. Cell Death Dis. 2017, 8, e2774. [Google Scholar] [CrossRef] [Green Version]
- Cao, B.Q.; Tan, F.; Zhan, J.; Lai, P.H. Mechanism underlying treatment of ischemic stroke using acupuncture: Transmission and regulation. Neural Regen. Res. 2021, 16, 944–954. [Google Scholar] [CrossRef]
- Israelsen, W.J.; Dayton, T.L.; Davidson, S.M.; Fiske, B.P.; Hosios, A.M.; Bellinger, G.; Li, J.; Yu, Y.; Sasaki, M.; Horner, J.W.; et al. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell 2013, 155, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Hyun, J.; Park, J.; Jung, S.; Oh, Y.; Kim, Y.; Ryu, S.H.; Kim, S.H.; Jeong, E.I.; Jo, D.G.; et al. Aberrant role of pyruvate kinase M2 in the regulation of gamma-secretase and memory deficits in Alzheimer’s disease. Cell Rep. 2021, 37, 110102. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Semenza, G.L. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol. Metab. 2012, 23, 560–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Lu, M.; Mei, M.; Wang, H.; Han, Z.; Chen, M.; Yao, H.; Song, N.; Ding, X.; Ding, J.; et al. Pyridoxine induces glutathione synthesis via PKM2-mediated Nrf2 transactivation and confers neuroprotection. Nat. Commun. 2020, 11, 941. [Google Scholar] [CrossRef] [Green Version]
- Christofk, H.R.; Vander Heiden, M.G.; Wu, N.; Asara, J.M.; Cantley, L.C. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 2008, 452, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.B.; Min, J.K.; Kim, J.G.; Cap, K.C.; Islam, R.; Hossain, A.J.; Dogsom, O.; Hamza, A.; Mahmud, S.; Choi, D.R.; et al. Multiple functions of pyruvate kinase M2 in various cell types. J. Cell. Physiol. 2022, 237, 128–148. [Google Scholar] [CrossRef] [PubMed]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Le, Z.; Yanxiang Guo, J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierre, K.; Pellerin, L. Monocarboxylate transporters in the central nervous system: Distribution, regulation and function. J. Neurochem. 2005, 94, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Vijay, N.; Morris, M.E. Role of monocarboxylate transporters in drug delivery to the brain. Curr. Pharm. Des. 2014, 20, 1487–1498. [Google Scholar] [CrossRef] [Green Version]
- Pellerin, L.; Pellegri, G.; Bittar, P.G.; Charnay, Y.; Bouras, C.; Martin, J.L.; Stella, N.; Magistretti, P.J. Evidence supporting the existence of an activity-dependent astrocyte-neuron lactate shuttle. Dev. Neurosci. 1998, 20, 291–299. [Google Scholar] [CrossRef]
- Murakami, R.; Chiba, Y.; Nishi, N.; Matsumoto, K.; Wakamatsu, K.; Yanase, K.; Uemura, N.; Nonaka, W.; Ueno, M. Immunoreactivity of receptor and transporters for lactate located in astrocytes and epithelial cells of choroid plexus of human brain. Neurosci. Lett. 2021, 741, 135479. [Google Scholar] [CrossRef]
- Philp, N.J.; Yoon, H.; Lombardi, L. Mouse MCT3 gene is expressed preferentially in retinal pigment and choroid plexus epithelia. Am. J. Physiol. Cell Physiol. 2001, 280, C1319–C1326. [Google Scholar] [CrossRef]
- Deitmer, J.W.; Theparambil, S.M.; Ruminot, I.; Noor, S.I.; Becker, H.M. Energy Dynamics in the Brain: Contributions of Astrocytes to Metabolism and pH Homeostasis. Front. Neurosci. 2019, 13, 1301. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Pellerin, L. Astrocytes Couple Synaptic Activity to Glucose Utilization in the Brain. News Physiol. Sci. 1999, 14, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Gladden, L.B. Lactate metabolism: A new paradigm for the third millennium. J. Physiol. 2004, 558, 5–30. [Google Scholar] [CrossRef] [PubMed]
- Schurr, A. Lactate: The ultimate cerebral oxidative energy substrate? J. Cereb. Blood Flow Metab. 2006, 26, 142–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uehara, T.; Sumiyoshi, T.; Itoh, H.; Kurata, K. Lactate production and neurotransmitters; evidence from microdialysis studies. Pharmacol. Biochem. Behav. 2008, 90, 273–281. [Google Scholar] [CrossRef]
- Suzuki, A.; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 2011, 144, 810–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oddo, M.; Levine, J.M.; Frangos, S.; Maloney-Wilensky, E.; Carrera, E.; Daniel, R.T.; Levivier, M.; Magistretti, P.J.; LeRoux, P.D. Brain lactate metabolism in humans with subarachnoid hemorrhage. Stroke 2012, 43, 1418–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holloway, R.; Zhou, Z.; Harvey, H.B.; Levasseur, J.E.; Rice, A.C.; Sun, D.; Hamm, R.J.; Bullock, M.R. Effect of lactate therapy upon cognitive deficits after traumatic brain injury in the rat. Acta Neurochir. 2007, 149, 919–927. [Google Scholar] [CrossRef]
- Ros, J.; Pecinska, N.; Alessandri, B.; Landolt, H.; Fillenz, M. Lactate reduces glutamate-induced neurotoxicity in rat cortex. J. Neurosci. Res. 2001, 66, 790–794. [Google Scholar] [CrossRef]
- Annoni, F.; Peluso, L.; Gouvêa Bogossian, E.; Creteur, J.; Zanier, E.R.; Taccone, F.S. Brain Protection after Anoxic Brain Injury: Is Lactate Supplementation Helpful? Cells 2021, 10, 1714. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. Osteoarthr. Cartil. 2012, 20, 256–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, B.S.; Choi, B.Y.; Kho, A.R.; Lee, S.H.; Hong, D.K.; Jeong, J.H.; Kang, D.H.; Park, M.K.; Suh, S.W. An Inhibitor of the Sodium-Hydrogen Exchanger-1 (NHE-1), Amiloride, Reduced Zinc Accumulation and Hippocampal Neuronal Death after Ischemia. Int. J. Mol. Sci. 2020, 21, 4232. [Google Scholar] [CrossRef]
- Kho, A.R.; Choi, B.Y.; Lee, S.H.; Hong, D.K.; Kang, B.S.; Lee, S.H.; Suh, S.W. Administration of an Acidic Sphingomyelinase (ASMase) Inhibitor, Imipramine, Reduces Hypoglycemia-Induced Hippocampal Neuronal Death. Cells 2022, 11, 667. [Google Scholar] [CrossRef]
- Tsenter, J.; Beni-Adani, L.; Assaf, Y.; Alexandrovich, A.G.; Trembovler, V.; Shohami, E. Dynamic changes in the recovery after traumatic brain injury in mice: Effect of injury severity on T2-weighted MRI abnormalities, and motor and cognitive functions. J. Neurotrauma 2008, 25, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Luh, C.; Gierth, K.; Timaru-Kast, R.; Engelhard, K.; Werner, C.; Thal, S.C. Influence of a brief episode of anesthesia during the induction of experimental brain trauma on secondary brain damage and inflammation. PLoS ONE 2011, 6, e19948. [Google Scholar] [CrossRef] [PubMed]
- Brayer, S.W.; Ketcham, S.; Zou, H.; Hurwitz, M.; Henderson, C.; Fuletra, J.; Kumar, K.; Skidmore, E.; Thiels, E.; Wagner, A.K. Developing a clinically relevant model of cognitive training after experimental traumatic brain injury. Neurorehabil. Neural Repair 2015, 29, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Bartsch, T.; Dohring, J.; Reuter, S.; Finke, C.; Rohr, A.; Brauer, H.; Deuschl, G.; Jansen, O. Selective neuronal vulnerability of human hippocampal CA1 neurons: Lesion evolution, temporal course, and pattern of hippocampal damage in diffusion-weighted MR imaging. J. Cereb. Blood Flow Metab. 2015, 35, 1836–1845. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Michaelis, E.K. Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2010, 2, 12. [Google Scholar] [CrossRef]
- Attwell, D.; Laughlin, S.B. An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 2001, 21, 1133–1145. [Google Scholar] [CrossRef]
- Gjedde, A.; Marrett, S.; Vafaee, M. Oxidative and nonoxidative metabolism of excited neurons and astrocytes. J. Cereb. Blood Flow Metab. 2002, 22, 1–14. [Google Scholar] [CrossRef]
- Rothman, D.L.; Behar, K.L.; Hyder, F.; Shulman, R.G. In vivo NMR studies of the glutamate neurotransmitter flux and neuroenergetics: Implications for brain function. Annu. Rev. Physiol. 2003, 65, 401–427. [Google Scholar] [CrossRef]
- Shulman, R.G.; Rothman, D.L.; Behar, K.L.; Hyder, F. Energetic basis of brain activity: Implications for neuroimaging. Trends Neurosci. 2004, 27, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Cater, H.L.; Benham, C.D.; Sundstrom, L.E. Neuroprotective role of monocarboxylate transport during glucose deprivation in slice cultures of rat hippocampus. J. Physiol. 2001, 531, 459–466. [Google Scholar] [CrossRef]
- Cater, H.L.; Chandratheva, A.; Benham, C.D.; Morrison, B., 3rd; Sundstrom, L.E. Lactate and glucose as energy substrates during, and after, oxygen deprivation in rat hippocampal acute and cultured slices. J. Neurochem. 2003, 87, 1381–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, S. Lactate Shuttles in Neuroenergetics-Homeostasis, Allostasis and Beyond. Front. Neurosci. 2017, 11, 43. [Google Scholar] [CrossRef] [Green Version]
- Park, M.K.; Choi, B.Y.; Kho, A.R.; Lee, S.H.; Hong, D.K.; Jeong, J.H.; Kang, D.H.; Kang, B.S.; Suh, S.W. Effects of Transient Receptor Potential Cation 5 (TRPC5) Inhibitor, NU6027, on Hippocampal Neuronal Death after Traumatic Brain Injury. Int. J. Mol. Sci. 2020, 21, 8256. [Google Scholar] [CrossRef] [PubMed]
- Dalleau, S.; Baradat, M.; Gueraud, F.; Huc, L. Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 2013, 20, 1615–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mali, V.R.; Palaniyandi, S.S. Regulation and therapeutic strategies of 4-hydroxy-2-nonenal metabolism in heart disease. Free Radic. Res. 2014, 48, 251–263. [Google Scholar] [CrossRef]
- Mihalas, B.P.; De Iuliis, G.N.; Redgrove, K.A.; McLaughlin, E.A.; Nixon, B. The lipid peroxidation product 4-hydroxynonenal contributes to oxidative stress-mediated deterioration of the ageing oocyte. Sci. Rep. 2017, 7, 6247. [Google Scholar] [CrossRef] [Green Version]
- Pillon, N.J.; Croze, M.L.; Vella, R.E.; Soulere, L.; Lagarde, M.; Soulage, C.O. The lipid peroxidation by-product 4-hydroxy-2-nonenal (4-HNE) induces insulin resistance in skeletal muscle through both carbonyl and oxidative stress. Endocrinology 2012, 153, 2099–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, J.S.; Li, L.P.; Zhu, X.M. Effects of moxibustion pretreating on SOD and MDA in the rat of global brain ischemia. J. Tradit. Chin. Med. 2008, 28, 289–292. [Google Scholar] [CrossRef]
- Kuhn, J.; Meissner, C.; Oehmichen, M. Microtubule-associated protein 2 (MAP2)--a promising approach to diagnosis of forensic types of hypoxia-ischemia. Acta Neuropathol. 2005, 110, 579–586. [Google Scholar] [CrossRef]
- Schurr, A.; Payne, R.S.; Tseng, M.T.; Miller, J.J.; Rigor, B.M. The glucose paradox in cerebral ischemia. New insights. Ann. New York Acad. Sci. 1999, 893, 386–390. [Google Scholar] [CrossRef]
- Genc, S.; Kurnaz, I.A.; Ozilgen, M. Astrocyte-neuron lactate shuttle may boost more ATP supply to the neuron under hypoxic conditions--in silico study supported by in vitro expression data. BMC Syst. Biol. 2011, 5, 162. [Google Scholar] [CrossRef] [Green Version]
- Lottes, R.G.; Newton, D.A.; Spyropoulos, D.D.; Baatz, J.E. Lactate as substrate for mitochondrial respiration in alveolar epithelial type II cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L953–L961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brisson, L.; Banski, P.; Sboarina, M.; Dethier, C.; Danhier, P.; Fontenille, M.J.; Van Hee, V.F.; Vazeille, T.; Tardy, M.; Falces, J.; et al. Lactate Dehydrogenase B Controls Lysosome Activity and Autophagy in Cancer. Cancer Cell 2016, 30, 418–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosienko, V.; Teschemacher, A.G.; Kasparov, S. Is L-lactate a novel signaling molecule in the brain? J. Cereb. Blood Flow Metab. 2015, 35, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lezi, E.; Swerdlow, R.H. Lactate’s effect on human neuroblastoma cell bioenergetic fluxes. Biochem. Pharmacol. 2016, 99, 88–100. [Google Scholar] [CrossRef] [Green Version]
- Won, S.J.; Jang, B.G.; Yoo, B.H.; Sohn, M.; Lee, M.W.; Choi, B.Y.; Kim, J.H.; Song, H.K.; Suh, S.W. Prevention of acute/severe hypoglycemia-induced neuron death by lactate administration. J. Cereb. Blood Flow Metab. 2012, 32, 1086–1096. [Google Scholar] [CrossRef] [Green Version]
- Horn, T.; Klein, J. Neuroprotective effects of lactate in brain ischemia: Dependence on anesthetic drugs. Neurochem. Int. 2013, 62, 251–257. [Google Scholar] [CrossRef]
- Roumes, H.; Dumont, U.; Sanchez, S.; Mazuel, L.; Blanc, J.; Raffard, G.; Chateil, J.F.; Pellerin, L.; Bouzier-Sore, A.K. Neuroprotective role of lactate in rat neonatal hypoxia-ischemia. J. Cereb. Blood Flow Metab. 2021, 41, 342–358. [Google Scholar] [CrossRef] [Green Version]
- Berthet, C.; Castillo, X.; Magistretti, P.J.; Hirt, L. New evidence of neuroprotection by lactate after transient focal cerebral ischaemia: Extended benefit after intracerebroventricular injection and efficacy of intravenous administration. Cerebrovasc. Dis. 2012, 34, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Schaar, K.L.; Brenneman, M.M.; Savitz, S.I. Functional assessments in the rodent stroke model. Exp. Transl. Stroke Med. 2010, 2, 13. [Google Scholar] [CrossRef] [Green Version]
- Vorhees, C.V.; Williams, M.T. Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 2006, 1, 848–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Choi, B.Y.; Kim, J.H.; Kho, A.R.; Sohn, M.; Song, H.K.; Choi, H.C.; Suh, S.W. Late treatment with choline alfoscerate (l-alpha glycerylphosphorylcholine, alpha-GPC) increases hippocampal neurogenesis and provides protection against seizure-induced neuronal death and cognitive impairment. Brain Res. 2017, 1654, 66–76. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, B.-S.; Choi, B.-Y.; Kho, A.-R.; Lee, S.-H.; Hong, D.-K.; Park, M.-K.; Lee, S.-H.; Lee, C.-J.; Yang, H.-W.; Woo, S.-Y.; et al. Effects of Pyruvate Kinase M2 (PKM2) Gene Deletion on Astrocyte-Specific Glycolysis and Global Cerebral Ischemia-Induced Neuronal Death. Antioxidants 2023, 12, 491. https://doi.org/10.3390/antiox12020491
Kang B-S, Choi B-Y, Kho A-R, Lee S-H, Hong D-K, Park M-K, Lee S-H, Lee C-J, Yang H-W, Woo S-Y, et al. Effects of Pyruvate Kinase M2 (PKM2) Gene Deletion on Astrocyte-Specific Glycolysis and Global Cerebral Ischemia-Induced Neuronal Death. Antioxidants. 2023; 12(2):491. https://doi.org/10.3390/antiox12020491
Chicago/Turabian StyleKang, Beom-Seok, Bo-Young Choi, A-Ra Kho, Song-Hee Lee, Dae-Ki Hong, Min-Kyu Park, Si-Hyun Lee, Chang-Juhn Lee, Hyeun-Wook Yang, Seo-Young Woo, and et al. 2023. "Effects of Pyruvate Kinase M2 (PKM2) Gene Deletion on Astrocyte-Specific Glycolysis and Global Cerebral Ischemia-Induced Neuronal Death" Antioxidants 12, no. 2: 491. https://doi.org/10.3390/antiox12020491